
Adaptive Mechanisms of Renal Bile Acid Transporters in a Rat Model of Carbon Tetrachloride-Induced Liver Cirrhosis

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and CCl4-Induced Liver Cirrhosis
2.2. Histology
2.3. Immunohistochemistry
2.4. Kidney and Liver Function Indexes
2.5. Luminex Analysis of Cytokines, Growth Factors and Urinary Markers
2.6. Overall Score of Decompensated Cirrhosis
2.7. Statistical Analysis
3. Results
3.1. Kidney and Liver Histopathological Alterations
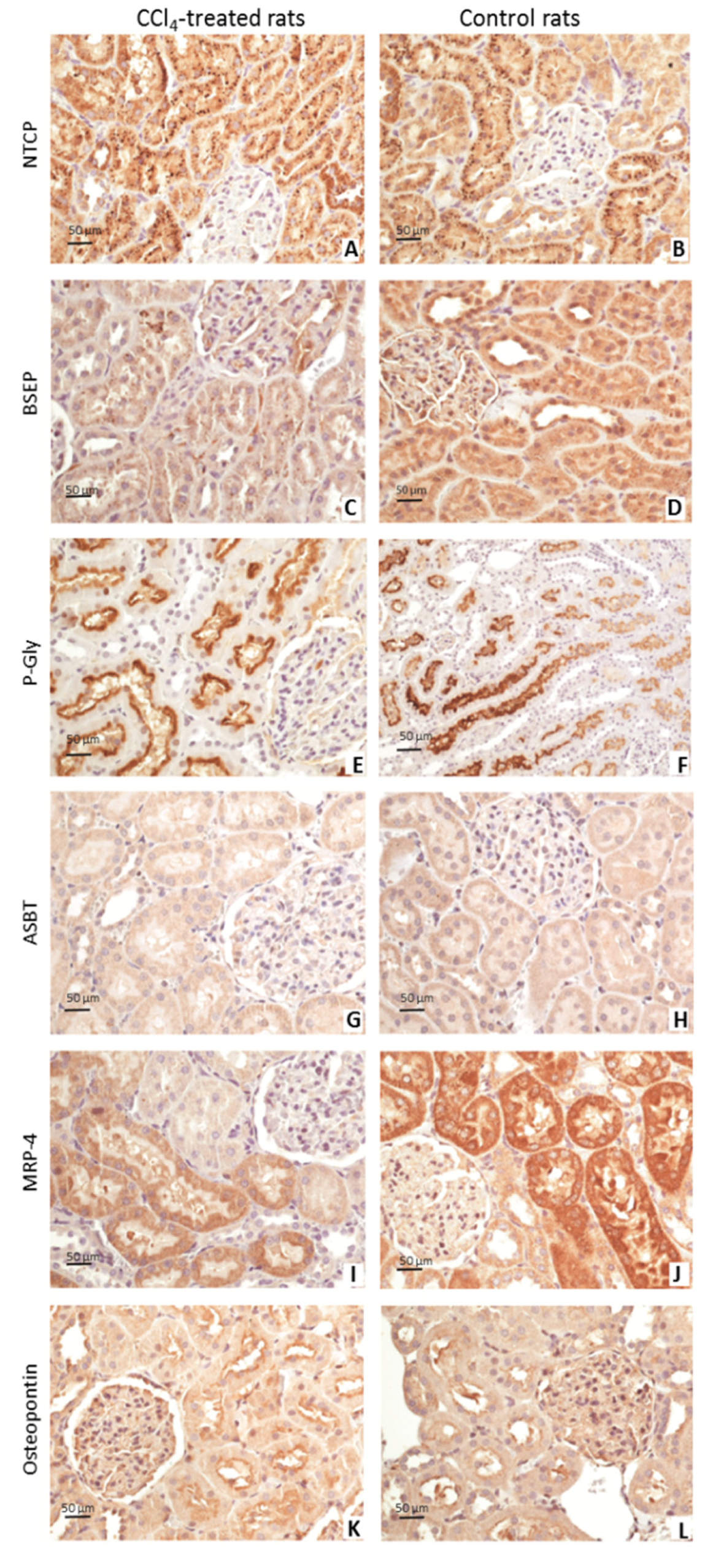
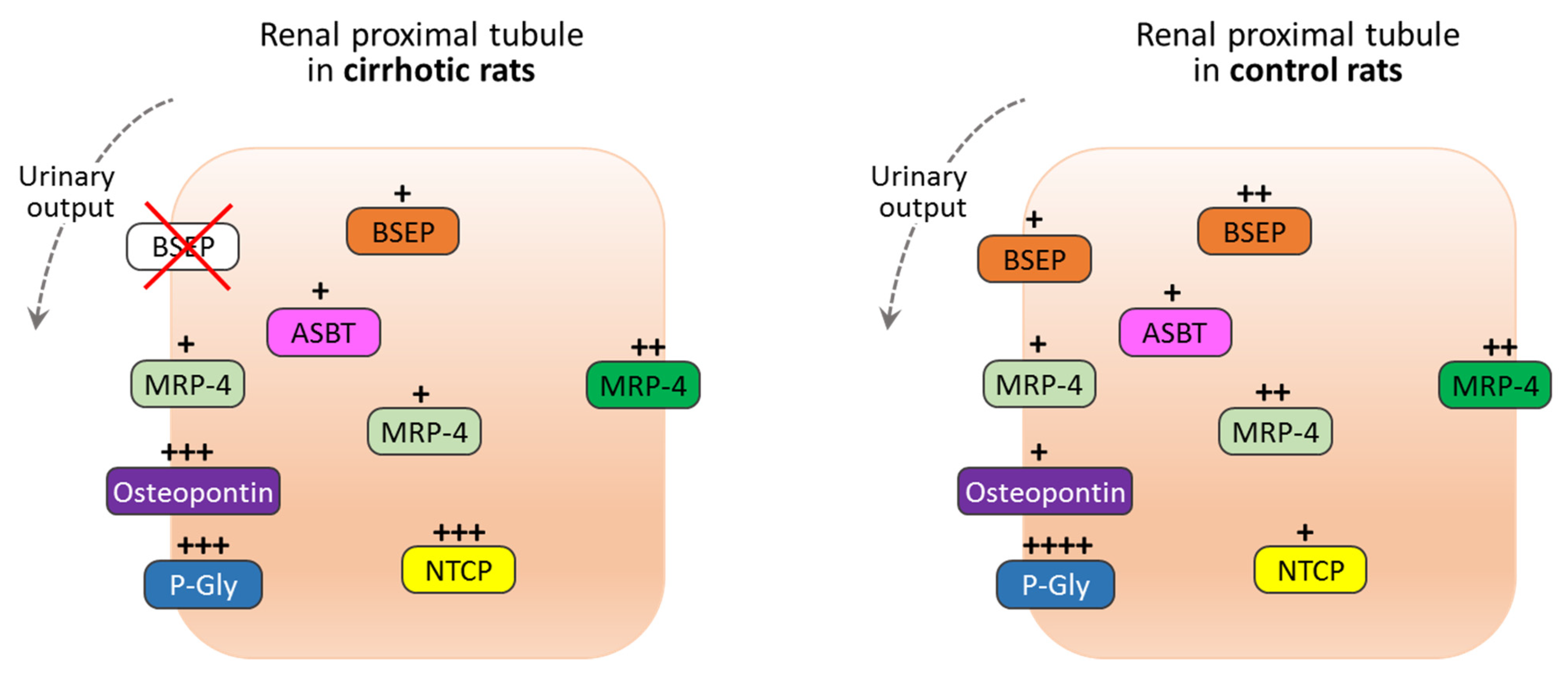
3.2. Immunohistochemistry Findings
3.3. Biomarkers of Kidney and Liver Injury
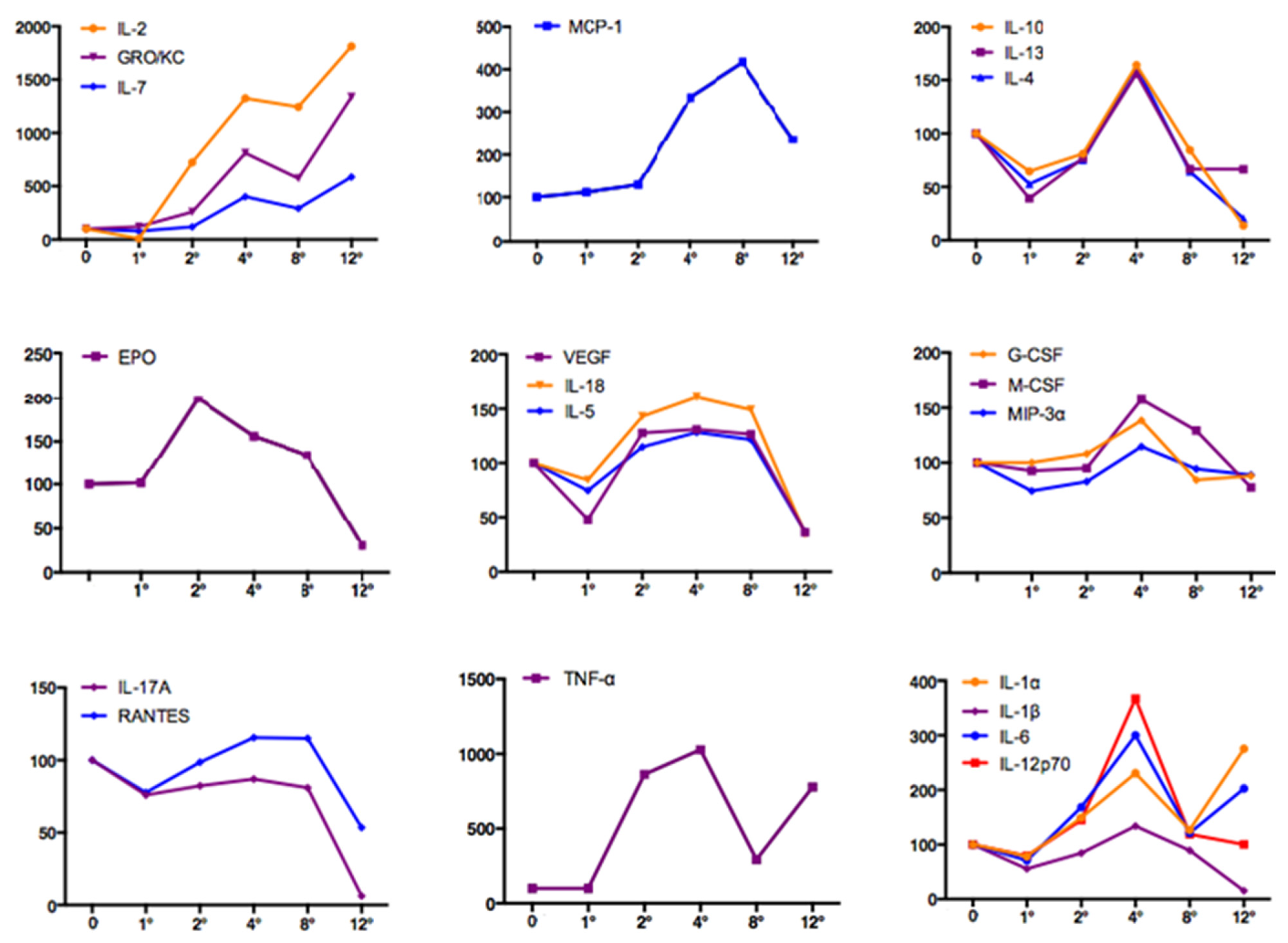
3.4. Systemic Inflammation and Early Kidney Injury
3.5. Overall Score
3.6. Correlations of Serum Creatinine with Blood and Urinary Biomarkers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Belcher, J.M.; Sanyal, A.J.; Peixoto, A.J.; Perazella, M.A.; Lim, J.; Thiessen-Philbrook, H.; Ansari, N.; Coca, S.G.; Garcia-Tsao, G.; Parikh, C.R.; et al. Kidney biomarkers and differential diagnosis of patients with cirrhosis and acute kidney injury. Hepatology 2013, 60, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Russ, K.B.; Stevens, T.M.; Singal, A.K. Acute Kidney Injury in Patients with Cirrhosis. J. Clin. Transl. Hepatol. 2015, 3, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Tsao, G.; Parikh, C.R.; Viola, A. Acute kidney injury in cirrhosis. Hepatology 2008, 48, 2064–2077. [Google Scholar] [CrossRef]
- Varga, Z.V.; Erdelyi, K.; Paloczi, J.; Cinar, R.; Zsengeller, Z.K.; Jourdan, T.; Matyas, C.; Nemeth, B.T.; Guillot, A.; Xiang, X.; et al. Disruption of R enal A rginine M etabolism P romotes K idney I njury in H epatorenal S yndrome in M ice. Hepatology 2018, 68, 1519–1533. [Google Scholar] [CrossRef] [Green Version]
- Carl, D.E.; Ghosh, S.S.; Gehr, T.W.; Abbate, A.; Toldo, S.; Sanyal, A.J. A model of acute kidney injury in mice with cirrhosis and infection. Liver Int. 2016, 36, 865–873. [Google Scholar] [CrossRef]
- Schrier, R.W.; Arroyo, V.; Bernardi, M.; Epstein, M.; Henriksen, J.H.; Rodés, J. Peripheral arterial vasodilation hypothesis: A proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology 1988, 8, 1151–1157. [Google Scholar] [CrossRef]
