
A Comprehensive Review on Neuroendocrine Neoplasms: Presentation, Pathophysiology and Management
 , , ,
, , ,  , ,
, ,  and
and
Abstract
: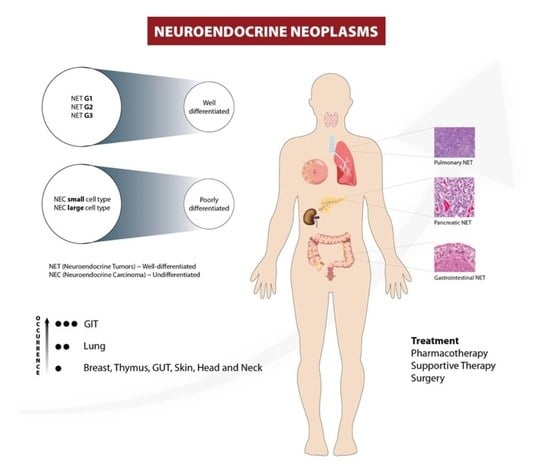
1. Introduction

2. Classification and Staging
3. Carcinoid Tumor NETs
4. Pulmonary NENs
5. Gastric Carcinoids: Gastric NETs Account for Approximately 7% of All Carcinoids and 1.8% of All Gastric Neoplasms [28] and Are further Divided into Four Subtypes
5.1. Duodenal NETs
5.2. Appendiceal NETs
5.3. Rectal NETs
6. Genitourinary NEN
7. Pancreatic NENs
7.1. Insulinomas
7.2. Glucagonomas
7.3. Gastrinoma
7.4. Somatostatinoma
7.5. VIPoma
7.6. Other Pancreatic NENs
8. Adrenocortical Tumors
9. Other Rare NENs
10. Genetics and Syndromes
10.1. Multiple Endocrine Neoplasia 2 (MEN 2)
10.2. Multiple Endocrine Neoplasia 4 (MEN 4)
10.3. Von Hippel–Lindau Syndrome
10.4. Neurofibromatosis 1 (NF 1)
10.5. Tuberous Sclerosis
10.6. Pheochromocytoma and Paraganglioma
11. Diagnosis
11.1. Imaging
11.1.1. CT
11.1.2. MRI
11.1.3. Ultrasonography and Related Applications
11.2. Tumor Markers
11.2.1. Specific Tumor Markers
11.2.2. Non-Specific Tumor Markers and Immunohistochemistry (IHC)

12. Management
12.1. Gastroenteropancreatic NETs (GEP NETs)
12.2. Pulmonary NETs
12.3. Evolving Therapy Targets for Other NETs and Future Perspectives
12.4. Supportive Management
13. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cives, M.; Strosberg, J.R. Gastroenteropancreatic Neuroendocrine Tumors. CA Cancer J. Clin. 2018, 68, 471–487. [Google Scholar] [CrossRef] [PubMed]
- Gaudenzi, G.; Carra, S.; Dicitore, A.; Cantone, M.C.; Persani, L.; Vitale, G. Fishing for Neuroendocrine Tumors. Endocr. Relat. Cancer 2020, 27, R163–R176. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.; Shen, C.; Halperin, D.M.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients with Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Oronsky, B.; Ma, P.C.; Morgensztern, D.; Carter, C.A. Nothing But NET: A Review of Neuroendocrine Tumors and Carcinomas. Neoplasia 2017, 19, 991–1002. [Google Scholar] [CrossRef]
- Yao, J.C.; Hassan, M.M.; Phan, A.T.; Dagohoy, C.G.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.N.; Rashid, A.; et al. One Hundred Years after “Carcinoid”: Epidemiology of and Prognostic Factors for Neuroendocrine Tumors in 35,825 Cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar] [CrossRef] [Green Version]
- Riihimäki, M.; Hemminki, A.; Sundquist, K.; Sundquist, J.; Hemminki, K. The Epidemiology of Metastases in Neuroendocrine Tumors. Int. J. Cancer 2016, 139, 2679–2686. [Google Scholar] [CrossRef]
- Klimstra, D.S.; Beltran, H.; Lilenbaum, R.; Lilenbaum, R.; Bergsland, E.K. The Spectrum of Neuroendocrine Tumors: Histologic Classification, Unique Features and Areas of Overlap. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Mete, O.; Uccella, S.; Basturk, O.; La Rosa, S.; Brosens, L.A.A.; Ezzat, S.; de Herder, W.W.; Klimstra, D.S.; Papotti, M.; et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr. Pathol. 2022, 33, 115–154. [Google Scholar] [CrossRef] [PubMed]
- Rothenstein, J.; Cleary, S.P.; Pond, G.R.; Dale, D.; Gallinger, S.; Moore, M.J.; Brierley, J.D.; Siu, L.L. Neuroendocrine Tumors of the Gastrointestinal Tract: A Decade of Experience at the Princess Margaret Hospital. Am. J. Clin. Oncol. 2008, 31, 64–70. [Google Scholar] [CrossRef]
- Silveira, F.; Basile, M.L.; Kuga, F.S.; Próspero, J.D.; Paes, R.A.P.; Paes, R.P.; Bernardi, F.D.C. Neuroendocrine Tumors: An Epidemiological Study of 250 Cases at a Tertiary Hospital. Rev. Assoc. Med. Bras. 2017, 63, 856–861. [Google Scholar] [CrossRef] [Green Version]
- Hitchcock, C.L.; Bland, K.I.; Laney, R.G.; Franzini, D.A.; Harris, B.A.; Copeland, E.M. Neuroendocrine (Merkel Cell) Carcinoma of the Skin. Its Natural History, Diagnosis, and Treatment. Ann. Surg. 1988, 207, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Baloch, Z.W.; LiVolsi, V.A. Neuroendocrine Tumors of the Thyroid Gland. Pathol. Patterns Rev. 2001, 115, S56–S67. [Google Scholar] [CrossRef] [PubMed]
- Berman, K.L.; Kirsch, J.; Bejarano, P.A.; Drexler, I.; Martinez, F. Primary Neuroendocrine Tumor of the Thymus: Radiological and Pathological Correlation. J. Radiol. Case Rep. 2020, 14, 1. [Google Scholar] [CrossRef]
- La Rosa, S.; Uccella, S. Classification of Neuroendocrine Neoplasms: Lights and Shadows. Rev. Endocr. Metab. Disord. 2020, 22, 527–538. [Google Scholar] [CrossRef]
- Vinik, A.I.; Chaya, C. Clinical Presentation and Diagnosis of Neuroendocrine Tumors. Hematol. Oncol. Clin. N. Am. 2016, 30, 21–48. [Google Scholar] [CrossRef] [PubMed]
- Legakis, I.; Legakis, I.; Legakis, I.; Saif, M.W.; Syrigos, K.N.; Syrigos, K. Therapeutic Challenges in Neuroendocrine Tumors. Anticancer Agents Med. Chem. 2017, 17, 902–919. [Google Scholar] [CrossRef]
- Amoroso, V.; Pavel, M.; Claps, M.; Roca, E.; Ravanelli, M.; Maroldi, R.; Öberg, K.; Berruti, A. IFN-α in Advanced Well-Differentiated Neuroendocrine Tumors: The Neglected Drug? Future Oncol. 2018, 14, 897–899. [Google Scholar] [CrossRef]
- Liu, I.H.; Kunz, P.L. Biologics in Gastrointestinal and Pancreatic Neuroendocrine Tumors. J. Gastrointest. Oncol. 2017, 8, 457. [Google Scholar] [CrossRef] [Green Version]
- Chow, Z.; Johnson, J.; Johnson, J.J.; Chauhan, A.; Izumi, T.; Cavnar, M.J.; Weiss, H.L.; Townsend, C.M.; Anthony, L.; Wasilchenko, C.; et al. PI3K/MTOR Dual Inhibitor PF-04691502 Is a Schedule-Dependent Radiosensitizer for Gastroenteropancreatic Neuroendocrine Tumors. Cells 2021, 10, 1261. [Google Scholar] [CrossRef]
- Fan, Y.; Ma, K.; Wang, C.; Ning, J.; Hu, Y.; Dong, D.; Dong, X.; Geng, Q.; Li, E.; Wu, Y. Prognostic Value of PD-L1 and PD-1 Expression in Pulmonary Neuroendocrine Tumors. OncoTargets Ther. 2016, 9, 6075–6082. [Google Scholar] [CrossRef] [Green Version]
- Kidd, M.; Modlin, I.M.; Bodei, L.; Drozdov, I. Decoding the Molecular and Mutational Ambiguities of Gastroenteropancreatic Neuroendocrine Neoplasm Pathobiology. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 131–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Hong, S.-M.; Ro, J.Y. Recent Updates on Grading and Classification of Neuroendocrine Tumors. Ann. Diagn. Pathol. 2017, 29, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Cancer.Net. Neuroendocrine Tumors: Grades. 2021. Available online: https://www.cancer.net/cancer-types/neuroendocrine-tumors/grades (accessed on 10 June 2023).
