
Over-Mutated Mitochondrial, Lysosomal and TFEB-Regulated Genes in Parkinson’s Disease

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Controls
2.2. Whole-Exome Sequencing
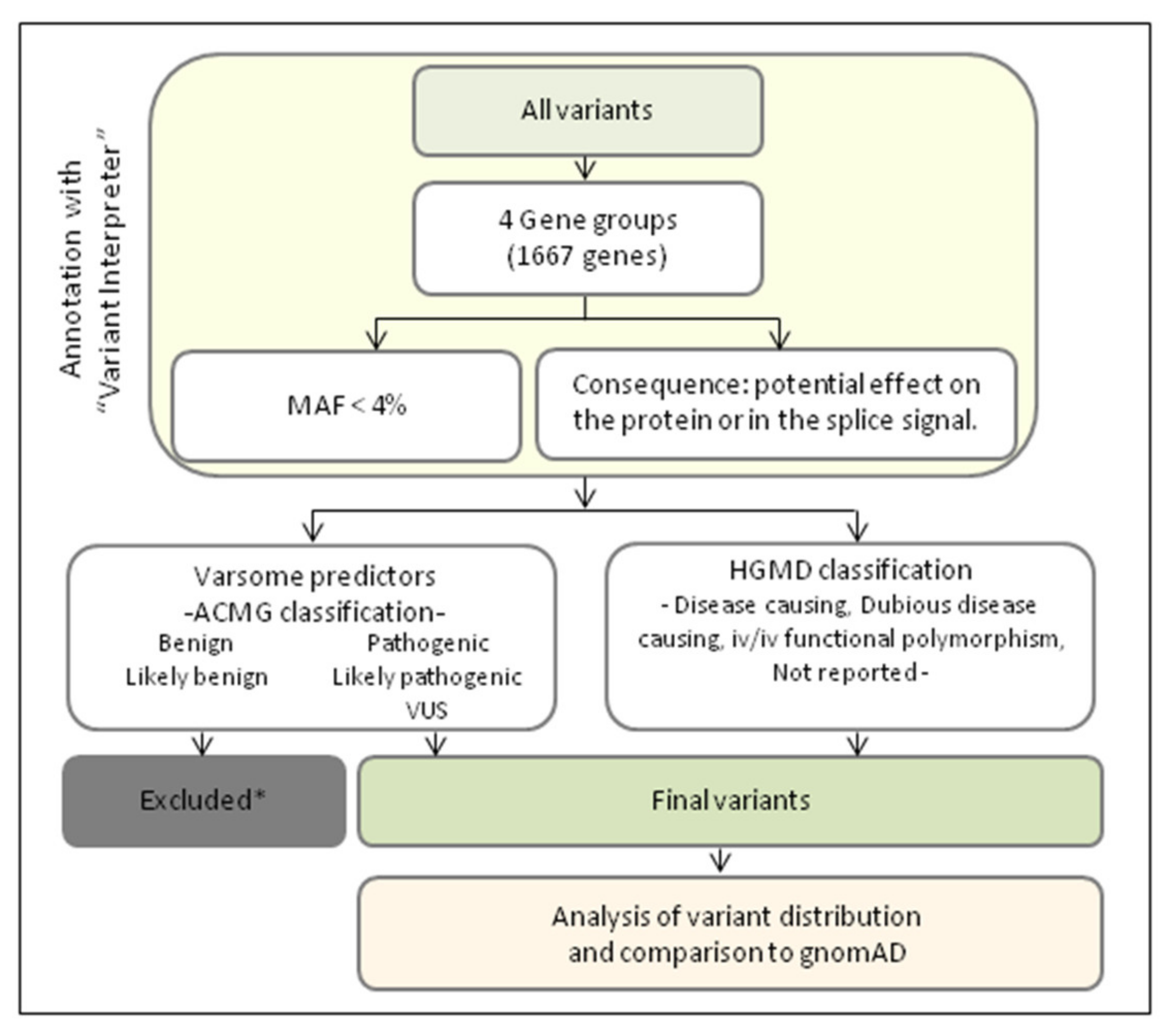
2.3. Genetic Data Analyses
2.4. Statistical Analysis
3. Results
3.1. Identification of Mutations in PD-Associated Genes
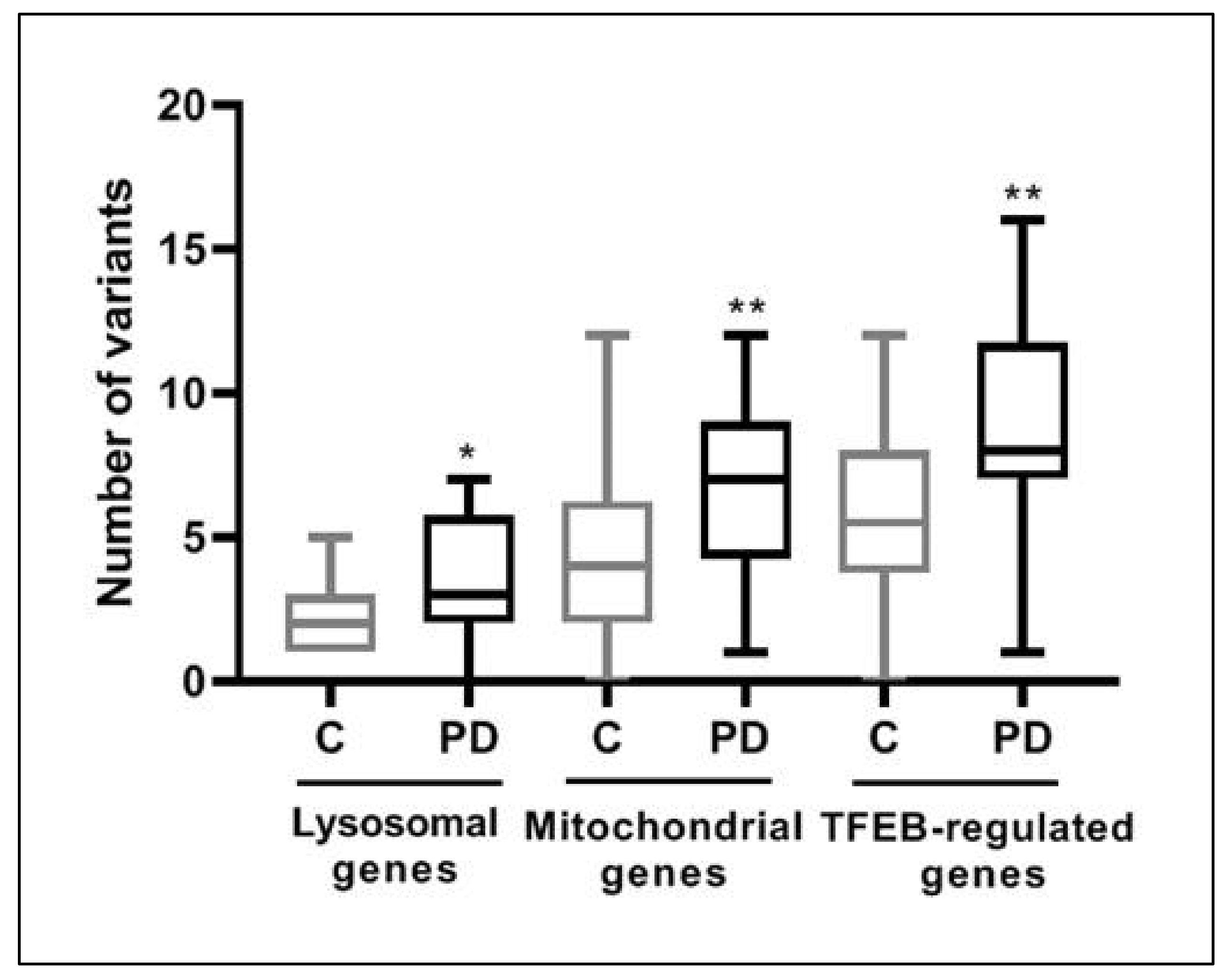
3.2. Over-Mutation in Lysosomal, Mitochondrial and TFEB-Regulated Genes in PD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic Perspective on the Role of the Autophagy-Lysosome Pathway in Parkinson Disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.G.; Reed, X.; Singleton, A.B. Genetics in Parkinson Disease: Mendelian versus Non-Mendelian Inheritance. J. Neurochem. 2016, 139, 59–74. [Google Scholar] [CrossRef]
- Billingsley, K.J.; Bandres-Ciga, S.; Saez-Atienzar, S.; Singleton, A.B. Genetic Risk Factors in Parkinson’s Disease. Cell Tissue Res. 2018, 373, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Ysselstein, D.; Shulman, J.M.; Krainc, D. Emerging Links between Pediatric Lysosomal Storage Diseases and Adult Parkinsonism. Mov. Disord. 2019, 34, 614–624. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef] [Green Version]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; Nalls, M.A.; Plagnol, V.; et al. Excessive Burden of Lysosomal Storage Disorder Gene Variants in Parkinson’s Disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef]
