
The Plasma DIA-Based Quantitative Proteomics Reveals the Pathogenic Pathways and New Biomarkers in Cervical Cancer and High Grade Squamous Intraepithelial Lesion
 and
and
Abstract
:1. Introduction
2. Materials and Methods
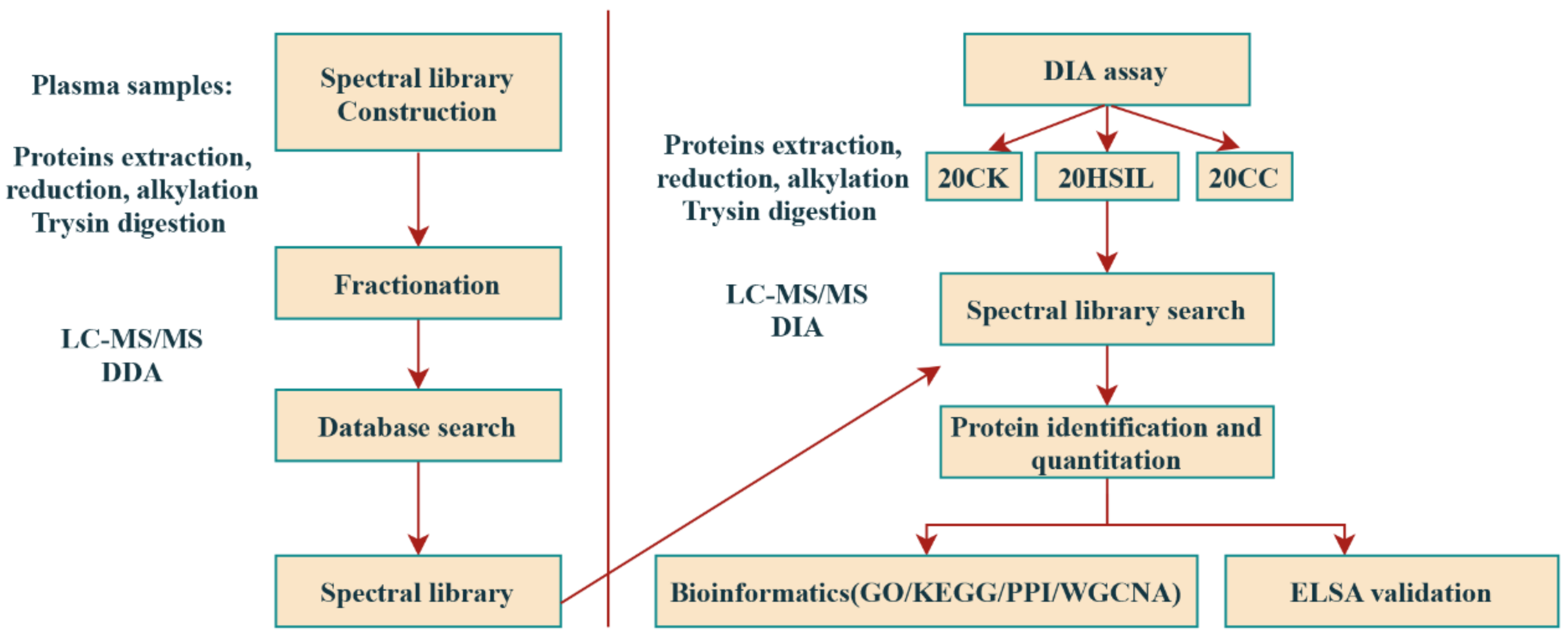
2.1. Study Design and Collection of Clinical Samples
2.2. Sample Preparation and Fractionation for DDA Library Generation