- Klimstra, D.S.; Modlin, I.; Coppola, D.; Lloyd, R.V.; Suster, S. The Pathologic Classification of Neuroendocrine Tumors: A Review of Nomenclature, Grading, and Staging Systems. Pancreas 2010, 39, 707–712. [Google Scholar] [CrossRef]
- Plöckinger, U.; Rindi, G.; Arnold, R.; Eriksson, B.; Krenning, E.P.; de Herder, W.W.; Goede, A.; Caplin, M.; Öberg, K.; Reubi, J.C. Guidelines for the Diagnosis and Treatment of Neuroendocrine Gastrointestinal Tumours. A Consensus Statement on Behalf of the European Neuroendocrine Tumour Society (ENETS). Neuroendocrinology 2004, 80, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th Edition of the AJCC Cancer Staging Manual and the Future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef]
- Jackson, A.S.; Rosenthal, A.; Rosenthal, A.; Cattoni, M.; Bograd, A.J.; Farivar, A.S.; Aye, R.W.; Vallières, E.; Louie, B.E. Staging System for Neuroendocrine Tumors of the Lung Needs to Incorporate Histologic Grade. Ann. Thorac. Surg. 2020, 109, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Pinchot, S.N.; Holen, K.; Sippel, R.S.; Chen, H. Carcinoid Tumors. Oncologist 2008, 13, 1255–1269. [Google Scholar] [CrossRef]
- McCormick, D. Carcinoid Tumors and Syndrome. Gastroenterol. Nurs. 2002, 25, 105–111. [Google Scholar] [CrossRef]
- Tempfer, C.B.; Tischoff, I.; Dogan, A.; Hilal, Z.; Schultheis, B.; Schultheis, B.; Kern, P.; Rezniczek, G.A. Neuroendocrine Carcinoma of the Cervix: A Systematic Review of the Literature. BMC Cancer 2018, 18, 530. [Google Scholar] [CrossRef] [Green Version]
- Belák, J.; Kudlác, M.; Kudlac, M.; Zak, V.; Cavarga, I.; Kocan, P.; Kocan, P.; Böör, A.; Stebnicky, M.; Somos, A.; et al. Surgical Management of Bronchopulmonary Carcinoid Tumors: Experience over 8 Years and Review of the Literature. Tumori J. 2010, 96, 84–89. [Google Scholar] [CrossRef]
- Hung, Y.P. Neuroendocrine Tumors of the Lung: Updates and Diagnostic Pitfalls. Surg. Pathol. Clin. 2019, 12, 1055–1071. [Google Scholar] [CrossRef]
- Gridelli, C.; Rossi, A.; Rossi, A.; Rossi, A.; Rossi, A.; Airoma, G.; Airoma, G.; Bianco, R.; Costanzo, R.; Daniele, B.; et al. Treatment of Pulmonary Neuroendocrine Tumours: State of the Art and Future Developments. Cancer Treat. Rev. 2013, 39, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, J.; Nicholson, A.G.; Ladas, G.P.; Goldstraw, P. Large Cell Neuroendocrine Carcinoma and Large Cell Carcinomas with Neuroendocrine Morphology of the Lung: Prognosis after Complete Resection and Systematic Nodal Dissection. Ann. Thorac. Surg. 2003, 75, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, B.I.; Kidd, M.; Chan, A.; Malfertheiner, M.V.; Modlin, I.M. Bronchopulmonary Neuroendocrine Tumors. Cancer 2008, 113, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Detterbeck, F.C. Management of Carcinoid Tumors. Ann. Thorac. Surg. 2010, 89, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Tabaksblat, E.M.; Langer, S.W.; Knigge, U.; Grønbæk, H.; Mortensen, J.; Petersen, R.H.; Federspiel, B.; Ladekarl, M. Diagnosis and Treatment of Bronchopulmonary Neuroendocrine Tumours: State of the Art. Acta Oncol. 2016, 55, 3–14. [Google Scholar] [CrossRef]
- Kim, J.Y.; Hong, S.-M. Recent Updates on Neuroendocrine Tumors from the Gastrointestinal and Pancreatobiliary Tracts. Arch. Pathol. Lab. Med. 2016, 140, 437–448. [Google Scholar] [CrossRef] [Green Version]
- Kwaan, M.R.; Goldberg, J.E.; Bleday, R. Rectal Carcinoid Tumors: Review of Results after Endoscopic and Surgical Therapy. Arch. Surg. 2008, 143, 471–475. [Google Scholar] [CrossRef]
- Witzigmann, H.; Loracher, C.; Geissler, F.; Wagner, T.; Tannapfel, A.; Uhlmann, D.; Caca, K.; Hauss, J.; Hehl, J. Neuroendocrine Tumours of the Duodenum. Langenbecks Arch. Surg. 2002, 386, 525–533. [Google Scholar] [CrossRef]
- Hoffmann, K.M.; Furukawa, M.; Jensen, R.T. Duodenal Neuroendocrine Tumors: Classification, Functional Syndromes, Diagnosis and Medical Treatment. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 675–697. [Google Scholar] [CrossRef]
- Rydzewska, G.; Fendler, W.; Sowa-Staszczak, A.; Stefańska, A. Gastrointestinal Neuroendocrine Tumors: Pathogenesis and Imaging. Nucl. Med. Rev. Cent. East. Eur. 2013, 16, 70–76. [Google Scholar]
- Thavaraputta, S.; Graham, S.; Rivas Mejia, A.M.; Lado-Abeal, J. Duodenal Somatostatinoma Presenting as Obstructive Jaundice with the Coexistence of a Gastrointestinal Stromal Tumour in Neurofibromatosis Type 1: A Case with Review of the Literature. BMJ Case Rep. 2019, 12, bcr-2018. [Google Scholar] [CrossRef] [PubMed]
- Cives, M.; Strosberg, J. Radionuclide Therapy for Neuroendocrine Tumors. Curr. Oncol. Rep. 2017, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Massironi, S.; Campana, D.; Partelli, S.; Panzuto, F.; Rossi, R.E.; Faggiano, A.; Brighi, N.; Falconi, M.; Rinzivillo, M.; Delle Fave, G.; et al. Heterogeneity of Duodenal Neuroendocrine Tumors: An Italian Multi-Center Experience. Ann. Surg. Oncol. 2018, 25, 3200–3206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonds, M.; Rocha, F.G.; Rocha, F.G.; Rocha, F.G. Neuroendocrine Tumors of the Pancreatobiliary and Gastrointestinal Tracts. Surg. Clin. N. Am. 2020, 100, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Stinner, B.; Rothmund, M. Neuroendocrine Tumours (Carcinoids) of the Appendix. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 729–738. [Google Scholar] [CrossRef]
- Robertson, R.G.; Geiger, W.J.; Davis, N.B. Carcinoid Tumors. Am. Fam. Physician 2006, 74, 429–434. [Google Scholar]
- Goto, K.; Kodama, T.; Matsuno, Y.; Yokose, T.; Asamura, H.; Kamiya, N.; Shimosato, Y.; Shimosato, Y. Clinicopathologic and DNA Cytometric Analysis of Carcinoid Tumors of the Thymus. Mod. Pathol. 2001, 14, 985–994. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.Y.; Ahmad, N.A. Rectal Carcinoids. Curr. Opin. Gastroenterol. 2006, 22, 529–535. [Google Scholar] [CrossRef]
- Yangong, H.; Shi, C.; Shahbaz, M.; Shahbaz, M.; Shahbaz, M.; Zhengchuan, N.; Wang, J.; Liang, B.; Ruliang, F.; Gao, H.; et al. Diagnosis and Treatment Experience of Rectal Carcinoid (a Report of 312 Cases). Int. J. Surg. 2014, 12, 408–411. [Google Scholar] [CrossRef] [Green Version]
- Northrop, J.A.; Lee, J.H. Large Bowel Carcinoid Tumors. Curr. Opin. Gastroenterol. 2007, 23, 74–78. [Google Scholar] [CrossRef] [PubMed]
- McDermott, F.D.; Heeney, A.; Courtney, D.; Mohan, H.M.; Winter, D.C. Rectal Carcinoids: A Systematic Review. Surg. Endosc. Other Interv. Tech. 2014, 28, 2020–2026. [Google Scholar] [CrossRef] [Green Version]
- Nesti, C.; Bräutigam, K.; Benavent, M.; Bernal, L.; Boharoon, H.; Botling, J.; Bouroumeau, A.; Brcic, I.; Brunner, M.; Cadiot, G.; et al. Hemicolectomy versus Appendectomy for Patients with Appendiceal Neuroendocrine Tumours 1–2 Cm in Size: A Retrospective, Europe-Wide, Pooled Cohort Study. Lancet Oncol. 2023, 24, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.E.; Lloyd, R.V.; Sippel, R.S.; Chen, H. Clinicopathologic Characteristics of Colonic Carcinoid Tumors. J. Surg. Res. 2013, 184, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehabeldin, A.N.; Ro, J.Y. Neuroendocrine Tumors of Genitourinary Tract: Recent Advances. Ann. Diagn. Pathol. 2019, 42, 48–58. [Google Scholar] [CrossRef]
- Fine, S.W. Neuroendocrine Lesions of the Genitourinary Tract. Adv. Anat. Pathol. 2007, 14, 286–296. [Google Scholar] [CrossRef]
- Teegavarapu, P.S.; Rao, P.; Matrana, M.R.; Cauley, D.H.; Wood, C.G.; Tannir, N.M. Neuroendocrine Tumors of the Kidney: A Single Institution Experience. Clin. Genitourin. Cancer 2014, 12, 422–427. [Google Scholar] [CrossRef] [Green Version]