- Hopfner, F.; Mueller, S.H.; Szymczak, S.; Junge, O.; Tittmann, L.; May, S.; Lohmann, K.; Grallert, H.; Lieb, W.; Strauch, K.; et al. Rare Variants in Specific Lysosomal Genes Are Associated with Parkinson’s Disease. Mov. Disord. 2020, 35, 1245–1248. [Google Scholar] [CrossRef]
- Koziorowski, D.; Figura, M.; Milanowski, Ł.M.; Szlufik, S.; Alster, P.; Madetko, N.; Friedman, A. Mechanisms of Neurodegeneration in Various Forms of Parkinsonism-Similarities and Differences. Cells 2021, 10, 656. [Google Scholar] [CrossRef]
- Chinta, S.; Mallajosyula, J.; Rane, A.; Andersen, J. Mitochondrial α-Synuclein Accumulation Impairs Complex I Function in Dopaminergic Neurons and Results in Increased Mitophagy in Vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [Green Version]
- Holper, L.; Ben-Shachar, D.; Mann, J. Multivariate Meta-Analyses of Mitochondrial Complex I and IV in Major Depressive Disorder, Bipolar Disorder, Schizophrenia, Alzheimer Disease, and Parkinson Disease. Neuropsychopharmacology 2019, 44, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Yarnall, A.J.; Granic, A.; Waite, S.; Hollingsworth, K.G.; Warren, C.; Vincent, A.E.; Turnbull, D.M.; Taylor, R.W.; Dodds, R.M.; Sayer, A.A. The Feasibility of Muscle Mitochondrial Respiratory Chain Phenotyping across the Cognitive Spectrum in Parkinson’s Disease. Exp. Gerontol. 2020, 138, 110997. [Google Scholar] [CrossRef] [PubMed]