- Neuhaus, T.J.; Stallmach, T.; Leumann, E.; Altorfer, J.; Braegger, C.P. Familial progressive tubulo-interstitial nephropathy and cholestatic liver disease—A newly recognized entity? Eur. J. Nucl. Med. Mol. Imaging 1997, 156, 723–726. [Google Scholar] [CrossRef]
- Popović-Rolović, M.; Kostić, M.; Sindjić, M.; Jovanovic, O.; Peco-Antic, A.; Krušcić, D. Progressive tubulointerstitial nephritis and chronic cholestatic liver disease. Pediatr. Nephrol. 1993, 7, 396–400. [Google Scholar] [CrossRef]
- Harris, H.; Carpenter, T.O.; Shanley, P.; Rosen, S.; Levey, R.H.; Harmon, W.E. Progressive tubulointerstitial renal disease in infancy with associated hepatic abnormalities. Am. J. Med. 1986, 81, 169–176. [Google Scholar] [CrossRef]
- Perreault, M.; Białek, A.; Trottier, J.; Verreault, M.; Caron, P.; Milkiewicz, P.; Barbier, O. Role of Glucuronidation for Hepatic Detoxification and Urinary Elimination of Toxic Bile Acids during Biliary Obstruction. PLoS ONE 2013, 8, e80994, Erratum in PLoS ONE 2014, 9, https://doi.org/10.1371/annotation/ef13ed51-6848-419d-94d8-1bb62e7bcf52. [Google Scholar] [CrossRef]
- Bomzon, A.; Holt, S.; Moore, K. Bile acids, oxidative stress, and renal function in biliary obstruction. Semin. Nephrol. 1997, 17, 549–562. [Google Scholar] [PubMed]
- Bernardi, M.; Moreau, R.; Angeli, P.; Schnabl, B.; Arroyo, V. Mechanisms of decompensation and organ failure in cirrhosis: From peripheral arterial vasodilation to systemic inflammation hypothesis. J. Hepatol. 2015, 63, 1272–1284. [Google Scholar] [CrossRef]
- Leung, N.; Croatt, A.J.; Haggard, J.J.; Grande, J.P.; Nath, K.A. Acute cholestatic liver disease protects against glycerol-induced acute renal failure in the rat. Kidney Int. 2001, 60, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollheimer, M.J.; Fickert, P.; Stieger, B. Chronic cholestatic liver diseases: Clues from histopathology for pathogenesis. Mol. Asp. Med. 2014, 37, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Qi, X.; Zhang, W.; Zhao, C.Y.; Cao, Y.; Wang, F.; Yang, C. Abnormal Expression of Urea Transporter Protein in a Rat Model of Hepatorenal Syndrome Induced by Succinylated Gelatin. Med. Sci. Monit. 2015, 21, 2905–2911. [Google Scholar] [CrossRef] [Green Version]