- Howitt, B.E.; Kelly, P.J.; McCluggage, W.G.; McCluggage, W.G. Pathology of Neuroendocrine Tumours of the Female Genital Tract. Curr. Oncol. Rep. 2017, 19, 59. [Google Scholar] [CrossRef]
- Conner, M.G.; Richter, H.E.; Moran, C.A.; Hameed, A.; Albores-Saavedra, J. Small Cell Carcinoma of the Cervix: A Clinicopathologic and Immunohistochemical Study of 23 Cases. Ann. Diagn. Pathol. 2002, 6, 345–348. [Google Scholar] [CrossRef]
- Katabathina, V.S.; Vikram, R.; Vikram, R.; Olaoya, A.; Paspulati, R.M.; Nicolas, M.M.; Rao, P.; Zaheer, A.; Prasad, S.R. Neuroendocrine Neoplasms of the Genitourinary Tract in Adults: Cross-Sectional Imaging Spectrum. Abdom. Radiol. 2017, 42, 1472–1484. [Google Scholar] [CrossRef]
- Iyoda, A.; Azuma, Y.; Sano, A. Neuroendocrine Tumors of the Lung: Clinicopathological and Molecular Features. Surg. Today 2020, 50, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- Parbhu, S.K.; Adler, D.G. Pancreatic Neuroendocrine Tumors: Contemporary Diagnosis and Management. Hosp. Pract. 2016, 44, 109–119. [Google Scholar] [CrossRef]
- Townsend, C.M., Jr. Vasoactive Intestinal Peptide. In Sabiston Textbook of Surgery; Elsevier: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Prosapio, J.G.; Jialal, I.; Sankar, P. Physiology, Gastrin; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Gao, L.; Natov, N.S.; Natov, N.S.; Daly, K.P.; Masud, F.N.; Masud, F.; Chaudhry, S.; Sterling, M.J.; Sterling, M.J.; Saif, M.W. An Update on the Management of Pancreatic Neuroendocrine Tumors. Anticancer Drugs 2018, 29, 597–612. [Google Scholar] [CrossRef]
- Guilmette, J.; Nose, V. Neoplasms of the Neuroendocrine Pancreas: An Update in the Classification, Definition, and Molecular Genetic Advances. Adv. Anat. Pathol. 2019, 26, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Basturk, O.; Yang, Z.; Tang, L.H.; Hruban, R.H.; Adsay, N.V.; McCall, C.M.; Krasinskas, A.M.; Jang, K.-T.; Frankel, W.L.; Balci, S.; et al. The High-Grade (WHO G3) Pancreatic Neuroendocrine Tumor Category Is Morphologically and Biologically Heterogenous and Includes Both Well Differentiated and Poorly Differentiated Neoplasms. Am. J. Surg. Pathol. 2015, 39, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Pavel, M.; Öberg, K.; Falconi, M.; Krenning, E.P.; Sundin, A.; Perren, A.; Berruti, A. Gastroenteropancreatic Neuroendocrine Neoplasms: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2020, 31, 844–860. [Google Scholar] [CrossRef] [PubMed]
- Hijioka, S.; Hosoda, W.; Morizane, C.; Mizuno, N.; Hara, K.; Okusaka, T. The Diagnosis and Treatment of Pancreatic NEN-G3-A Focus on Clinicopathological Difference of NET-G3 and NEC G3. J. Pancreas 2018, 3, 346–353. [Google Scholar]
- Fang, J.M.; Fang, J.M.; Shi, J.; Shi, J. A Clinicopathologic and Molecular Update of Pancreatic Neuroendocrine Neoplasms with a Focus on the New World Health Organization Classification. Arch. Pathol. Lab. Med. 2019, 143, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Badarna, M.; Percik, R.; Aharon-Hananel, G.; Aharon-Hananel, G.; Uri, I.; Uri, I.; Tirosh, A.; Tirosh, A. Anatomic Site as Prognostic Marker of Pancreatic Neuroendocrine Tumors: A Cohort Study. Eur. J. Endocrinol. 2019, 181, 325–330. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Caplin, M.E.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; Pavel, M.E.; et al. 177Lu-Dotatate plus Long-Acting Octreotide versus High-dose Long-Acting Octreotide in Patients with Midgut Neuroendocrine Tumours (NETTER-1): Final Overall Survival and Long-Term Safety Results from an Open-Label, Randomised, Controlled, Phase 3 Trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef]
- Chivukula, S.V.; Tierney, J.F.; Hertl, M.; Poirier, J.; Keutgen, X.M. Operative Resection in Early Stage Pancreatic Neuroendocrine Tumors in the United States: Are We over- or Undertreating Patients? Surgery 2020, 167, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, F.; Anastasopoulou, C. Insulinoma; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rahman, S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Adegoke, E.O.; Rahman, M.A.; Rahman, M.A.; Rahman, M.A.; Rahman, A.; et al. Role of Insulin in Health and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef] [PubMed]
- Mathew, P.; Thoppil, D. Hypoglycemia; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Karanth, J.B.; Pai, V.; Maribashetti, K. Pancreatic Neuroendocrine Tumour—Insulinoma Masquerading as a Psychiatric Illness. Case Rep. 2022, 15, e249698. [Google Scholar] [CrossRef] [PubMed]
- Giannis, D.; Moris, D.; Karachaliou, G.S.; Tsilimigras, D.; Karaolanis, G.; Papalampros, A.; Felekouras, E. Insulinomas: From Diagnosis to Treatment. A Review of the Literature. J. Buon 2020, 25, 1302–1314. [Google Scholar]
- Liao, J.; Ding, F.; Luo, W.; Nie, X.; He, Y.; Li, G.-X. Using the Secretion Ratios of Insulin and C-Peptide During the 2-h Oral Glucose Tolerance Test to Diagnose Insulinoma. Dig. Dis. Sci. 2020, 66, 1533–1539. [Google Scholar] [CrossRef]
- Hirshberg, B.; Hirshberg, B.; Hirshberg, B.; Livi, A.; Bartlett, D.L.; Libutti, S.K.; Hr, A.; Doppman, J.L.; Skarulis, M.C.; Gorden, P.; et al. Forty-Eight-Hour Fast: The Diagnostic Test for Insulinoma. J. Clin. Endocrinol. Metab. 2000, 85, 3222–3226. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, H.; Li, F.; Fang, C.; Zhen, J.; He, Q.; Liu, M. Ectopic Insulinoma Diagnosed by 68Ga-Exendin-4 PET/CT. Medicine 2021, 100, e25076. [Google Scholar] [CrossRef]
- Aida, A.; Noto, H. Diagnosis and Treatment Course of Insulinoma Presenting as Hypoglycemia Unawareness Using a Factory-Calibrated Continuous Glucose Monitoring System. Am. J. Case Rep. 2022, 23, e936723. [Google Scholar] [CrossRef]
- John, A.M.; Schwartz, R.A. Glucagonoma Syndrome: A Review and Update on Treatment. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 2016–2022. [Google Scholar] [CrossRef]
- Rix, I.; Nexøe-Larsen, C.; Bergmann, N.C.; Lund, A.; Knop, F.K. Glucagon Physiology; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Toberer, F.; Hartschuh, W.; Wiedemeyer, K. Glucagonoma-Associated Necrolytic Migratory Erythema: The Broad Spectrum of the Clinical and Histopathological Findings and Clues to the Diagnosis. Am. J. Dermatopathol. 2019, 41, e29–e32. [Google Scholar] [CrossRef]
- Ferrara, G.; Ingordo, I.; Ingordo, V. Pseudoglucagonoma Syndrome: Description of an ‘Idiopatic’ Case. Australas. J. Dermatol. 2020, 61, e403–e405. [Google Scholar] [CrossRef]
- Jansen, T.J.P.; van Lith, S.A.M.; Boss, M.; Brom, M.; Joosten, L.; Béhé, M.; Buitinga, M.; Gotthardt, M. Exendin-4 Analogs in Insulinoma Theranostics. J. Label. Comp. Radiopharm. 2019, 62, 656–672. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.-F.; Zheng, S.-S. Glucagonoma Syndrome with Necrolytic Migratory Erythema as Initial Manifestation. Hepatobiliary Pancreat. Dis. Int. 2021, 20, 598–600. [Google Scholar] [CrossRef]
- Cao, X.; Wang, X.; Lu, Y.; Zhao, B.; Shi, J.; Guan, Q.; Zhang, X. Spleen-Preserving Distal Pancreatectomy and Lymphadenectomy for Glucagonoma Syndrome. Medicine 2019, 98, e17037. [Google Scholar] [CrossRef]
- Li, M.L.; Norton, J.A. Gastrinoma. Curr. Treat. Options Oncol. 2001, 2, 337–346. [Google Scholar] [CrossRef]
- Libutti, S.K.; Alexander, H.R. Gastrinoma: Sporadic and Familial Disease. Surg. Oncol. Clin. 2006, 15, 479–496. [Google Scholar] [CrossRef]