- Gaare, J.J.; Nido, G.S.; Sztromwasser, P.; Knappskog, P.M.; Dahl, O.; Lund-Johansen, M.; Maple-Grødem, J.; Alves, G.; Tysnes, O.B.; Johansson, S.; et al. Rare Genetic Variation in Mitochondrial Pathways Influences the Risk for Parkinson’s Disease. Mov. Disord. 2018, 33, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; Pereira, S.; Klein, C.; Grünewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Parkinsons Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef]
- Moors, T.; Paciotti, S.; Chiasserini, D.; Calabresi, P.; Parnetti, L.; Beccari, T.; van de Berg, W.D.J. Lysosomal Dysfunction and α-Synuclein Aggregation in Parkinson’s Disease: Diagnostic Links. Mov. Disord. 2016, 31, 791–801. [Google Scholar] [CrossRef]
- Decressac, M.; Björklund, A. TFEB: Pathogenic Role and Therapeutic Target in Parkinson Disease. Autophagy 2013, 9, 1244–1246. [Google Scholar] [CrossRef] [Green Version]
- Moors, T.E.; Hoozemans, J.J.M.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; Van De Berg, W.D.J. Therapeutic Potential of Autophagy-Enhancing Agents in Parkinson’s Disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.H.; Park, J.H.; Chung, K.C. The Central Regulator P62 between Ubiquitin Proteasome System and Autophagy and Its Role in the Mitophagy and Parkinson’s Disease. BMB Rep. 2020, 53, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS Clinical Diagnostic Criteria for Parkinson’s Disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Broad Institute. MitoCarta: An Inventory of Mammalian Mitochondrial Genes. Available online: https://www.broadinstitute.org/scientific-community/science/programs/metabolic-disease-program/publications/mitocarta/mitocarta-in-0 (accessed on 10 December 2021).