2.3. Data Dependent Acquisition (DDA) Mass Spectrometry Assay
2.4. Mass Spectrometry Assay for Data Independent Acquisition (DIA)
2.5. Mass Spectrometry Data Analysis
2.6. Bioinformatical and Statistical Analysis
2.6.1. Subcellular Localization
2.6.2. Domain Annotation
2.6.3. GO Annotation
2.6.4. KEGG Annotation
2.6.5. Enrichment Analysis
2.6.6. Protein-Protein Interaction Analysis
2.6.7. WGCNA Analysis (Weighted Gene Co-Expression Network Analysis)
2.6.8. ELISA
2.6.9. Statistical Analysis
3. Results
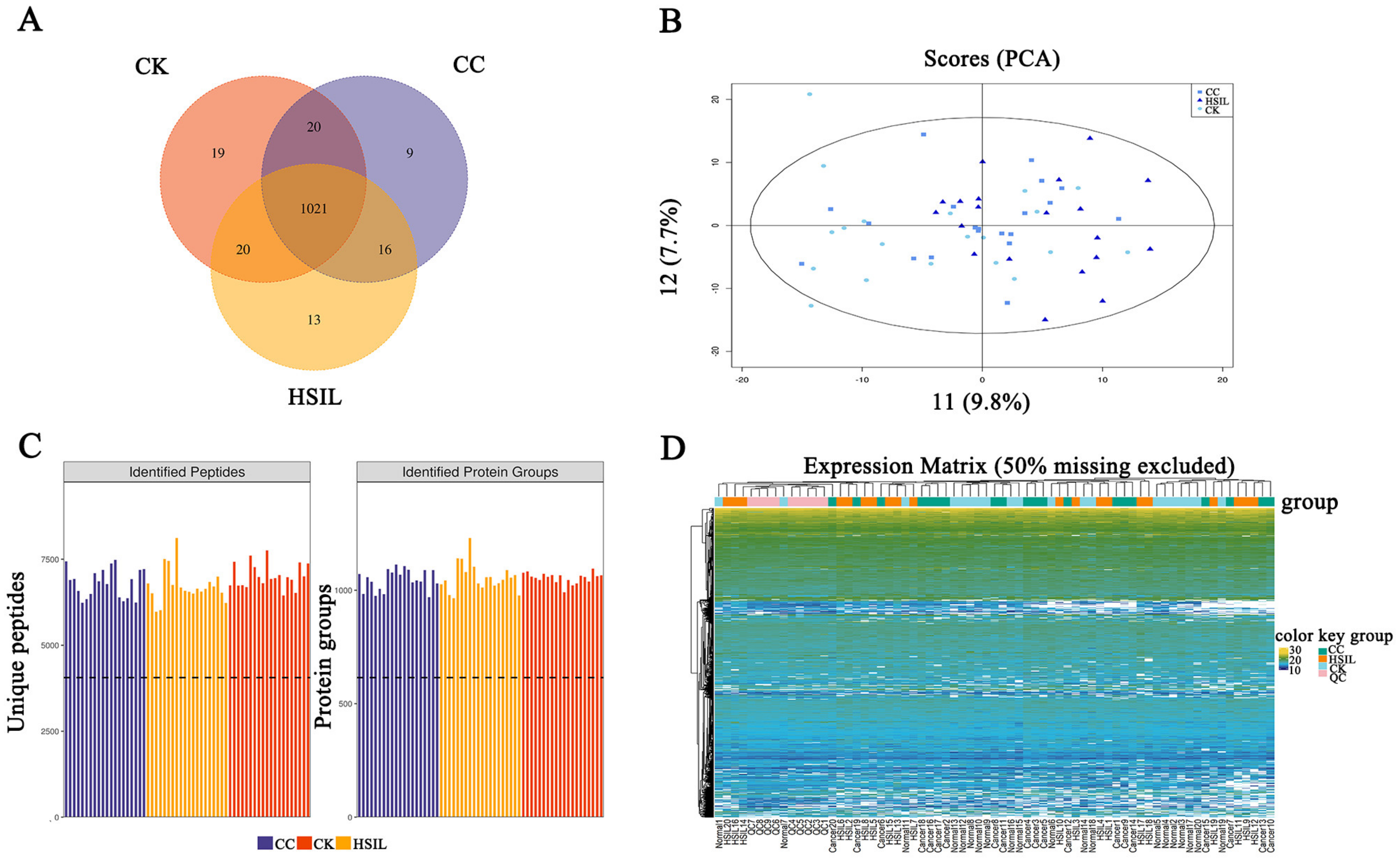
3.1. An Overview of the Quantitative Proteomics Analysis
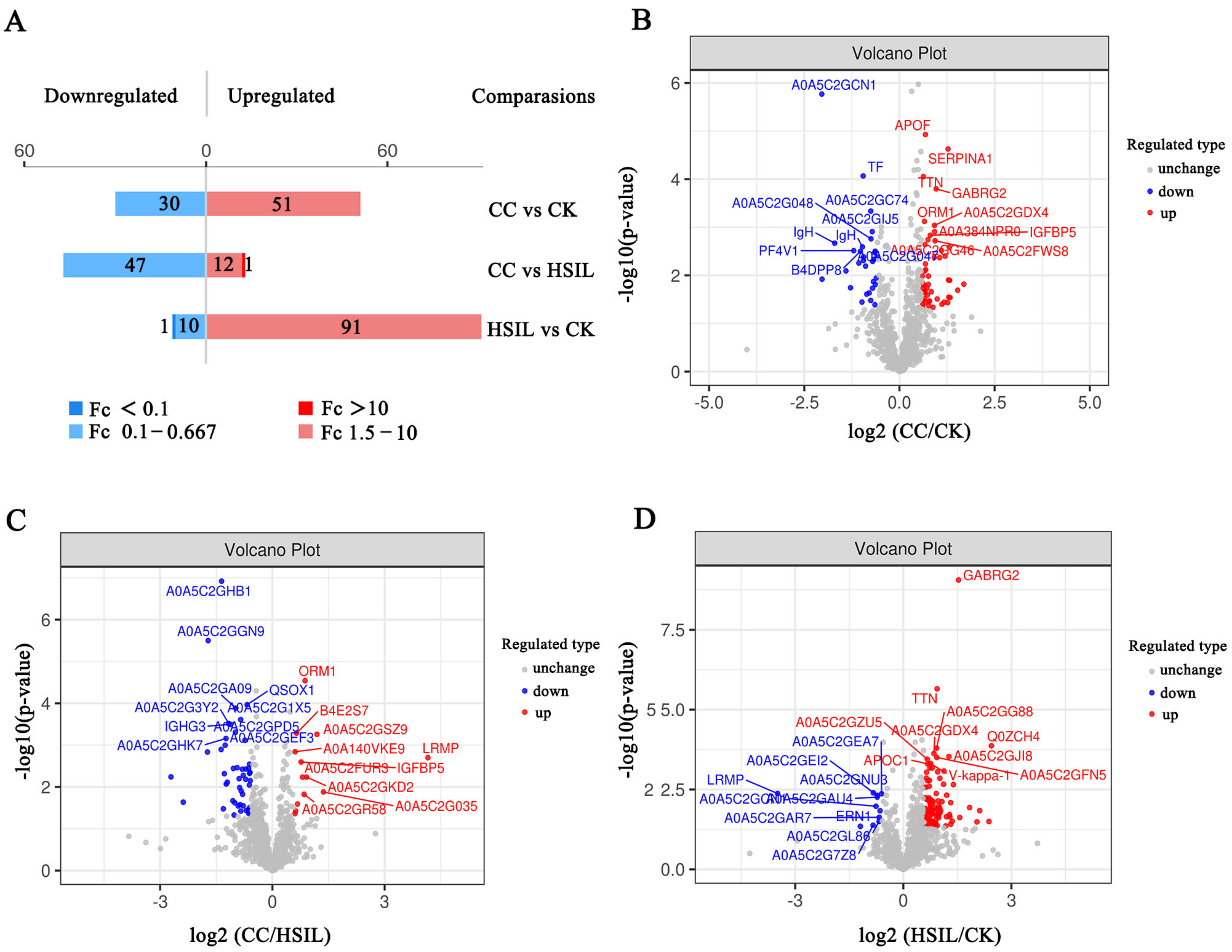
3.2. Identification of Differentially Expressed Proteins
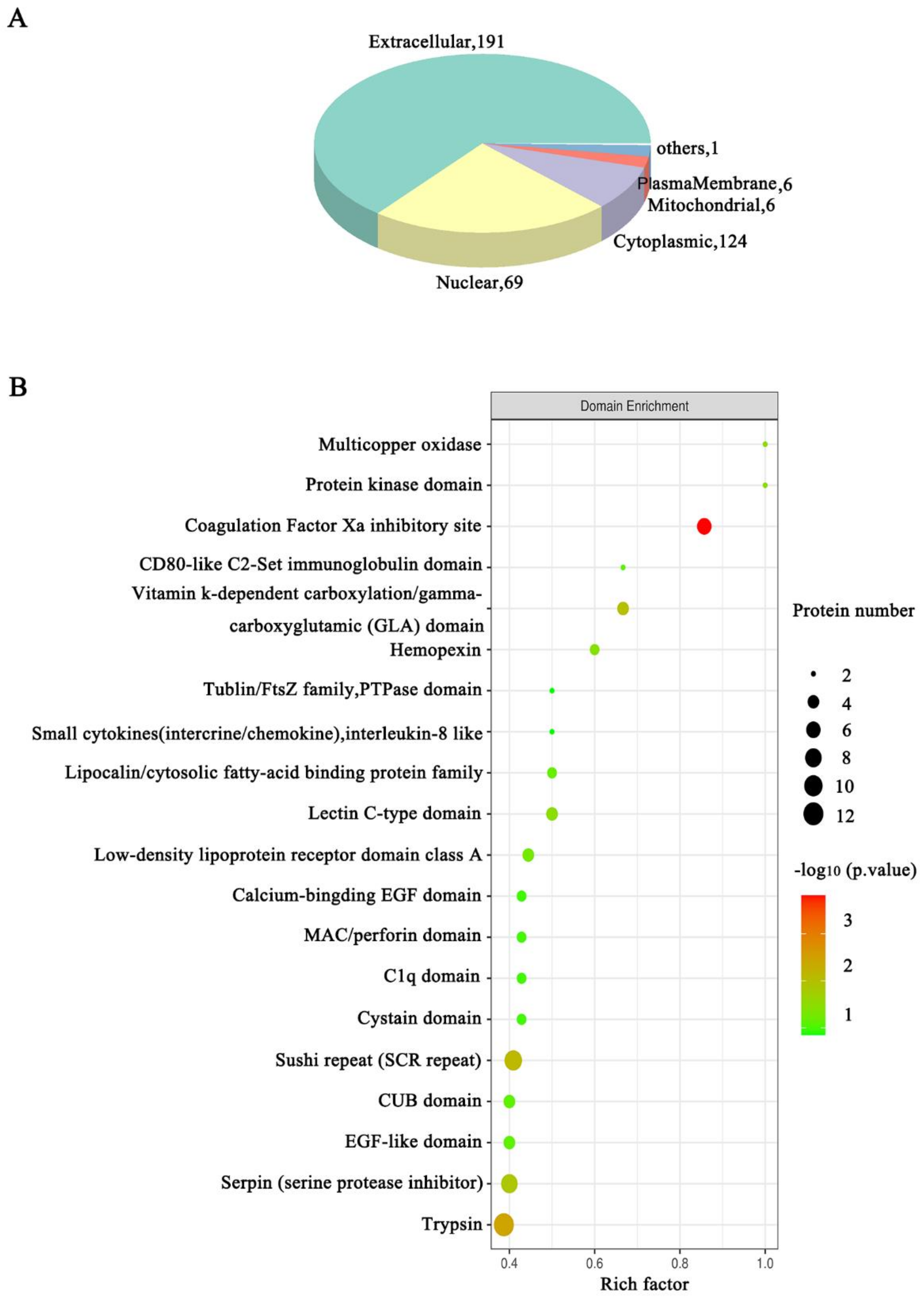