- Rafieian, S.; Vahedi, M.; Jahanbin, B.; Ghasemloee, A. Primary Thoracic Gastrinoma Causing Zollinger-Ellison Syndrome. Indian. J. Thorac. Cardiovasc. Surg. 2021, 37, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.E.; Elvevi, A.; Citterio, D.; Coppa, J.; Invernizzi, P.; Mazzaferro, V.; Massironi, S. Gastrinoma and Zollinger Ellison Syndrome: A Roadmap for the Management between New and Old Therapies. World J. Gastroenterol. 2021, 27, 5890–5907. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Igarashi, H.; Uehara, H.; Jensen, R.T. Pharmacotherapy of Zollinger–Ellison Syndrome. Expert. Opin. Pharmacother. 2013, 14, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goebel, S.U.; Peghini, P.L.; Goldsmith, P.K.; Spiegel, A.M.; Gibril, F.; Raffeld, M.; Jensen, R.T.; Serrano, J. Expression of the Calcium-Sensing Receptor in Gastrinomas. J. Clin. Endocrinol. Metab. 2000, 85, 4131–4137. [Google Scholar] [CrossRef]
- DiGregorio, N.; Sharma, S. Physiology, Secretin; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Guarnotta, V.; Martini, C.; Davì, M.V.; Pizza, G.; Colao, A.; Faggiano, A. The Zollinger-Ellison Syndrome: Is There a Role for Somatostatin Analogues in the Treatment of the Gastrinoma? Endocrine 2018, 60, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.A.; Foster, D.S.; Ito, T.; Jensen, R.T. Gastrinomas. Endocrinol. Metab. Clin. N. Am. 2018, 47, 577–601. [Google Scholar] [CrossRef]
- Sandru, F.; Carsote, M.; Valea, A.; Albu, S.; Petca, R.-C.; Dumitrascu, M. Somatostatinoma: Beyond Neurofibromatosis Type 1 (Review). Exp. Ther. Med. 2020, 20, 3383–3388. [Google Scholar] [CrossRef]
- Martin, S.; Fica, S.; Parfeni, O.; Popa, L.; Manuc, T.; Rizea, O.; Lupescu, I.; Gherghe, M.; Becheanu, G.; Croitoru, A. Somatostatinoma and Neurofibromatosis Type 1-A Case Report and Review of the Literature. Diagnostics 2020, 10, 620. [Google Scholar] [CrossRef]
- Ito, T.; Igarashi, H.; Jensen, R.T. Pancreatic Neuroendocrine Tumors: Clinical Features, Diagnosis and Medical Treatment: Advances. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 737–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandee, W.T.; Hofland, J.; de Herder, W.W. Vasoactive Intestinal Peptide Tumor (VIPoma); MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Krejs, G.J. Vipoma Syndrome. Am. J. Med. 1987, 82, 37–48. [Google Scholar] [CrossRef]
- Shrikhande, S.V.; Sirohi, B.; Goel, M.; Barreto, S.G. Pancreatic Neuroendocrine Tumors. Indian J. Gastroenterol. 2013, 32, 3–17. [Google Scholar] [CrossRef]
- Siddappa, P.K.; Vege, S.S. Vasoactive Intestinal Peptide–Secreting Tumors. Pancreas 2019, 48, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Brugel, M.; Walter, T.; Goichot, B.; Smith, D.; Lepage, C.; Cao, C.D.; Hautefeuille, V.; Rebours, V.; Cadiot, G.; de Mestier, L. Efficacy of Treatments for VIPoma: A GTE Multicentric Series. Pancreatology 2021, 21, 1531–1539. [Google Scholar] [CrossRef]
- Romano, L.; Giuliani, A.; Vicentini, V.; Schietroma, M.; Schietroma, M.; Schietroma, M.; Carlei, F.; Carlei, F.; Carlei, F. Basics for Surgeons about the Immunohistochemistry Role in Pancreatic NETs Diagnosis. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 2548–2553. [Google Scholar] [CrossRef]
- Young, K.; Iyer, R.; Morganstein, D.; Chau, I.; Cunningham, D.; Starling, N. Pancreatic Neuroendocrine Tumors: A Review. Future Oncol. 2015, 11, 853–864. [Google Scholar] [CrossRef]
- Yang, M.; Ke, N.-W.; Zhang, Y.; Tan, C.-L.; Tian, B.-L.; Liu, X.-B.; Huang, W.; Nunes, Q.; Sutton, R. Functional and Non-Functional Pancreatic Neuroendocrine Tumours: ENETS or AJCC TNM Staging System? Oncotarget 2017, 8, 82784–82795. [Google Scholar] [CrossRef] [PubMed]
- Erickson, L.A.; Rivera, M.; Zhang, J. Adrenocortical Carcinoma. Adv. Anat. Pathol. 2014, 21, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Shimizu, Y.; Uketa, S.; Utsunomiya, N.; Kida, K.; Ishihara, M.; Hashimoto, K.; Kanamaru, S. Primary Small Cell Neuroendocrine Carcinoma of Adrenal Gland. Int. Cancer Conf. J. 2019, 8, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Lee, K.S.; Kim, H.Y.; Kim, Y.K.; Kim, B.-T.; Chung, M.J.; Yi, C.A.; Kwon, G.Y. Integrated PET-CT for the Characterization of Adrenal Gland Lesions in Cancer Patients: Diagnostic Efficacy and Interpretation Pitfalls. RadioGraphics 2006, 26, 1811–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, M.W.; Hemal, A.K.; Allaf, M.E. International Consultation on Urological Diseases and European Association of Urology International Consultation on Minimally Invasive Surgery in Urology: Laparoscopic and Robotic Adrenalectomy. BJU Int. 2017, 119, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.B.; Kimura, K.; Chapurin, N.; Saab Chalhoub, M.; Mehrad, M.; Langerman, A.; Mannion, K.; Netterville, J.; Rohde, S.; Sinard, R.; et al. Neuroendocrine Carcinomas of the Head and Neck: A Small Case Series. Am. J. Otolaryngol. 2021, 42, 102992. [Google Scholar] [CrossRef] [PubMed]
- Ruffini, E.; Oliaro, A.; Novero, D.; Campisi, P.; Filosso, P.L. Neuroendocrine Tumors of the Thymus. Thorac. Surg. Clin. 2011, 21, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Moura, M.M.; Cabrera, R.A.; Esteves, S.; Cavaco, B.M.; Soares, P.; Leite, V. Correlation of Molecular Data with Histopathological and Clinical Features in a Series of 66 Patients with Medullary Thyroid Carcinoma. J. Endocrinol. Investig. 2021, 44, 1837–1846. [Google Scholar] [CrossRef]
- Gallo, M.; Campione, S.; Di Vito, V.; Fortunati, N.; Lo Calzo, F.; Messina, E.; Ruggeri, R.M.; Faggiano, A.; Colao, A.A.L. Primary Neuroendocrine Neoplasms of the Breast: Still Open Issues. Front. Endocrinol. 2021, 11, 610230. [Google Scholar] [CrossRef]
- Eftekhari, F.; Wallace, S.; Silva, E.G.; Lenzi, R. Merkel Cell Carcinoma of the Skin: Imaging and Clinical Features in 93 Cases. Br. J. Radiol. 1996, 69, 226–233. [Google Scholar] [CrossRef]
- Mafficini, A.; Scarpa, A. Genetics and Epigenetics of Gastroenteropancreatic Neuroendocrine Neoplasms. Endocr. Rev. 2019, 40, 506–536. [Google Scholar] [CrossRef]
- Lewis, M.A.; Lewis, M.A.; Lewis, M.A. Hereditary Syndromes in Neuroendocrine Tumors. Curr. Treat. Options Oncol. 2020, 21, 50. [Google Scholar] [CrossRef]
- Al-Salameh, A.; Al-Salameh, A.; Cadiot, G.; Calender, A.; Goudet, P.; Chanson, P. Clinical Aspects of Multiple Endocrine Neoplasia Type 1. Nat. Rev. Endocrinol. 2021, 17, 207–224. [Google Scholar] [CrossRef]
- Moline, J.; Eng, C. Multiple Endocrine Neoplasia Type 2: An Overview. Genet. Med. 2011, 13, 755–764. [Google Scholar] [CrossRef]
- Xie, R.; Fu, K.-I.; Fu, K.-I.; Chen, S.-M.; Tuo, B.; Wu, H. Neurofibromatosis Type 1-Associated Multiple Rectal Neuroendocrine Tumors: A Case Report and Review of the Literature. World J. Gastroenterol. 2018, 24, 3806–3812. [Google Scholar] [CrossRef]
- Jensen, R.T.; Berna, M.J.; Berna, M.J.; Berna, M.J.; Bingham, D.B.; Norton, J.A. Inherited Pancreatic Endocrine Tumor Syndromes: Advances in Molecular Pathogenesis, Diagnosis, Management, and Controversies. Cancer 2008, 113, 1807–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishbein, L. Pheochromocytoma and Paraganglioma: Genetics, Diagnosis, and Treatment. Hematol. Oncol. Clin. N. Am. 2016, 30, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Turaga, K.K.; Kvols, L.K. Recent Progress in the Understanding, Diagnosis, and Treatment of Gastroenteropancreatic Neuroendocrine Tumors. CA Cancer J. Clin. 2011, 61, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Al-Nahhas, A. Nuclear Medicine Imaging of Neuroendocrine Tumours. Clin. Med. 2012, 12, 377–380. [Google Scholar] [CrossRef] [Green Version]