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta 2.0: An Updated Inventory of Mammalian Mitochondrial Proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR Network Reveals an Integrated Control of Cellular Clearance Pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VarSome. The Human Genomics Community. Available online: https://varsome.com (accessed on 10 December 2021).
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- QIAGEN. Digital Insights—Login. Available online: https://apps.ingenuity.com/ingsso/login (accessed on 10 December 2021).
- gnomAD. Available online: https://gnomad.broadinstitute.org (accessed on 10 December 2021).
- Sharma, M.; Maraganore, D.M.; Ioannidis, J.P.A.; Riess, O.; Aasly, J.O.; Annesi, G.; Abahuni, N.; Bentivoglio, A.R.; Brice, A.; Van Broeckhoven, C.; et al. Role of Sepiapterin Reductase Gene at the PARK3 Locus in Parkinson’s Disease. Neurobiol. Aging 2011, 32, 2108.e1–2108.e5. [Google Scholar] [CrossRef]
- Scuderi, C.; Borgione, E.; Castello, F.; Lo Giudice, M.; Santa Paola, S.; Giambirtone, M.; Di Blasi, F.D.; Elia, M.; Amato, C.; Città, S.; et al. The in Cis T251I and P587L POLG1 Base Changes: Description of a New Family and Literature Review. Neuromuscul. Disord. 2015, 25, 333–339. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA Maintenance Defects. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef]
- Davidzon, G.; Greene, P.; Mancuso, M.; Klos, K.J.; Ahlskog, J.E.; Hirano, M.; DiMauro, S. Early-Onset Familial Parkinsonism Due to POLG Mutations. Ann. Neurol. 2006, 59, 859–862. [Google Scholar] [CrossRef]
- Ma, L.; Mao, W.; Xu, E.; Cai, Y.; Wang, C.; Chhetri, J.K.; Chan, P. Novel POLG Mutation in a Patient with Early-Onset Parkinsonism, Progressive External Ophthalmoplegia and Optic Atrophy. Int. J. Neurosci. 2020, 130, 319–321. [Google Scholar] [CrossRef]
- Lamantea, E.; Tiranti, V.; Bordoni, A.; Toscano, A.; Bono, F.; Servidei, S.; Papadimitriou, A.; Spelbrink, H.; Silvestri, L.; Casari, G.; et al. Mutations of Mitochondrial DNA Polymerase Gamma A Are a Frequent Cause of Autosomal Dominant or Recessive Progressive External Ophthalmoplegia. Ann. Neurol. 2002, 52, 211–219. [Google Scholar] [CrossRef]
- Mancuso, M.; Filosto, M.; Oh, S.J.; DiMauro, S. A Novel Polymerase Gamma Mutation in a Family with Ophthalmoplegia, Neuropathy, and Parkinsonism. Arch. Neurol. 2004, 61, 1777–1779. [Google Scholar] [CrossRef] [PubMed]
- Luoma, P.; Melberg, A.; Rinne, J.O.; Kaukonen, J.A.; Nupponen, N.N.; Chalmers, R.M.; Oldfors, P.A.; Rautakorpi, I.; Peltonen, P.L.; Majamaa, P.K.; et al. Parkinsonism, Premature Menopause, and Mitochondrial DNA Polymerase Gamma Mutations: Clinical and Molecular Genetic Study. Lancet 2004, 364, 875–882. [Google Scholar] [CrossRef]