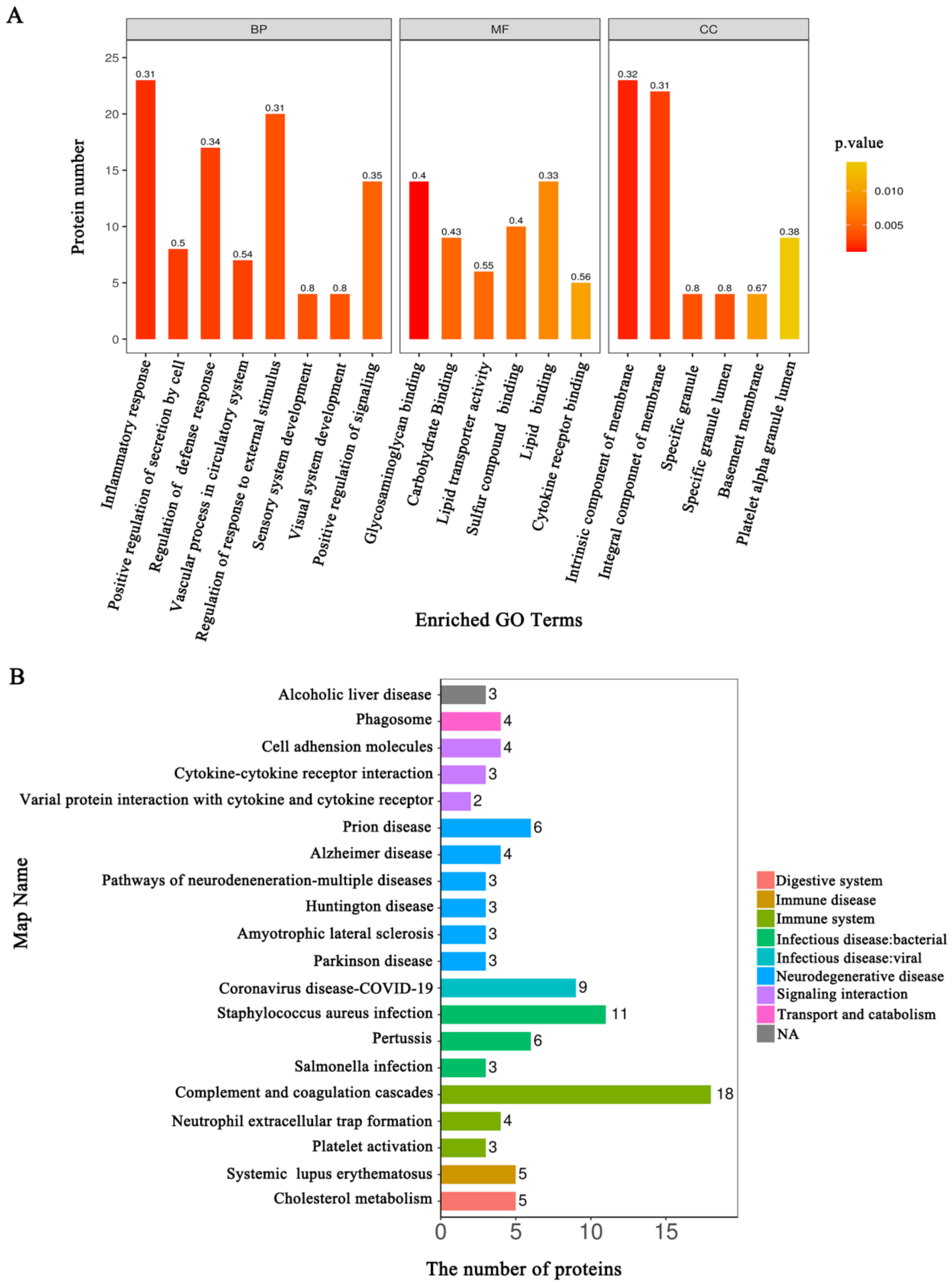
3.3. Functional Annotation Analysis of the DEPs Related to the HSIL and Cervical Cancer Occurrence
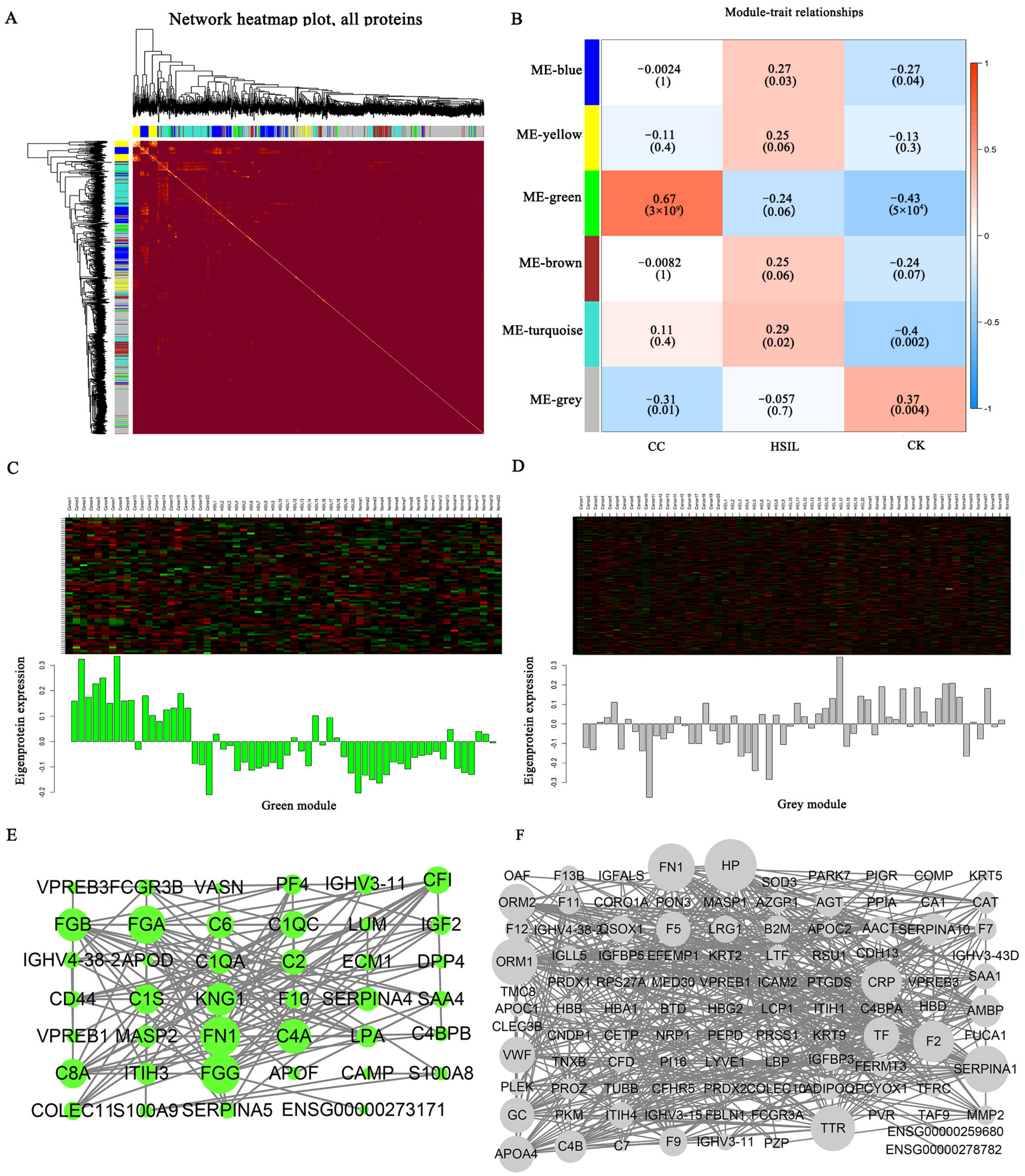
3.4. Protein-Protein Interaction (PPI) Network and Weighted Gene Co-Expression Network Analysis (WGCNA)
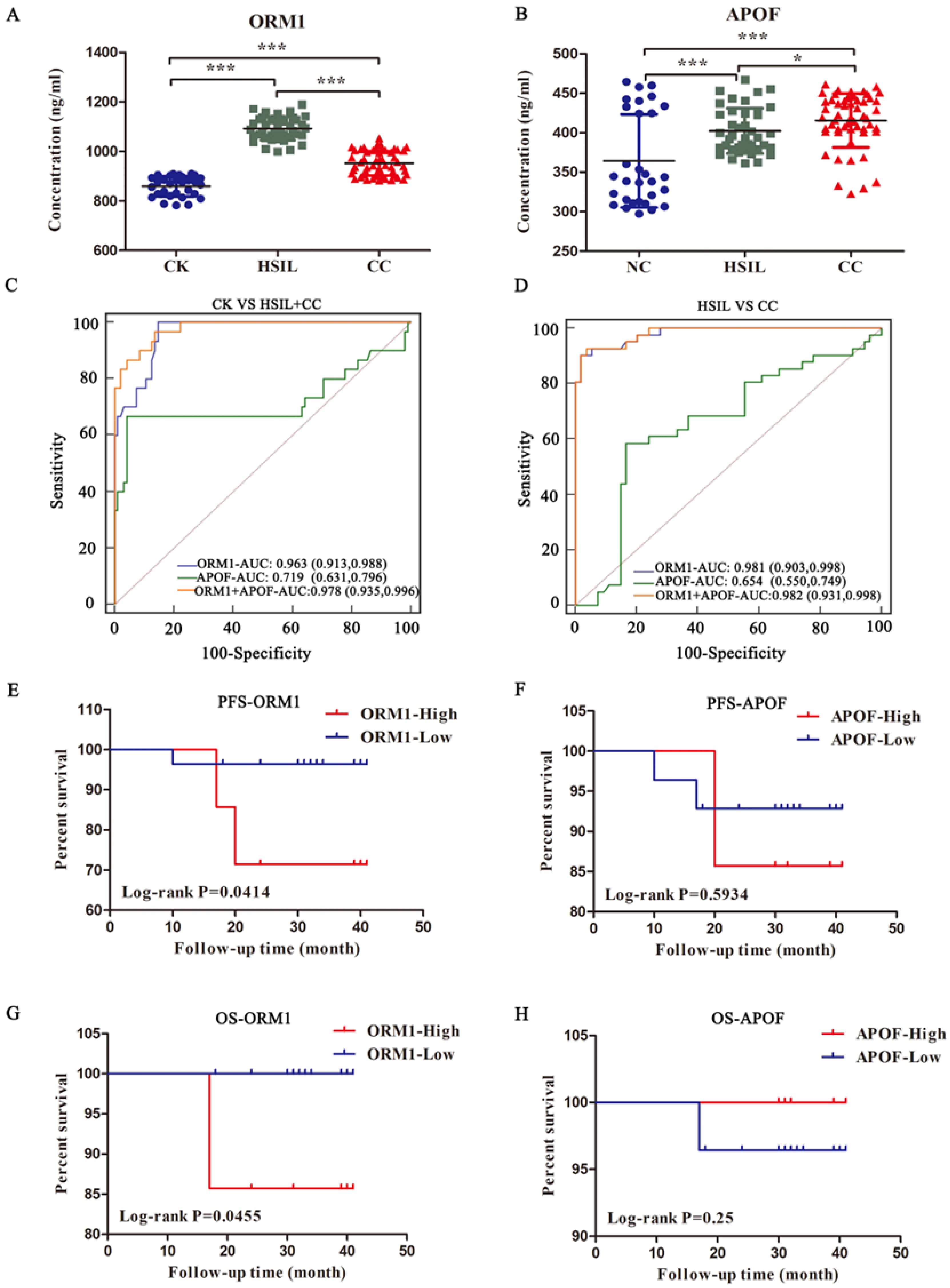
3.5. Validation of the Proteomic Data by ELSIA
3.6. Relationship between ORM1/APOF Expression and the Clinicopathological Factors as well as Survival Prognosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Cancer Report. 2020. Available online: https://publications.iarc.fr/Non-Series-Publications/World-Cancer-Reports/World-Cancer-Report-Cancer-Research-For-Cancer-Prevention-2020 (accessed on 1 February 2020).