- Rayamajhi, S.J.; Rayamajhi, S.J.; Rayamajhi, S.J.; Lee, J.E.; Mittal, B.R.; Jessop, A.; Chasen, B.; Bhosale, P.R. Cross Sectional and Nuclear Medicine Imaging of Pancreatic Insulinomas. Abdom. Radiol. 2017, 42, 531–543. [Google Scholar] [CrossRef]
- Andreassen, M.; Ilett, E.; Wiese, D.; Slater, E.P.; Klose, M.; Hansen, C.P.; Gercke, N.; Langer, S.W.; Kjaer, A.; Maurer, E.; et al. Surgical Management, Preoperative Tumor Localization, and Histopathology of 80 Patients Operated on for Insulinoma. J. Clin. Endocrinol. Metab. 2019, 104, 6129–6138. [Google Scholar] [CrossRef] [PubMed]
- Sundin, A.; Arnold, R.; Baudin, E.; Cwikla, J.B.; Eriksson, B.; Fanti, S.; Fazio, N.; Giammarile, F.; Hicks, R.J.; Kjaer, A.; et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: Radiological, Nuclear Medicine and Hybrid Imaging. Neuroendocrinology 2017, 105, 212–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niederle, B.; Pape, U.-F.; Costa, F.; Gross, D.; Kelestimur, F.; Knigge, U.; Öberg, K.; Pavel, M.; Perren, A.; Toumpanakis, C.; et al. ENETS Consensus Guidelines Update for Neuroendocrine Neoplasms of the Jejunum and Ileum. Neuroendocrinology 2016, 103, 125–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, M.; Decristoforo, C.; Kendler, D.; Dobrozemsky, G.; Heute, D.; Uprimny, C.; Kovacs, P.; Von Guggenberg, E.; Bale, R.; Virgolini, I.J. 68Ga-DOTA-Tyr3-Octreotide PET in Neuroendocrine Tumors: Comparison with Somatostatin Receptor Scintigraphy and CT. J. Nucl. Med. 2007, 48, 508–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Jensen, R.T. Molecular Imaging in Neuroendocrine Tumors. Curr. Opin. Endocrinol. Diabetes Obes. 2016, 24, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofland, J.; Kaltsas, G.; de Herder, W.W. Advances in the Diagnosis and Management of Well-Differentiated Neuroendocrine Neoplasms. Endocr. Rev. 2020, 41, 371–403. [Google Scholar] [CrossRef] [Green Version]
- Puli, S.R. Diagnostic Accuracy of Endoscopic Ultrasound in Pancreatic Neuroendocrine Tumors: A Systematic Review and Meta Analysis. World J. Gastroenterol. 2013, 19, 3678. [Google Scholar] [CrossRef]
- Perren, A.; Couvelard, A.; Scoazec, J.-Y.; Costa, F.; Borbath, I.; Delle Fave, G.; Gorbounova, V.; Gross, D.; Grossman, A.; Jensen, R.T.; et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: Pathology—Diagnosis and Prognostic Stratification. Neuroendocrinology 2017, 105, 196–200. [Google Scholar] [CrossRef]
- Hassan, S.A.; Palaskas, N.L.; Agha, A.M.; Iliescu, C.; Lopez-Mattei, J.; Chen, C.; Zheng, H.; Yusuf, S.W. Carcinoid Heart Disease: A Comprehensive Review. Curr. Cardiol. Rep. 2019, 21, 140. [Google Scholar] [CrossRef]
- Lamberts, S.W.J.; Hofland, L.J.; Nobels, F.R.E. Neuroendocrine Tumor Markers. Front. Neuroendocr. 2001, 22, 309–339. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, B.; Öberg, K.; Stridsberg, M. Tumor Markers in Neuroendocrine Tumors. Digestion 2000, 62, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Nobels, F.R.E.; Kwekkeboom, D.J.; Coopmans, W.; Schoenmakers, C.H.H.; Lindemans, J.; De Herder, W.W.; Krenning, E.P.; Bouillon, R.; Lamberts, S.W.J. Chromogranin A as Serum Marker for Neuroendocrine Neoplasia: Comparison with Neuron-Specific Enolase and the α-Subunit of Glycoprotein Hormones. J. Clin. Endocrinol. Metab. 1997, 82, 2622–2628. [Google Scholar] [CrossRef] [Green Version]
- Iacangelo, A.L.; Eiden, L.E. Chromogranin A: Current Status as a Precursor for Bioactive Peptides and a Granulogenic/Sorting Factor in the Regulated Secretory Pathway. Regul. Pept. 1995, 58, 65–88. [Google Scholar] [CrossRef] [Green Version]
- Taupenot, L.; Harper, K.L.; O’Connor, D.T. The Chromogranin–Secretogranin Family. N. Engl. J. Med. 2003, 348, 1134–1149. [Google Scholar] [CrossRef]
- Baudin, E.; Bidart, J.M.; Bachelot, A.; Ducreux, M.; Elias, D.; Ruffié, P.; Schlumberger, M. Impact of Chromogranin A Measurement in the Work-up of Neuroendocrine Tumors. Ann. Oncol. 2001, 12, S79–S82. [Google Scholar] [CrossRef]
- Hsiao, R.J.; Parmer, R.J.; Takiyyuddin, M.A.; O’Connor, D.T. Chromogranin A Storage and Secretion: Sensitivity and Specificity for the Diagnosis of Pheochromocytoma. Medicine 1991, 70, 33–45. [Google Scholar]
- Baudin, E.; Gigliotti, A.; Ducreux, M.; Ropers, J.; Comoy, E.; Sabourin, J.C.; Bidart, J.M.; Cailleux, A.F.; Bonacci, R.; Ruffié, P.; et al. Neuron-Specific Enolase and Chromogranin A as Markers of Neuroendocrine Tumours. Br. J. Cancer 1998, 78, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Janson, E.T.; Holmberg, L.; Stridsberg, M.; Eriksson, B.; Theodorsson, E.; Wilander, E.; Öberg, K. Carcinoid Tumors: Analysis of Prognostic Factors and Survival in 301 Patients from a Referral Center. Ann. Oncol. 1997, 8, 685–690. [Google Scholar] [CrossRef]
- Aparicio, T.; Ducreux, M.; Baudin, E.; Sabourin, J.-C.; De Baere, T.; Mitry, E.; Schlumberger, M.; Rougier, P. Antitumour Activity of Somatostatin Analogues in Progressive Metastatic Neuroendocrine Tumours. Eur. J. Cancer 2001, 37, 1014–1019. [Google Scholar] [CrossRef]
- Kimura, N.; Miura, W.; Noshiro, T.; Mizunashi, K.; Hanew, K.; Shimizu, K.; Watanabe, T.; Shibukawa, S.; Sohn, H.E.; Abe, K.; et al. Plasma Chromogranin A in Pheochromocytoma, Primary Hyperparathyroidism and Pituitary Adenoma in Comparison with Catecholamine, Parathyroid Hormone and Pituitary Hormones. Endocr. J. 1997, 44, 319–327. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, D.T.; Deftos, L.J. Secretion of Chromogranin A by Peptide-Producing Endocrine Neoplasms. N. Engl. J. Med. 1986, 314, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Drivsholm, L.; Paloheimo, L.I.; Østerlind, K. Chromogranin A, a Significant Prognostic Factor in Small Cell Lung Cancer. Br. J. Cancer 1999, 81, 667–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öberg, K. Carcinoid Tumors: Molecular Genetics, Tumor Biology, and Update of Diagnosis and Treatment. Curr. Opin. Oncol. 2002, 14, 38–45. [Google Scholar] [CrossRef]
- Stridsberg, M.; Eriksson, B.; Oberg, K.; Janson, E.T. A Comparison between Three Commercial Kits for Chromogranin A Measurements. J. Endocrinol. 2003, 177, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulkin, B.L.; Thompson, N.W.; Shapiro, B.; Francis, I.R.; Sisson, J.C. Pheochromocytomas: Imaging with 2-[Fluorine-18]Fluoro-2-Deoxy-d-Glucose PET. Radiology 1999, 212, 35–41. [Google Scholar] [CrossRef]
- Kaltsas, G.A.; Besser, G.M.; Grossman, A.B. The Diagnosis and Medical Management of Advanced Neuroendocrine Tumors. Endocr. Rev. 2004, 25, 458–511. [Google Scholar] [CrossRef]
- Kwekkeboom, D.J.; Kam, B.L.; van Essen, M.; Teunissen, J.J.M.; van Eijck, C.H.J.; Valkema, R.; de Jong, M.; de Herder, W.W.; Krenning, E.P. Somatostatin Receptor-Based Imaging and Therapy of Gastroenteropancreatic Neuroendocrine Tumors. Endocr. Relat. Cancer 2010, 17, R53–R73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Carbonero, R.; Rinke, A.; Valle, J.W.; Fazio, N.; Caplin, M.; Gorbounova, V.; O’Connor, J.; Eriksson, B.; Sorbye, H.; Kulke, M.; et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Neoplasms: Systemic Therapy—Chemotherapy. Neuroendocrinology 2017, 105, 281–294. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Sharma, P.; Singh, H.; Bal, C. PET/CT Imaging of Neuroendocrine Tumors with 68 Gallium-Labeled Somatostatin Analogues: An Overview and Single Institutional Experience from India. Indian J. Nucl. Med. 2014, 29, 2. [Google Scholar] [CrossRef]