- Domenicali, M.; Caraceni, P.; Giannone, F.; Baldassarre, M.; Lucchetti, G.; Quarta, C.; Patti, C.; Catani, L.; Nanni, C.; Lemoli, R.M.; et al. A novel model of CCl4-induced cirrhosis with ascites in the mouse. J. Hepatol. 2009, 51, 991–999. [Google Scholar] [CrossRef]
- Chen, T.-M.; Subeq, Y.-M.; Lee, R.-P.; Chiou, T.-W.; Hsu, B.-G. Single dose intravenous thioacetamide administration as a model of acute liver damage in rats. Int. J. Exp. Pathol. 2008, 89, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Laleman, W.; Elst, I.V.; Zeegers, M.; Servaes, R.; Libbrecht, L.; Roskams, T.; Fevery, J.; Nevens, F. A stable model of cirrhotic portal hypertension in the rat: Thioacetamide revisited. Eur. J. Clin. Investig. 2006, 36, 242–249. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G.; NC3Rs Reporting Guidelines Working Group. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef]
- Jiménez, W.; Clària, J.; Arroyo, V.; Rodés, J. Carbon tetrachloride induced cirrhosis in rats: A useful tool for investigating the pathogenesis of ascites in chronic liver disease. J. Gastroenterol. Hepatol. 1992, 7, 90–97. [Google Scholar] [CrossRef]
- Domenicali, M.; Caraceni, P.; Principe, A.; Pertosa, A.M.; Ros, J.; Chieco, P.; Trevisani, F.; Jiménez, W.; Bernardi, M. A novel sodium overload test predicting ascites decompensation in rats with CCl4-induced cirrhosis. J. Hepatol. 2005, 43, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuch, N.; Reichel, C.; Stieger, B.; Cattori, V.; Fattinger, K.E.; Landmann, L.; Meier, P.J.; Kullak-Ublick, G.A. Effect of phenobarbital on the expression of bile salt and organic anion transporters of rat liver. J. Hepatol. 2001, 34, 881–887. [Google Scholar] [CrossRef] [Green Version]
- Csonka, T.; Murnyák, B.; Szepesi, R.; Bencze, J.; Bognár, L.; Klekner, A.; Hortobágyi, T. Assessment of candidate immunohistochemical prognostic markers of meningioma recurrence. Folia Neuropathol. 2016, 2, 114–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishak, K.; Baptista, A.; Bianchi, L.; Callea, F.; De Groote, J.; Gudat, F.; Denk, H.; Desmet, V.; Korb, G.; Macsween, R.N.; et al. Histological grading and staging of chronic hepatitis. J. Hepatol. 1995, 22, 696–699. [Google Scholar] [CrossRef]