- Hudson, G.; Schaefer, A.M.; Taylor, R.W.; Tiangyou, W.; Gibson, A.; Venables, G.; Griffiths, P.; Burn, D.J.; Turnbull, D.M.; Chinnery, P.F. Mutation of the Linker Region of the Polymerase Gamma-1 (POLG1) Gene Associated with Progressive External Ophthalmoplegia and Parkinsonism. Arch. Neurol. 2007, 64, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifati, V.; Dekker, M.C.J.; Vanacore, N.; Fabbrini, G.; Squitieri, F.; Marconi, R.; Antonini, A.; Brustenghi, P.; Dalla Libera, A.; De Mari, M.; et al. Autosomal Recessive Early Onset Parkinsonism Is Linked to Three Loci: PARK2, PARK6, and PARK7. Neurol. Sci. 2002, 23, s59–s60. [Google Scholar] [CrossRef]
- Illés, A.; Csabán, D.; Grosz, Z.; Balicza, P.; Gézsi, A.; Molnár, V.; Bencsik, R.; Gál, A.; Klivényi, P.; Molnar, M.J. The Role of Genetic Testing in the Clinical Practice and Research of Early-Onset Parkinsonian Disorders in a Hungarian Cohort: Increasing Challenge in Genetic Counselling, Improving Chances in Stratification for Clinical Trials. Front. Genet. 2019, 10, 1061. [Google Scholar] [CrossRef] [PubMed]
- Ilyechova, E.Y.; Miliukhina, I.V.; Karpenko, M.N.; Orlov, I.A.; Puchkova, L.V.; Samsonov, S.A. Case of Early-Onset Parkinson’s Disease in a Heterozygous Mutation Carrier of the ATP7B Gene. J. Pers. Med. 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bento, C.F.; Ashkenazi, A.; Jimenez-Sanchez, M.; Rubinsztein, D.C. The Parkinson’s Disease-Associated Genes ATP13A2 and SYT11 Regulate Autophagy via a Common Pathway. Nat. Commun. 2016, 7, 11803. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Trajkovic, K.; Tsunemi, T.; Krainc, D. Parkin Modulates Endosomal Organization and Function of the Endo-Lysosomal Pathway. J. Neurosci. 2016, 36, 2425–2437. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Han, S.; Choi, I.; Kim, B.; Park, S.P.; Joe, E.H.; Suh, Y.H. Interplay between Leucine-Rich Repeat Kinase 2 (LRRK2) and P62/SQSTM-1 in Selective Autophagy. PLoS ONE 2016, 11, e0163029. [Google Scholar] [CrossRef] [Green Version]
- Greenamyre, J.T.; Sherer, T.B.; Betarbet, R.; Panov, A.V. Complex I and Parkinson’s Disease. IUBMB Life 2001, 52, 135–141. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of Mitochondrial Complex I Induces Progressive Parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Illés, A.; Balicza, P.; Gál, A.; Pentelényi, K.; Csabán, D.; Gézsi, A.; Molnár, V.; Molnár, M.J. Hereditary Parkinson’s Disease as a New Clinical Manifestation of the Damaged POLG Gene. Orv. Hetil. 2020, 161, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Burke, E.A.; Frucht, S.J.; Thompson, K.; Wolfe, L.A.; Yokoyama, T.; Bertoni, M.; Huang, Y.; Sincan, M.; Adams, D.R.; Taylor, R.W.; et al. Biallelic Mutations in Mitochondrial Tryptophanyl-TRNA Synthetase Cause Levodopa-Responsive Infantile-Onset Parkinsonism. Clin. Genet. 2018, 93, 712–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Mutations Classification | Patient | Gene | Heredity | Nucleotide Change | Predicted Effect on Protein | ACMG Classification/Varsome 1 | HGMD Classification |