- Hofman, M.S.; Hicks, R.J. Changing Paradigms with Molecular Imaging of Neuroendocrine Tumors. Discov. Med. 2012, 14, 71–81. [Google Scholar]
- Alevroudis, E.; Spei, M.-E.; Chatziioannou, S.N.; Tsoli, M.; Wallin, G.; Kaltsas, G.; Daskalakis, K. Clinical Utility of 18F-FDG PET in Neuroendocrine Tumors Prior to Peptide Receptor Radionuclide Therapy: A Systematic Review and Meta-Analysis. Cancers 2021, 13, 1813. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; Valle, J.W.; Eriksson, B.; Rinke, A.; Caplin, M.; Chen, J.; Costa, F.; Falkerby, J.; Fazio, N.; Gorbounova, V.; et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Neoplasms: Systemic Therapy—Biotherapy and Novel Targeted Agents. Neuroendocrinology 2017, 105, 266–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlman, H.; Kölby, L.; Lundell, L.; Olbe, L.; Wängberg, B.; Granérus, G.; Grimelius, L.; Nilsson, O. Clinical Management of Gastric Carcinoid Tumors. Digestion 1994, 55, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.T.; Wang, H.; Yao, J.C.; Lee, J.H.; Perrier, N.D.; Pisters, P.W.T.; Lee, J.E.; Evans, D.B. Carcinoid Tumors of the Duodenum. Surgery 2005, 138, 971–978. [Google Scholar] [CrossRef]
- Delle Fave, G.; Kwekkeboom, D.J.; Van Cutsem, E.; Rindi, G.; Kos-Kudla, B.; Knigge, U.; Sasano, H.; Tomassetti, P.; Salazar, R.; Ruszniewski, P. ENETS Consensus Guidelines for the Management of Patients with Gastroduodenal Neoplasms. Neuroendocrinology 2012, 95, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Grozinsky-Glasberg, S.; Kaltsas, G.; Gur, C.; Gal, E.; Thomas, D.; Fichman, S.; Alexandraki, K.; Barak, D.; Glaser, B.; Shimon, I.; et al. Long-Acting Somatostatin Analogues Are an Effective Treatment for Type 1 Gastric Carcinoid Tumours. Eur. J. Endocrinol. 2008, 159, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Sutton, R.; Doran, H.E.; Williams, E.M.I.; Vora, J.; Vinjamuri, S.; Evans, J.; Campbell, F.; Raraty, M.G.T.; Ghaneh, P.; Hartley, M.; et al. Surgery for Midgut Carcinoid. Endocr. Relat. Cancer 2003, 10, 469–481. [Google Scholar] [CrossRef]
- Landry, C.S.; Lin, H.Y.; Phan, A.; Charnsangavej, C.; Abdalla, E.K.; Aloia, T.; Nicolas Vauthey, J.; Katz, M.H.G.; Yao, J.C.; Fleming, J.B. Resection of At-Risk Mesenteric Lymph Nodes Is Associated with Improved Survival in Patients with Small Bowel Neuroendocrine Tumors. World J. Surg. 2013, 37, 1695–1700. [Google Scholar] [CrossRef] [Green Version]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of 177 Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, R.A.; Beyer, D.T.; Chauhan, A.; Boudreaux, J.P.; Wang, Y.-Z.; Woltering, E.A. The Role of Capecitabine/Temozolomide in Metastatic Neuroendocrine Tumors. Oncologist 2016, 21, 671–675. [Google Scholar] [CrossRef] [Green Version]
- Crespo, G.; Jiménez-Fonseca, P.; Custodio, A.; López, C.; Carmona-Bayonas, A.; Alonso, V.; Navarro, M.; Aller, J.; Sevilla, I.; Grande, E.; et al. Capecitabine and Temozolomide in Grade 1/2 Neuroendocrine Tumors: A Spanish Multicenter Experience. Future Oncol. 2017, 13, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Kvols, L.K.; Oberg, K.E.; O’Dorisio, T.M.; Mohideen, P.; de Herder, W.W.; Arnold, R.; Hu, K.; Zhang, Y.; Hughes, G.; Anthony, L.; et al. Pasireotide (SOM230) Shows Efficacy and Tolerability in the Treatment of Patients with Advanced Neuroendocrine Tumors Refractory or Resistant to Octreotide LAR: Results from a Phase II Study. Endocr. Relat. Cancer 2012, 19, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litvak, A.; Pietanza, M.C. Bronchial and Thymic Carcinoid Tumors. Hematol. Oncol. Clin. N. Am. 2016, 30, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Benson, A.B.; Huynh, L.; Duh, M.S.; Goldman, J.; Sahai, V.; Rademaker, A.W.; Kulke, M.H. Clinical Benefits of Above-Standard Dose of Octreotide LAR in Patients with Neuroendocrine Tumors for Control of Carcinoid Syndrome Symptoms: A Multicenter Retrospective Chart Review Study. Oncologist 2014, 19, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Ezziddin, S.; Logvinski, T.; Yong-Hing, C.; Ahmadzadehfar, H.; Fischer, H.-P.; Palmedo, H.; Bucerius, J.; Reinhardt, M.J.; Biersack, H.-J. Factors Predicting Tracer Uptake in Somatostatin Receptor and MIBG Scintigraphy of Metastatic Gastroenteropancreatic Neuroendocrine Tumors. J. Nucl. Med. 2006, 47, 223–233. [Google Scholar]
- Pape, U.-F.; Perren, A.; Niederle, B.; Gross, D.; Gress, T.; Costa, F.; Arnold, R.; Denecke, T.; Plöckinger, U.; Salazar, R.; et al. ENETS Consensus Guidelines for the Management of Patients with Neuroendocrine Neoplasms from the Jejuno-Ileum and the Appendix Including Goblet Cell Carcinomas. Neuroendocrinology 2012, 95, 135–156. [Google Scholar] [CrossRef]
- Castellano, D.; Bajetta, E.; Panneerselvam, A.; Saletan, S.; Kocha, W.; O’Dorisio, T.; Anthony, L.B.; Hobday, T. Everolimus Plus Octreotide Long-Acting Repeatable in Patients with Colorectal Neuroendocrine Tumors: A Subgroup Analysis of the Phase III RADIANT-2 Study. Oncologist 2013, 18, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.C.; Grant, C.S.; Salomao, D.R.; Fletcher, J.G.; Takahashi, N.; Fidler, J.L.; Levy, M.J.; Huebner, M. Small, Nonfunctioning, Asymptomatic Pancreatic Neuroendocrine Tumors (PNETs): Role for Nonoperative Management. Surgery 2012, 152, 965–974. [Google Scholar] [CrossRef]
- Crippa, S. Surgical Management of Insulinomas. Arch. Surg. 2012, 147, 261. [Google Scholar] [CrossRef] [Green Version]
- Tamburrino, D.; Spoletini, G.; Partelli, S.; Muffatti, F.; Adamenko, O.; Crippa, S.; Falconi, M. Surgical Management of Neuroendocrine Tumors. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 93–102. [Google Scholar] [CrossRef]
- Bartsch, D.K.; Fendrich, V.; Langer, P.; Celik, I.; Kann, P.H.; Rothmund, M. Outcome of Duodenopancreatic Resections in Patients with Multiple Endocrine Neoplasia Type 1. Ann. Surg. 2005, 242, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.L.; Falconi, M.; Waldmann, J.; Boninsegna, L.; Fendrich, V.; Goretzki, P.K.; Langer, P.; Kann, P.H.; Partelli, S.; Bartsch, D.K. Partial Pancreaticoduodenectomy Can Provide Cure for Duodenal Gastrinoma Associated with Multiple Endocrine Neoplasia Type 1. Ann. Surg. 2013, 257, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in Metastatic Enteropancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Kvols, L.K.; O’Connell, M.J.; Rubin, J. Treatment of Neuroendocrine Carcinomas with Combined Etoposide and Cisplatin. Evidence of Major Therapeutic Activity in the Anaplastic Variants of These Neoplasms. Cancer 1991, 68, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Kouvaraki, M.A.; Ajani, J.A.; Hoff, P.; Wolff, R.; Evans, D.B.; Lozano, R.; Yao, J.C. Fluorouracil, Doxorubicin, and Streptozocin in the Treatment of Patients with Locally Advanced and Metastatic Pancreatic Endocrine Carcinomas. J. Clin. Oncol. 2004, 22, 4762–4771. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Fine, R.L.; Choi, J.; Nasir, A.; Coppola, D.; Chen, D.-T.; Helm, J.; Kvols, L. First-Line Chemotherapy with Capecitabine and Temozolomide in Patients with Metastatic Pancreatic Endocrine Carcinomas. Cancer 2011, 117, 268–275. [Google Scholar] [CrossRef]
- Frilling, A.; Clift, A.K. Therapeutic Strategies for Neuroendocrine Liver Metastases. Cancer 2015, 121, 1172–1186. [Google Scholar] [CrossRef]