- Bedossa, P.; Poynard, T. An algorithm for the grading of activity in chronic hepatitis C. Hepatology 1996, 24, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, O.; Valentini, C.; Donati, G.; Comai, G.; Cuna, V.; Capelli, I.; Angelini, M.L.; Moretti, M.I.; Angeletti, A.; Piscaglia, F.; et al. Hepatorenal syndrome: Update on diagnosis and treatment. World J. Nephrol. 2015, 4, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Velez, J.C.Q.; Therapondos, G.; Juncos, L.A. Reappraising the spectrum of AKI and hepatorenal syndrome in patients with cirrhosis. Nat. Rev. Nephrol. 2019, 16, 137–155. [Google Scholar] [CrossRef]
- Bernardi, M.; Bassi, S.L.; Arienti, V.; De Collibus, C.; Scialpi, C.; Boriani, L.; Zanzani, S.; Caraceni, P.; Trevisani, F. Systemic and regional hemodynamics in pre-ascitic cirrhosis: Effects of posture. J. Hepatol. 2003, 39, 502–508. [Google Scholar] [CrossRef]
- Bernardl, M.; Di Marco, C.; Trevisani, F.; Fornalé, L.; Andreoné, P.; Cursaro, C.; Baraldini, M.; Ligabue, A.; Tamé, M.R.; Gasbarrini, G. Renal sodium retention during upright posture in preascitic cirrhosis. Gastroenterology 1993, 105, 188–193. [Google Scholar] [CrossRef]
- Recknagel, R.O.; Glende, E.A.; Dolak, J.A.; Waller, R.L. Mechanisms of carbon tetrachloride toxicity. Pharmacol. Ther. 1989, 43, 139–154. [Google Scholar] [CrossRef]
- Holmes, T.W. The Histologic Lesion of Cholemic Nephrosis. J. Urol. 1953, 70, 677–685. [Google Scholar] [CrossRef]
- Haessler, H.; Rous, P.; Broun, G.O. The Renal Elimination of Bilirubin. J. Exp. Med. 1922, 35, 533–552. [Google Scholar] [CrossRef]
- Levy, M.; Finestone, H. Renal response to four hours of biliary obstruction in the dog. Am. J. Physiol. Physiol. 1983, 244, F516–F525. [Google Scholar] [CrossRef] [PubMed]
- Topuzlu, C.; Stahl, W.M. Effect of Bile Infusion on the Dog Kidney. N. Engl. J. Med. 1966, 274, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Gollan, J.L.; Billing, B.H.; Huang, S.N. Ultrastructural changes in the isolated rat kidney induced by conjugated bilirubin and bile acids. Br. J. Exp. Pathol. 1976, 57, 571–581. [Google Scholar]