|---|---|---|---|---|---|---|---|
| Monogenic-PD | 25 | LRRK2 | AD | c.6566A>G | p.Tyr2189Cys | Likely benign | Disease-causing variant |
| 3 | PRKN | AR | c.635G>A | p.Cys212Tyr | Likely Pathogenic | Disease-causing variant | |
| PRKN | AR | c.155delA | p.Asn52MetfsTer29 | Pathogenic | Disease-causing variant | ||
| Other-PD | 2 | GIGYF2 | AD | c.3167C>G | p.Ser1056Cys | VUS | Not reported |
| 4 | GIGYF2 | AD | c.658C>T | p.Arg220Cys | VUS | Not reported | |
| 8 | ATXN2 | AD | c.542_543insACA | p.Gln188dup | VUS | Not reported | |
| 10 | ATP7B | AR | c.4301C>T | p.Thr1434Met | VUS | Dubious disease-causing variant | |
| ATP7B | AR | c.1301A>G | p.Asn434Ser | VUS | Not reported | ||
| 12 | ATXN2 | AD | c.2937 + 4A>C | p.? | VUS | Not reported | |
| 20 | ATXN2 | AD | c.519_520delGC | p.Gln174AlafsTer75 | Likely pathogenic | Not reported | |
| 24 | DCTN1 | AD | c.2968C>T | p.Arg990Cys | VUS | Not reported | |
| 28 | SPR | AD; AR | c.448A>G | p.Arg150Gly | Pathogenic | Disease-causing variant | |
| 32 | POLG | AR | c.752C>T | p.Thr251Ile | VUS | Disease-causing variant | |
| POLG | AR | c.1760C>T | p.Pro587Leu | Likely pathogenic | Disease-causing variant |
| TFEB-Regulated Genes | Non-TFEB-Regulated Genes | Number of Genes | |
|---|---|---|---|
| PD-associated genes | - | ATP7B, ATXN2, C19orf12, PRKN, SPR, SYNJ1 | 6 |
| Lysosomal genes | ATP13A2, CD63, CTBS, GBA, HEXA, MPO, PS1, SMPD1, TMEM192, UNC13D | GNPTAB | 11 |
| Mitochondrial genes | - | ABCB6, CYP27A1, MFN2, MUTYH, NDUFB3, NDUFV1, POLG, SCO2, SLC25A46, TXNDR, WARS2 | 11 |
| Other genes | CRY1, CSPG4, FAM83G, PSEN2, RAD9A, SQSTM1, SEMA3D, TNFAIP3 | - | 8 |
| Number of genes | 18 | 18 | 36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segur-Bailach, E.; Ugarteburu, O.; Tort, F.; Texido, L.; Painous, C.; Compta, Y.; Martí, M.J.; Ribes, A.; Gort, L. Over-Mutated Mitochondrial, Lysosomal and TFEB-Regulated Genes in Parkinson’s Disease. J. Clin. Med. 2022, 11, 1749. https://doi.org/10.3390/jcm11061749
Segur-Bailach E, Ugarteburu O, Tort F, Texido L, Painous C, Compta Y, Martí MJ, Ribes A, Gort L. Over-Mutated Mitochondrial, Lysosomal and TFEB-Regulated Genes in Parkinson’s Disease. Journal of Clinical Medicine. 2022; 11(6):1749. https://doi.org/10.3390/jcm11061749
Chicago/Turabian StyleSegur-Bailach, Eulàlia, Olatz Ugarteburu, Frederic Tort, Laura Texido, Celia Painous, Yaroslau Compta, Maria José Martí, Antonia Ribes, and Laura Gort. 2022. "Over-Mutated Mitochondrial, Lysosomal and TFEB-Regulated Genes in Parkinson’s Disease" Journal of Clinical Medicine 11, no. 6: 1749. https://doi.org/10.3390/jcm11061749