- Frilling, A.; Li, J.; Malamutmann, E.; Schmid, K.-W.; Bockisch, A.; Broelsch, C.E. Treatment of Liver Metastases from Neuroendocrine Tumours in Relation to the Extent of Hepatic Disease. Br. J. Surg. 2009, 96, 175–184. [Google Scholar] [CrossRef]
- Kianmanesh, R.; Sauvanet, A.; Hentic, O.; Couvelard, A.; Lévy, P.; Vilgrain, V.; Ruszniewski, P.; Belghiti, J. Two-Step Surgery for Synchronous Bilobar Liver Metastases from Digestive Endocrine Tumors: A Safe Approach for Radical Resection. Ann. Surg. 2008, 247, 659–665. [Google Scholar] [CrossRef]
- Schnitzbauer, A.A.; Lang, S.A.; Goessmann, H.; Nadalin, S.; Baumgart, J.; Farkas, S.A.; Fichtner-Feigl, S.; Lorf, T.; Goralcyk, A.; Hörbelt, R.; et al. Right Portal Vein Ligation Combined with In Situ Splitting Induces Rapid Left Lateral Liver Lobe Hypertrophy Enabling 2-Staged Extended Right Hepatic Resection in Small-for-Size Settings. Ann. Surg. 2012, 255, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.E.; Burroughs, A.K.; Caplin, M.E. Liver Transplantation for Unresectable Neuroendocrine Tumor Liver Metastases. Ann. Surg. Oncol. 2014, 21, 2398–2405. [Google Scholar] [CrossRef]
- Elias, D.; Lefevre, J.H.; Duvillard, P.; Goéré, D.; Dromain, C.; Dumont, F.; Baudin, E. Hepatic Metastases from Neuroendocrine Tumors with a “Thin Slice” Pathological Examination. Ann. Surg. 2010, 251, 307–310. [Google Scholar] [CrossRef]
- Gurusamy, K.S.; Ramamoorthy, R.; Sharma, D.; Davidson, B.R. Liver Resection versus Other Treatments for Neuroendocrine Tumours in Patients with Resectable Liver Metastases. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]
- Yao, J.C.; Pavel, M.; Lombard-Bohas, C.; Van Cutsem, E.; Voi, M.; Brandt, U.; He, W.; Chen, D.; Capdevila, J.; de Vries, E.G.E.; et al. Everolimus for the Treatment of Advanced Pancreatic Neuroendocrine Tumors: Overall Survival and Circulating Biomarkers from the Randomized, Phase III RADIANT-3 Study. J. Clin. Oncol. 2016, 34, 3906–3913. [Google Scholar] [CrossRef]
- Ferolla, P.; Brizzi, M.P.; Meyer, T.; Mansoor, W.; Mazieres, J.; Do Cao, C.; Léna, H.; Berruti, A.; Damiano, V.; Buikhuisen, W.; et al. Efficacy and Safety of Long-Acting Pasireotide or Everolimus Alone or in Combination in Patients with Advanced Carcinoids of the Lung and Thymus (LUNA): An Open-Label, Multicentre, Randomised, Phase 2 Trial. Lancet Oncol. 2017, 18, 1652–1664. [Google Scholar] [CrossRef]
- Imhof, A.; Brunner, P.; Marincek, N.; Briel, M.; Schindler, C.; Rasch, H.; Mäcke, H.R.; Rochlitz, C.; Müller-Brand, J.; Walter, M.A. Response, Survival, and Long-Term Toxicity after Therapy with the Radiolabeled Somatostatin Analogue [90Y-DOTA]-TOC in Metastasized Neuroendocrine Cancers. J. Clin. Oncol. 2011, 29, 2416–2423. [Google Scholar] [CrossRef]
- Della Corte, C.M.; Sen, T.; Gay, C.M.; Ramkumar, K.; Diao, L.; Cardnell, R.J.; Rodriguez, B.L.; Stewart, C.A.; Papadimitrakopoulou, V.A.; Gibson, L.; et al. STING Pathway Expression Identifies NSCLC with an Immune-Responsive Phenotype. J. Thorac. Oncol. 2020, 15, 777–791. [Google Scholar] [CrossRef]
- Andrini, E.; Marchese, P.V.; De Biase, D.; Mosconi, C.; Siepe, G.; Panzuto, F.; Ardizzoni, A.; Campana, D.; Lamberti, G. Large Cell Neuroendocrine Carcinoma of the Lung: Current Understanding and Challenges. J. Clin. Med. 2022, 11, 1461. [Google Scholar] [CrossRef]
- Caplin, M.E.; Baudin, E.; Ferolla, P.; Filosso, P.; Garcia-Yuste, M.; Lim, E.; Oberg, K.; Pelosi, G.; Perren, A.; Rossi, R.E.; et al. Pulmonary Neuroendocrine (Carcinoid) Tumors: European Neuroendocrine Tumor Society Expert Consensus and Recommendations for Best Practice for Typical and Atypical Pulmonary Carcinoids. Ann. Oncol. 2015, 26, 1604–1620. [Google Scholar] [CrossRef]
- Jensen, R.T. Carcinoid and Pancreatic Endocrine Tumors: Recent Advances in Molecular Pathogenesis, Localization, and Treatment. Curr. Opin. Oncol. 2000, 12, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Öberg, K.; Eriksson, B. The Role of Interferons in the Management of Carcinoid Tumours. Br. J. Haematol. 1991, 79, 74–77. [Google Scholar] [CrossRef]
- Kulke, M.H.; Lenz, H.-J.; Meropol, N.J.; Posey, J.; Ryan, D.P.; Picus, J.; Bergsland, E.; Stuart, K.; Tye, L.; Huang, X.; et al. Activity of Sunitinib in Patients with Advanced Neuroendocrine Tumors. J. Clin. Oncol. 2008, 26, 3403–3410. [Google Scholar] [CrossRef]
- Faivre, S.; Niccoli, P.; Castellano, D.; Valle, J.W.; Hammel, P.; Raoul, J.-L.; Vinik, A.; Van Cutsem, E.; Bang, Y.-J.; Lee, S.-H.; et al. Sunitinib in Pancreatic Neuroendocrine Tumors: Updated Progression-Free Survival and Final Overall Survival from a Phase III Randomized Study. Ann. Oncol. 2017, 28, 339–343. [Google Scholar] [CrossRef]
- Deguelte, S.; Perrier, M.; Hammoutene, C.; Cadiot, G.; Kianmanesh, R. Surgery and Perioperative Management in Small Intestinal Neuroendocrine Tumors. J. Clin. Med. 2020, 9, 2319. [Google Scholar] [CrossRef]
- Herrera-Martínez, A.D.; Hofland, J.; Hofland, L.J.; Brabander, T.; Eskens, F.A.L.M.; Gálvez Moreno, M.A.; Luque, R.M.; Castaño, J.P.; de Herder, W.W.; Feelders, R.A. Targeted Systemic Treatment of Neuroendocrine Tumors: Current Options and Future Perspectives. Drugs 2019, 79, 21–42. [Google Scholar] [CrossRef] [Green Version]
- Katz, S.C.; Donkor, C.; Glasgow, K.; Pillarisetty, V.G.; Gönen, M.; Espat, N.J.; Klimstra, D.S.; D’Angelica, M.I.; Allen, P.J.; Jarnagin, W.; et al. T Cell Infiltrate and Outcome Following Resection of Intermediate-Grade Primary Neuroendocrine Tumours and Liver Metastases. HPB 2010, 12, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Tarhini, A.; Lo, E.; Minor, D.R. Releasing the Brake on the Immune System: Ipilimumab in Melanoma and Other Tumors. Cancer Biother. Radiopharm. 2010, 25, 601–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reck, M.; Bondarenko, I.; Luft, A.; Serwatowski, P.; Barlesi, F.; Chacko, R.; Sebastian, M.; Lu, H.; Cuillerot, J.-M.; Lynch, T.J. Ipilimumab in Combination with Paclitaxel and Carboplatin as First-Line Therapy in Extensive-Disease-Small-Cell Lung Cancer: Results from a Randomized, Double-Blind, Multicenter Phase 2 Trial. Ann. Oncol. 2013, 24, 75–83. [Google Scholar] [CrossRef]
- Ahmed, M. Gastrointestinal Neuroendocrine Tumors in 2020. World J. Gastrointest. Oncol. 2020, 12, 791–807. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neuroendocrine Neoplasm | Classification Category | Mitotic Count | Ki-67 Index | Other Features |
|---|---|---|---|---|
| 1. Gastrointestinal tract and pancreato-biliary tract neoplasms | ||||
| a. Well-differentiated neuroendocrine neoplasm (NET) | Grade 1, NET | Less than 2 mitoses/2 mm2 | Less than 3% | - |
| Grade 2, NET | 2 to 20 mitoses/2 mm2 | 3 to 20% | - | |
| Grade 3, NET | More than 20 mitoses/2 mm2 | More than 20% | - | |
| b. Poorly differentiated neuroendocrine carcinoma (NEC) | Small cell NECs | More than 20 mitoses/2 mm2 | More than 20% (often more than 70%) | Small cell cytomorphology |
| Large cell NECs | More than 20 mitoses/2 mm2 | More than 20% (often more than 70%) | Large cell cytomorphology | |
| 2. Upper airway, digestive tract and salivary gland neoplasms (head and neck) | ||||
| a. Well-differentiated neuroendocrine neoplasm (NET) | Grade 1, NET | Less than 2 mitoses/2 mm2 | Less than 20% | No necrosis |
| Grade 2, NET | 2–10 mitoses/2 mm2 | Less than 20% | Necrosis | |
| Grade 3, NET | More than 10 mitoses/2 mm2 | More than 20% | - | |
| b. Poorly differentiated neuroendocrine carcinomas (NECs) | Small cell NECs | More than 10 mitoses/2 mm2 | More than 20% (often more than 70%) | Small cell cytomorphology |