- Rivera-Huizar, S.; Rincón-Sánchez, A.R.; Covarrubias-Pinedo, A.; Islas-Carbajal, M.C.; Gabriel-Ortíz, G.; Pedraza-Chaverrí, J.; Álvarez-Rodríguez, A.; Meza-García, E.; Armendáriz-Borunda, J. Renal dysfunction as a consequence of acute liver damage by bile duct ligation in cirrhotic rats. Exp. Toxicol. Pathol. 2006, 58, 185–195. [Google Scholar] [CrossRef]
- Uslu, A.; Taşli, F.A.; Nart, A.; Postaci, H.; Aykas, A.; Bati, H.; Coşkun, Y. Human kidney histopathology in acute obstructive jaundice. Eur. J. Gastroenterol. Hepatol. 2010, 22, 1458–1465. [Google Scholar] [CrossRef]
- Van Slambrouck, C.; Salem, F.; Meehan, S.M.; Chang, A. Bile cast nephropathy is a common pathologic finding for kidney injury associated with severe liver dysfunction. Kidney Int. 2013, 84, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Krones, E.; Wagner, M.; Eller, K.; Rosenkranz, A.R.; Trauner, M.; Fickert, P. Bile Acid-Induced Cholemic Nephropathy. Dig. Dis. 2015, 33, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Karpen, S.J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. [Google Scholar] [CrossRef] [Green Version]
- Meier, P.J.; Stieger, B. Bile Salt Transporters. Annu. Rev. Physiol. 2002, 64, 635–661. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Boyer, J.L. Bile Salt Transporters: Molecular Characterization, Function, and Regulation. Physiol. Rev. 2003, 83, 633–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballatori, N.; Christian, W.V.; Lee, J.Y.; Dawson, P.A.; Soroka, C.J.; Boyer, J.L.; Madejczyk, M.S.; Li, N. OSTα-OSTβ: A major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 2005, 42, 1270–1279. [Google Scholar] [CrossRef]
- Kullak-Ublick, G.E.R.D.A.; Stieger, B.R.U.N.O.; Hagenbuch, B.R.U.N.O.; Meier, P.E.T.E.R.J. Hepatic Transport of Bile Salts. Semin. Liver Dis. 2000, 20, 273–292. [Google Scholar] [CrossRef] [Green Version]
- Zollner, G.; Trauner, M. Mechanisms of Cholestasis. Clin. Liver Dis. 2008, 12, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Rudman, D.; Kendall, F.E. Bile Acid Content of Human Serum. I. Serum Bile Acids in Patients with Hepatic Disease. J. Clin. Investig. 1957, 36, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauli-Magnus, C.; Stieger, B.; Meier, Y.; Kullak-Ublick, G.A.; Meier, P.J. Enterohepatic transport of bile salts and genetics of cholestasis. J. Hepatol. 2005, 43, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Karpen, S.J.; Sun, A.-Q.; Kudish, B.; Hagenbuch, B.; Meier, P.J.; Ananthanarayanan, M.; Suchy, F.J. Multiple Factors Regulate the Rat Liver Basolateral Sodium-dependent Bile Acid Cotransporter Gene Promoter. J. Biol. Chem. 1996, 271, 15211–15221. [Google Scholar] [CrossRef] [Green Version]