- Hu, H.; Zhao, J.; Yu, W.; Zhao, J.; Wang, Z.; Jin, L.; Yu, Y.; Han, L.; Wang, L.; Zhu, H.; et al. Human papillomavirus DNA, HPV L1 capsid protein and p16(INK4a) protein as markers to predict cervical lesion progression. Arch. Gynecol. Obstet. 2019, 299, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Borella, F.; Carosso, A.R.; Cosma, S.; Preti, M.; Collemi, G.; Cassoni, P.; Bertero, L.; Benedetto, C. Gut Microbiota and Gynecological Cancers: A Summary of Pathogenetic Mechanisms and Future Directions. ACS Infect. Dis. 2021, 7, 987–1009. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Huang, B.; Zhang, K.; Liu, M.; Xu, T. Long non-coding RNA in cervical cancer: From biology to therapeutic opportunity. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 127, 110209. [Google Scholar] [CrossRef] [PubMed]
- Sadri Nahand, J.; Moghoofei, M.; Salmaninejad, A.; Bahmanpour, Z.; Karimzadeh, M.; Nasiri, M.; Mirzaei, H.R.; Pourhanifeh, M.H.; Bokharaei-Salim, F.; Mirzaei, H. Pathogenic role of exosomes and microRNAs in HPV-mediated inflammation and cervical cancer: A review. Int. J. Cancer 2020, 146, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Revathidevi, S.; Murugan, A.K.; Nakaoka, H.; Inoue, I.; Munirajan, A.K. APOBEC: A molecular driver in cervical cancer pathogenesis. Cancer Lett. 2021, 496, 104–116. [Google Scholar] [CrossRef]
- Shamseddine, A.A.; Burman, B.; Lee, N.Y.; Zamarin, D. Tumor Immunity and Immunotherapy for HPV-Related Cancers. Cancer Discov. 2021, 11, 1896–1912. [Google Scholar] [CrossRef]
- Saw, P.E.; Xu, X.; Chen, J.; Song, E.W. Non-coding RNAs: The new central dogma of cancer biology. Sci. China Life Sci. 2021, 64, 22–50. [Google Scholar] [CrossRef]
- Piergentili, R.; Zaami, S.; Cavaliere, A.F.; Signore, F.; Scambia, G.; Mattei, A.; Marinelli, E.; Gulia, C.; Perelli, F. Non-Coding RNAs as Prognostic Markers for Endometrial Cancer. Int. J. Mol. Sci. 2021, 22, 3151. [Google Scholar] [CrossRef]
- Cavaliere, A.F.; Perelli, F.; Zaami, S.; Piergentili, R.; Mattei, A.; Vizzielli, G.; Scambia, G.; Straface, G.; Restaino, S.; Signore, F. Towards Personalized Medicine: Non-Coding RNAs and Endometrial Cancer. Healthcare 2021, 9, 965. [Google Scholar] [CrossRef]
- Zhao, D.; Ren, C.; Yao, Y.; Wang, Q.; Li, F.; Li, Y.; Jiang, A.; Wang, G. Identifying prognostic biomarkers in endometrial carcinoma based on ceRNA network. J. Cell. Biochem. 2020, 121, 2437–2446. [Google Scholar] [CrossRef]
- Ludwig, C.; Gillet, L.; Rosenberger, G. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Kang, Y.; Li, L.; Zhao, G.; Sun, J.; Liu, Z. Proteome analysis of rainbow trout (Oncorhynchus mykiss) liver responses to chronic heat stress using DIA/SWATH. J. Proteom. 2021, 233, 104079. [Google Scholar] [CrossRef]
- Meng, K.; Lu, S.; Yan, X.; Sun, Y.; Gao, J.; Wang, Y.; Yin, X.; Sun, Z.; He, Q.Y. Quantitative Mitochondrial Proteomics Reveals ANXA7 as a Crucial Factor in Mitophagy. J. Proteome Res. 2020, 19, 1275–1284. [Google Scholar] [CrossRef]
- Rai, A.J.; Gelfand, C.A.; Haywood, B.C.; Warunek, D.J.; Yi, J.; Schuchard, M.D.; Mehigh, R.J.; Cockrill, S.L.; Scott, G.B.; Tammen, H.; et al. HUPO Plasma Proteome Project specimen collection and handling: Towards the standardization of parameters for plasma proteome samples. Proteomics 2005, 5, 3262–3277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef]
- Zhao, Q.; He, Y.; Wang, X.L.; Zhang, Y.X.; Wu, Y.M. Differentially expressed proteins among normal cervix, cervical intraepithelial neoplasia and cervical squamous cell carcinoma. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2015, 17, 620–631. [Google Scholar] [CrossRef]
- Bao, H.; Li, X.; Cao, Z.; Huang, Z.; Chen, L.; Wang, M.; Hu, J.; Li, W.; Sun, H.; Jiang, X.; et al. Identification of COPA as a potential prognostic biomarker and pharmacological intervention target of cervical cancer by quantitative proteomics and experimental verification. J. Transl. Med. 2022, 20, 18. [Google Scholar] [CrossRef]
- Franciosi, M.L.M.; do Carmo, T.I.T. Inflammatory profile in cervical cancer: Influence of purinergic signaling and possible therapeutic targets. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2022, 71, 555–564. [Google Scholar] [CrossRef]