| Large cell NECs | More than 10 mitoses/2 mm2 | More than 20% (often more than 55%) | Large cell cytomorphology | |
| 3. Thymus and lung neoplasms | ||||
| a. Well-differentiated neuroendocrine tumor (NET) | Grade 1, typical carcinoid/NET | Less than 2 mitoses/2 mm2 | - | No necrosis seen |
| Grade 2, atypical carcinoid/NET | 2 to 10 mitoses/2 mm2 | - | Necrosis (usually punctate) | |
| Carcinoids/NETs with mitotic counts and/or Ki-67 proliferation index on the higher side | More than 10 mitoses per 2 mm2 | More than 30% | Atypical carcinoid morphology | |
| b. Poorly-differentiated neuroendocrine carcinomas (NECs) | Small cell (lung) carcinomas | More than 10 mitoses/2 mm2 | - | Necrosis and small cell cytomorphology often seen |
| Large cell NECs | More than 10 mitoses/2 mm2 | - | Virtually almost always large cell cyto-morphology and necrosis seen | |
| 4. Thyroid neoplasms | ||||
| a. Medullary thyroid carcinomas (MTCs) | MTC, low-grade | Less than 5 mitoses/2 mm2 | - | No necrosis seen |
| MTC, high-grade | More than or equal to 5 mitoses/2 mm2 | More than or equal to 5% OR | Necrosis | |
| S.N. | Type of NEN | Clinical Diagnosis | Lab Diagnosis | Imaging | Treatment Options | Upcoming Evidence/Clinical Trials/Evolving Drug Targets |
|---|---|---|---|---|---|---|
| 1. | Pulmonary NENs | Central tumors: cough, hemoptysis and recurrent pneumonia. Peripheral tumors: non-specific symptoms and complications including, dysphagia, SVC syndrome and hoarseness of voice [4,30,34,35,36]. | High levels of ACTH or ADH AND 5 HT [153]. | CT scan: lesions around the central bronchi in typical carcinoids and SCLC, and peripheral lesions in atypical carcinoids and LCNEC. Calcifications present. Octreotide scan (111In-pentetreotide) detects both TC and AC [33]. | Medical: with somatostatin analogs(SSA), everolimus, PRRT, and chemotherapy [147,192,193,194,195]. Surgical: with bronchial resection plus LND [33]. | Identification of newer targets including the stimulator of interferon gene (STING) pathway [196], mutations in the epithelial growth factor receptor (EGFR), Phosphoinositol-3 kinase (PI3K), mTOR, VEGF and human epidermal growth factor receptor 2 (HER-2) [197]. |
| 2. | GIT NENs | Gastric: heartburn, peptic ulcers and diarrhea [28,29]. Duodenal: abdominal pain, diarrhea, gastrointestinal bleeding and jaundice [43]. Appendiceal: abdominal pain [46]. Rectal: abdominal pain, hematochezia, change in bowel movements and pruritis [49,50]. | Gastrin/somatostatin/histamine/peptide YY positive [153]. | Endoscopy with biopsy and endoscopic ultrasound are required for diagnosis and staging. CT scan and MRI are performed to identify distant metastasis [208]. | Medical: with SSA, PRRTs, everolimus, IFN alpha chemotherapy [131,157,161,167,168,169]. Surgical: with bowel resection + LND, hemicolectomy if ileal involvement [165,166]. | Currently, registered clinical trials are aimed at understanding the efficacy of already available treatments in different combinations and comparisons between them, for instance, PRRT vs. temozolomide/capecitabine or everolimus, or sunitinib. Sequential treatments are also being compared [202]. |
| 3. | Genitourinary NENs | Females: abnormal uterine bleeding is seen in cervical and endometrial tumors [57,58,59,60,61]. Males: urinary frequency, urgency and hematuria in prostate tumors. Testicular mass with or without pain. Bladder tumors: cause hematuria or urinary obstruction [55,56,60]. Renal NENs: Flank pain and hematuria [56,57]. | CgA or Synaptophysin or NSE positive [153]. | CT scan, MRI and octreotide scan are required for diagnosis and surveillance of lesions. FDG-PET is useful for staging [61]. | Medical: unresectable tumors are treated with radiotherapy and platinum-based chemotherapy. Surgical: for resectable tumors, oncological resection with neoadjuvant chemotherapy [56,57,60,61]. | A new combination of chemotherapy agents, BXCL701-Pembrolizumab, is being studied for efficacy in small cell NETs of prostate and adenocarcinoma phenotype (phase Ib/II—NCT03910660) [204]. |
| 4. | Pancreatic NENs | Insulinomas: tremors, palpitations, diaphoresis, syncope, confusion, anxiety, visual changes and coma [62,76]. Glucagonomas: migratory erythema diarrhea, weight loss, diabetes mellitus, DVT, anemia, and stomatitis [83,85]. Gastrinoma: Heartburn, abdominal pain, diarrhea and weight loss. Strictures, bleeding and perforation [93]. Somatostatinomas: Weight loss, abdominal pain, diarrhea or statorrhea, diabetes mellitus and cholelithiasis [100]. VIPomas: Watery diarrhea, hypocalcemia, flushing and glucose intolerance [103]. | Insulin levels in the blood must be greater than or equal to 36 pmol/L [78]. Glucagon level of more than 500 to 1000 pg/mL (normal 50 to 150 pg/mL) [62]. Fasting serum gastrin level of 1000 pg/mL [93]. A fasting plasma hormone concentration of more than 3-fold the normal [62]. VIP elevations (>200 pg/dL) are present [104]. | Demarcated hyper or hypovascular homogenous tumor on CT scan and endoscopic ultrasound [70]. | Medical: with SSA, everolimus, sunitinib and chemotherapy [157,181,184]. Surgical: G1 via pancreaticoduodenectomy, pancreatectomy + splenectomy pr debulking surgery [176,177,178]. | Research is being performed to determine the efficacy of 177Lu-DOTATOC (Lutathera), a new therapeutic agent for PRRT in gastroenteropancreatic NETs. Various phase 2 and phase 3 clinical trials for different combination therapies for pancreatic NETs are also in place [204]. The presence of tumor-infiltrating lymphocytes (TILs) in pancreatic NETs is thought to be a predictor of survival; hence, modulating the TIL density may be another treatment option [204]. |
| 5. | Rare NENs | Head and neck NENs: dysphagia, hoarseness of voice and epistaxis [114]. Thymus NENs: cough, dyspnea, chest pain, weight loss and chronic fever [116]. Thyroid NENs: thyroid nodule and cervical lymphadenopathy [117]. Breast NENs: solitary breast nodule, axillary lymphadenopathy, nipple discharge and retraction [118]. Skin NENs: Warts or blister [119]. | CgA or Synaptophysin or NSE positive [153]. | CT scan and MRI help identify adrenal lesions and FDG-PET scan distinguishes malignant from benign lesions. | Based on the site of involvement the tumor is managed and some tumors can also present as part of genetic syndromes and are treated with them. | Newer targets: programmed death ligand-1 (PDL1) expression and cytotoxic T lymphocyte antigen-4 are being reported in SCLC and melanoma [20,205,206,207]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sultana, Q.; Kar, J.; Verma, A.; Sanghvi, S.; Kaka, N.; Patel, N.; Sethi, Y.; Chopra, H.; Kamal, M.A.; Greig, N.H. A Comprehensive Review on Neuroendocrine Neoplasms: Presentation, Pathophysiology and Management. J. Clin. Med. 2023, 12, 5138. https://doi.org/10.3390/jcm12155138
Sultana Q, Kar J, Verma A, Sanghvi S, Kaka N, Patel N, Sethi Y, Chopra H, Kamal MA, Greig NH. A Comprehensive Review on Neuroendocrine Neoplasms: Presentation, Pathophysiology and Management. Journal of Clinical Medicine. 2023; 12(15):5138. https://doi.org/10.3390/jcm12155138
Chicago/Turabian StyleSultana, Qamar, Jill Kar, Amogh Verma, Shreya Sanghvi, Nirja Kaka, Neil Patel, Yashendra Sethi, Hitesh Chopra, Mohammad Amjad Kamal, and Nigel H. Greig. 2023. "A Comprehensive Review on Neuroendocrine Neoplasms: Presentation, Pathophysiology and Management" Journal of Clinical Medicine 12, no. 15: 5138. https://doi.org/10.3390/jcm12155138