- Geier, A.; Dietrich, C.G.; Voigt, S.; Kim, S.; Gerloff, T.; Kullak-Ublick, G.A.; Lorenzen, J.; Matern, S.; Gartung, C. Effects of proinflammatory cytokines on rat organic anion transporters during toxic liver injury and cholestasis. Hepatology 2003, 38, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.A.; Miyake, J.H.; Hui, T.Y.; Spann, N.J. Regulation of cholesterol-7α-hydroxylase: BAREly missing a SHP. J. Lipid Res. 2002, 43, 533–543. [Google Scholar] [CrossRef]
- Shah, N.; Dhar, D.; Mohammed, F.E.Z.; Habtesion, A.; Davies, N.A.; Jover-Cobos, M.; Macnaughtan, J.; Sharma, V.; Damink, S.W.O.; Mookerjee, R.P.; et al. Prevention of acute kidney injury in a rodent model of cirrhosis following selective gut decontamination is associated with reduced renal TLR4 expression. J. Hepatol. 2012, 56, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.-G.; Huang, H.-C.; Lee, F.-Y.; Chan, C.-Y.; Lee, J.-Y.; Lee, S.-D. Kinetics of cytokine expression in cirrhotic rats. J. Chin. Med. Assoc. 2011, 74, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCl4-Treated Rats | Total Activity Stage [24] | Fibrosis Stage [24] | METAVIR Stage [25] |
|---|---|---|---|
| 1 | 5 | 6 | F4 |
| 2 | 5 | 6 | F4 |
| 3 | 5 | 6 | F4 |
| 4 | 5 | 6 | F4 |
| 5 | 8 | 6 | F4 |
| 6 | 8 | 6 | F4 |
| 7 | 8 | 5 | F3 |
| 8 | 8 | 6 | F4 |
| 9 | 3 | 6 | F4 |
| 10 | 3 | 6 | F4 |
| 11 | 3 | 6 | F4 |
| 12 | 4 | 6 | F4 |
| 13 | 2 | 6 | F4 |
| 14 | 4 | 6 | F4 |
| 15 | 4 | 6 | F4 |
| 16 | 6 | 6 | F4 |
| 17 | 4 | 5 | F3 |
| 18 | 3 | 6 | F4 |
| 19 | 2 | 6 | F4 |
| 20 | 2 | 4 | F3 |
| 21 | 2 | 6 | F4 |
| Control rats | |||
| 22 | 0 | 0 | F0 |
| 23 | 0 | 0 | F0 |
| 24 | 0 | 0 | F0 |
| 25 | 0 | 0 | F0 |
| 26 | 0 | 0 | F0 |
| CCl4-Treated Rats (n = 21) | Control Rats (n = 5) | p-Value | |
|---|---|---|---|
| Free BA in plasma, µmol/L | 16 ± 1.5 | 0.92 ± 0.03 | <0.0001 |
| Free BA in urines, µmol/L | 81 ± 11.6 | 0.12 ± 0.06 | <0.0001 |