- Xu, H.H.; Xie, Y.Y.; Jun, G.; Yang, Z.; Han, Q.Y. Dynamic changes of soluble HLA-G and cytokine plasma levels in cervical cancer patients: Potential role in cancer progression and immunotherapy. J. Cancer Res. Clin. Oncol. 2022, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Wang, Y. Effects of Th17 cells and IL-17 in the progression of cervical carcinogenesis with high-risk human papillomavirus infection. Cancer Med. 2018, 7, 297–306. [Google Scholar] [CrossRef]
- Choi, J.U.; Lee, N.K.; Seo, H.; Chung, S.W.; Al-Hilal, T.A.; Park, S.J.; Kweon, S.; Min, N.; Kim, S.K.; Ahn, S.; et al. Anticoagulation therapy promotes the tumor immune-microenvironment and potentiates the efficacy of immunotherapy by alleviating hypoxia. J. Immunother. Cancer 2021, 9, e002332. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Rafalski, V.A.; Meyer-Franke, A.; Adams, R.A.; Poda, S.B.; Rios Coronado, P.E.; Pedersen, L.; Menon, V.; Baeten, K.M.; Sikorski, S.L.; et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat. Immunol. 2018, 19, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Graf, C.; Wilgenbus, P.; Pagel, S.; Pott, J.; Marini, F.; Reyda, S.; Kitano, M.; Macher-Göppinger, S.; Weiler, H.; Ruf, W. Myeloid cell-synthesized coagulation factor X dampens antitumor immunity. Sci. Immunol. 2019, 4, eaaw8405. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, S.; You, X.; Shi, X.; Liu, L. The role of low density lipoprotein receptor-related protein 11 as a tumor promoter in cervical cancer. Cancer Manag. Res. 2019, 11, 8081–8093. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.; Zheng, R.; Yu, C.; Su, Y.; Yan, X. Predictive role of serum cholesterol and triglycerides in cervical cancer survival. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2021, 31, 171–176. [Google Scholar] [CrossRef]
- Block, J. Alzheimer’s disease might depend on enabling pathogens which do not necessarily cross the blood-brain barrier. Med. Hypotheses 2019, 125, 129–136. [Google Scholar] [CrossRef]
- Charakorn, C.; Thadanipon, K.; Chaijindaratana, S.; Rattanasiri, S.; Numthavaj, P.; Thakkinstian, A. The association between serum squamous cell carcinoma antigen and recurrence and survival of patients with cervical squamous cell carcinoma: A systematic review and meta-analysis. Gynecol. Oncol. 2018, 150, 190–200. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Zhu, J.; Wu, Y.; Wen, H.; Xia, L.; Wu, X.; Ju, X. Predictive value of preoperative serum squamous cell carcinoma antigen (SCC-Ag) level on tumor recurrence in cervical squamous cell carcinoma patients treated with radical surgery: A single-institution study. Eur. J. Surg. Oncol. 2020, 46, 131–138. [Google Scholar] [CrossRef]
- Harima, Y.; Ariga, T. Clinical value of serum biomarkers, squamous cell carcinoma antigen and apolipoprotein C-II in follow-up of patients with locally advanced cervical squamous cell carcinoma treated with radiation: A multicenter prospective cohort study. PLoS ONE 2021, 16, e0259235. [Google Scholar] [CrossRef] [PubMed]
- Gaarenstroom, K.N.; Kenter, G.G.; Bonfrer, J.M.; Korse, C.M.; Van de Vijver, M.J.; Fleuren, G.J.; Trimbos, J.B. Can initial serum cyfra 21-1, SCC antigen, and TPA levels in squamous cell cervical cancer predict lymph node metastases or prognosis? Gynecol. Oncol. 2000, 77, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, J. Discovery and validation of novel biomarkers for detection of cervical cancer. Cancer Med. 2021, 10, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, L.; Zhou, Y.; Ge, W.; Wang, H.; Wu, R.; Liu, W.; Chen, H.; Xiao, Q.; Cai, X.; et al. Stratification of follicular thyroid tumours using data-independent acquisition proteomics and a comprehensive thyroid tissue spectral library. Mol. Oncol. 2022, 16, 1611–1624. [Google Scholar] [CrossRef]
- Jia, B.; Zhao, X.; Wu, D.; Dong, Z.; Chi, Y.; Zhao, J.; Wu, M.; An, T.; Wang, Y.; Zhuo, M.; et al. Identification of serum biomarkers to predict pemetrexed/platinum chemotherapy efficacy for advanced lung adenocarcinoma patients by data-independent acquisition (DIA) mass spectrometry analysis with parallel reaction monitoring (PRM) verification. Transl. Lung Cancer Res. 2021, 10, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, Z.; Sun, Z.; Zhang, X.; Lu, L.; Wang, Y.; Zhang, M. S100A9 and ORM1 serve as predictors of therapeutic response and prognostic factors in advanced extranodal NK/T cell lymphoma patients treated with pegaspargase/gemcitabine. Sci. Rep. 2016, 6, 23695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Yu, Z.; Chen, P.; Lin, G.; Li, T.; Hou, L.; Du, Y.; Tan, W. The increased excretion of urinary orosomucoid 1 as a useful biomarker for bladder cancer. Am. J. Cancer Res. 2016, 6, 331–340. [Google Scholar] [PubMed]