| Taurine-conjugated BA in plasma, µmol/L | 10 ± 5 | 2.1 ± 1.3 | 0.021 |
| Taurine-conjugated BA in urines, µmol/L | 112 ± 20 | 0.06 ± 0.01 | <0.0001 |
| Total BA in plasma, µmol/L | 26 ± 17 | 2.8 ± 1.2 | 0.0062 |
| Total BA in urines, µmol/L | 101 ± 11.2 | 0.26 ± 0.2 | <0.0001 |
| CCl4-Treated Rats (n = 21) | Control Rats (n = 5) | ||||||
|---|---|---|---|---|---|---|---|
| Putative Role in Kidney | Cytoplasm | Apical Membrane | BL Membrane | Cytoplasm | Apical Membrane | BL Membrane | |
| NTCP | Unknown | Intensive/granular | / | / | Weak/granular | / | / |
| BSEP | Unknown | Weak/granular | / | / | Moderate/granular | Weak | / |
| P-Gly | Unknown | / | Moderate/intensive | / | / | High/intensive | / |
| ASBT | Reabsorption of free bile acids from lumen to PTC | Weak | / | / | Weak | / | / |
| MRP4 | Transporter for free bile acids from cytoplasm of PTC to lumen | Weak | Weak | Moderate | Moderate | Weak | Intensive |
| Osteopontin | Transporter for free bile acids from cytoplasm of PTC to serum, through the basolateral membrane | / | Moderate/intensive | / | / | Weak | / |
| CCl4-Treated Rats | Histological Score (Activity + Fibrosis) | Biochemical Score | Ascites | Encephalopathy | Overall Score |
|---|---|---|---|---|---|
| 1 | 11 | 7 | 1 | 2 | 21 |
| 2 | 11 | 13 | 3 | 3 | 30 |
| 3 | 11 | 6 | 3 | 1 | 21 |
| 4 | 11 | 0 | 2 | 1 | 14 |
| 5 | 14 | 10 | 3 | 3 | 30 |
| 6 | 14 | 6 | 3 | 2 | 25 |
| 7 | 13 | 9 | 3 | 2 | 27 |
| 8 | 14 | 7 | 1 | 1 | 23 |
| 9 | 9 | 5 | 3 | 1 | 18 |
| 10 | 9 | 7 | 3 | 3 | 22 |
| 11 | 9 | 6 | 3 | 2 | 20 |
| 12 | 10 | 7 | 1 | 1 | 19 |
| 13 | 8 | 5 | 3 | 2 | 18 |
| 14 | 10 | 5 | 3 | 1 | 19 |
| 15 | 10 | 7 | 3 | 1 | 21 |
| 16 | 12 | 6 | 1 | 2 | 21 |
| 17 | 9 | 5 | 1 | 1 | 16 |
| 18 | 9 | 7 | 3 | 2 | 21 |
| 19 | 8 | 8 | 3 | 2 | 21 |
| 20 | 6 | 6 | 3 | 3 | 18 |
| 21 | 8 | 4 | 3 | 2 | 17 |
| Mean ± SD | 10.3 ± 2.1 | 6.5 ± 2.4 | 2.5 ± 0.9 | 1.8 ± 0.7 | 21.0 ± 4.1 |
| sCreat T1 | sCreat T2 | sCreat T4 | sCreat T8 | sCreat T12 | ||
|---|---|---|---|---|---|---|
| IL-1α | Correlation coefficient | −0.316 | 0.467 | 0.409 | 0.005 | 0.197 |
| p-value | 0.541 | 0.038 * | 0.073 | 0.982 | 0.391 | |
| IL-6 | Correlation coefficient | −0.566 | 0.424 | 0.451 | 0.084 | 0.181 |
| p-value | 0.242 | 0.063 | 0.046 * | 0.724 | 0.432 | |
| IL-12p70 | Correlation coefficient | −0.664 | 0.359 | 0.496 | 0.063 | −0.037 |
| p-value | 0.150 | 0.143 | 0.043 * | 0.792 | 0.873 | |
| IL-18 | Correlation coefficient | −0.566 | 0.381 | 0.466 | −0.112 | −0.234 |
| p-value | 0.242 | 0.108 | 0.051 ^ | 0.638 | 0.308 | |
| GRO/KC | Correlation coefficient | −0.310 | 0.250 | 0.468 | 0.178 | −0.033 |
| p-value | 0.550 | 0.288 | 0.038 * | 0.479 | 0.890 | |
| MCP-1 | Correlation coefficient | −0.655 | 0.449 | 0.360 | 0.422 | 0.325 |
| p-value | 0.158 | 0.047 * | 0.119 | 0.064 | 0.175 |
| sCreat T1 | sCreat T2 | sCreat T4 | sCreat T8 | sCreat T12 | ||
|---|---|---|---|---|---|---|
| IL-18 | Correlation coefficient | −0.100 | 0.647 | 0.098 | 0.194 | 0.200 |
| p-value | 0.873 | 0.002 * | 0.718 | 0.488 | 0.475 | |
| Osteopontin | Correlation coefficient | −0.600 | 0.622 | 0.242 | 0.089 | 0.047 |
| p-value | 0.285 | 0.003 * | 0.367 | 0.710 | 0.879 | |
| B2M | Correlation coefficient | 0.200 | 0.328 | 0.757 | 0.018 | 0.041 |
| p-value | 0.747 | 0.171 | 0.003 * | 0.946 | 0.884 | |
| NGAL | Correlation coefficient | 0.200 | 0.108 | 0.721 | 0.031 | −0.447 |
| p-value | 0.747 | 0.651 | 0.004 * | 0.904 | 0.095 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donadei, C.; Angeletti, A.; Cappuccilli, M.; Conti, M.; Conte, D.; Zappulo, F.; De Giovanni, A.; Malvi, D.; Aldini, R.; Roda, A.; et al. Adaptive Mechanisms of Renal Bile Acid Transporters in a Rat Model of Carbon Tetrachloride-Induced Liver Cirrhosis. J. Clin. Med. 2022, 11, 636. https://doi.org/10.3390/jcm11030636
Donadei C, Angeletti A, Cappuccilli M, Conti M, Conte D, Zappulo F, De Giovanni A, Malvi D, Aldini R, Roda A, et al. Adaptive Mechanisms of Renal Bile Acid Transporters in a Rat Model of Carbon Tetrachloride-Induced Liver Cirrhosis. Journal of Clinical Medicine. 2022; 11(3):636. https://doi.org/10.3390/jcm11030636
Chicago/Turabian StyleDonadei, Chiara, Andrea Angeletti, Maria Cappuccilli, Massimiliano Conti, Diletta Conte, Fulvia Zappulo, Alessio De Giovanni, Deborah Malvi, Rita Aldini, Aldo Roda, and et al. 2022. "Adaptive Mechanisms of Renal Bile Acid Transporters in a Rat Model of Carbon Tetrachloride-Induced Liver Cirrhosis" Journal of Clinical Medicine 11, no. 3: 636. https://doi.org/10.3390/jcm11030636