- Qiong, L.; Yin, J. Characterization of alpha-1-acid glycoprotein as a potential biomarker for breast cancer. Bioengineered 2022, 13, 5818–5826. [Google Scholar] [CrossRef]
- Pan, S.; Gao, Q.; Chen, Q.; Liu, P.; Tan, Y.; Liu, F.; Xu, H. Integrative analysis-based identification and validation of a prognostic immune cell infiltration-based model for patients with advanced gastric cancer. Int. Immunopharmacol. 2021, 101, 108258. [Google Scholar] [CrossRef]
- Wu, W.; Wang, Q.; Yin, F.; Yang, Z.; Zhang, W.; Gabra, H.; Li, L. Identification of proteomic and metabolic signatures associated with chemoresistance of human epithelial ovarian cancer. Int. J. Oncol. 2016, 49, 1651–1665. [Google Scholar] [CrossRef]
- Luo, Z.; Lei, H.; Sun, Y.; Liu, X.; Su, D.F. Orosomucoid, an acute response protein with multiple modulating activities. J. Physiol. Biochem. 2015, 71, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.B.; Zhou, B.X.; Ling, Y.B.; Xiong, Z.Y.; Li, R.X.; Zhong, Y.S.; Xu, M.X.; Lu, Y.; Liang, H.; Chen, G.H.; et al. Decreased expression of ApoF associates with poor prognosis in human hepatocellular carcinoma. Gastroenterol. Rep. 2019, 7, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Da, Z.; Gao, L.; Su, G.; Yao, J.; Fu, W.; Zhang, J.; Zhang, X.; Pei, Z.; Yue, P.; Bai, B.; et al. Bioinformatics combined with quantitative proteomics analyses and identification of potential biomarkers in cholangiocarcinoma. Cancer Cell Int. 2020, 20, 130. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.; Saraswat, M.; Syrjänen, S.; Tohmola, T.; Silén, R.; Randén-Brady, R.; Carpén, T.; Hagström, J.; Haglund, C.; Mattila, P.; et al. Comparing serum protein levels can aid in differentiating HPV-negative and -positive oropharyngeal squamous cell carcinoma patients. PLoS ONE 2020, 15, e0233974. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | No. | Expression of ORM1 | Expression of APOF | ||||
|---|---|---|---|---|---|---|---|
| Low | High | p-Value | Low | High | p-Value | ||
| Age | 0.6388 | 0.8869 | |||||
| ≤45 | 20 | 9 | 11 | 10 | 10 | ||
| >45 | 23 | 12 | 11 | 11 | 12 | ||
| Histology | 0.9667 | 0.9667 | |||||
| SCC | 40 | 20 | 20 | 20 | 20 | ||
| Adenocarcinoma | 3 | 1 | 2 | 1 | 2 | ||
| Differentiation | 0.6578 | 0.0946 | |||||
| Low-Moderate | 19 | 10 | 9 | 12 | 7 | ||
| Moderate-High | 24 | 11 | 13 | 9 | 15 | ||
| Clinical Stage | 0.0299 * | 0.9374 | |||||
| Stage I-II | 34 | 20 | 14 | 16 | 18 | ||
| Stage III-IV | 9 | 1 | 8 | 5 | 4 | ||
| Tumor Size | 0.7497 | 0.2117 | |||||
| <4 cm | 35 | 18 | 17 | 15 | 20 | ||
| ≥4 cm | 8 | 3 | 5 | 6 | 2 | ||
| LVSI | 0.897 | 0.5329 | |||||
| Negative | 25 | 12 | 13 | 11 | 13 | ||
| Positive | 18 | 9 | 9 | 10 | 8 | ||
| LNM | 0.0299 * | 0.9374 | |||||
| Negative | 34 | 20 | 14 | 16 | 18 | ||
| Positive | 9 | 1 | 8 | 5 | 4 | ||
| DSI | 0.8869 | 0.8869 | |||||
| <1/2 | 20 | 10 | 10 | 10 | 10 | ||
| ≥1/2 | 23 | 11 | 12 | 11 | 12 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.; Zhang, J.; Sun, Y.; Liu, L.; Guo, L.; Zhao, C.; Zhang, J.; Qian, Q.; Cui, B.; Zhang, Y. The Plasma DIA-Based Quantitative Proteomics Reveals the Pathogenic Pathways and New Biomarkers in Cervical Cancer and High Grade Squamous Intraepithelial Lesion. J. Clin. Med. 2022, 11, 7155. https://doi.org/10.3390/jcm11237155
Han S, Zhang J, Sun Y, Liu L, Guo L, Zhao C, Zhang J, Qian Q, Cui B, Zhang Y. The Plasma DIA-Based Quantitative Proteomics Reveals the Pathogenic Pathways and New Biomarkers in Cervical Cancer and High Grade Squamous Intraepithelial Lesion. Journal of Clinical Medicine. 2022; 11(23):7155. https://doi.org/10.3390/jcm11237155
Chicago/Turabian StyleHan, Sai, Junhua Zhang, Yu Sun, Lu Liu, Lingyu Guo, Chunru Zhao, Jiaxin Zhang, Qiuhong Qian, Baoxia Cui, and Youzhong Zhang. 2022. "The Plasma DIA-Based Quantitative Proteomics Reveals the Pathogenic Pathways and New Biomarkers in Cervical Cancer and High Grade Squamous Intraepithelial Lesion" Journal of Clinical Medicine 11, no. 23: 7155. https://doi.org/10.3390/jcm11237155