
Progress in the Pathogenesis and Treatment of Neuropsychiatric Systemic Lupus Erythematosus

 ,
,
Abstract
:1. Introduction
2. Pathogenesis of NPSLE
2.1. Ischemic Pathway
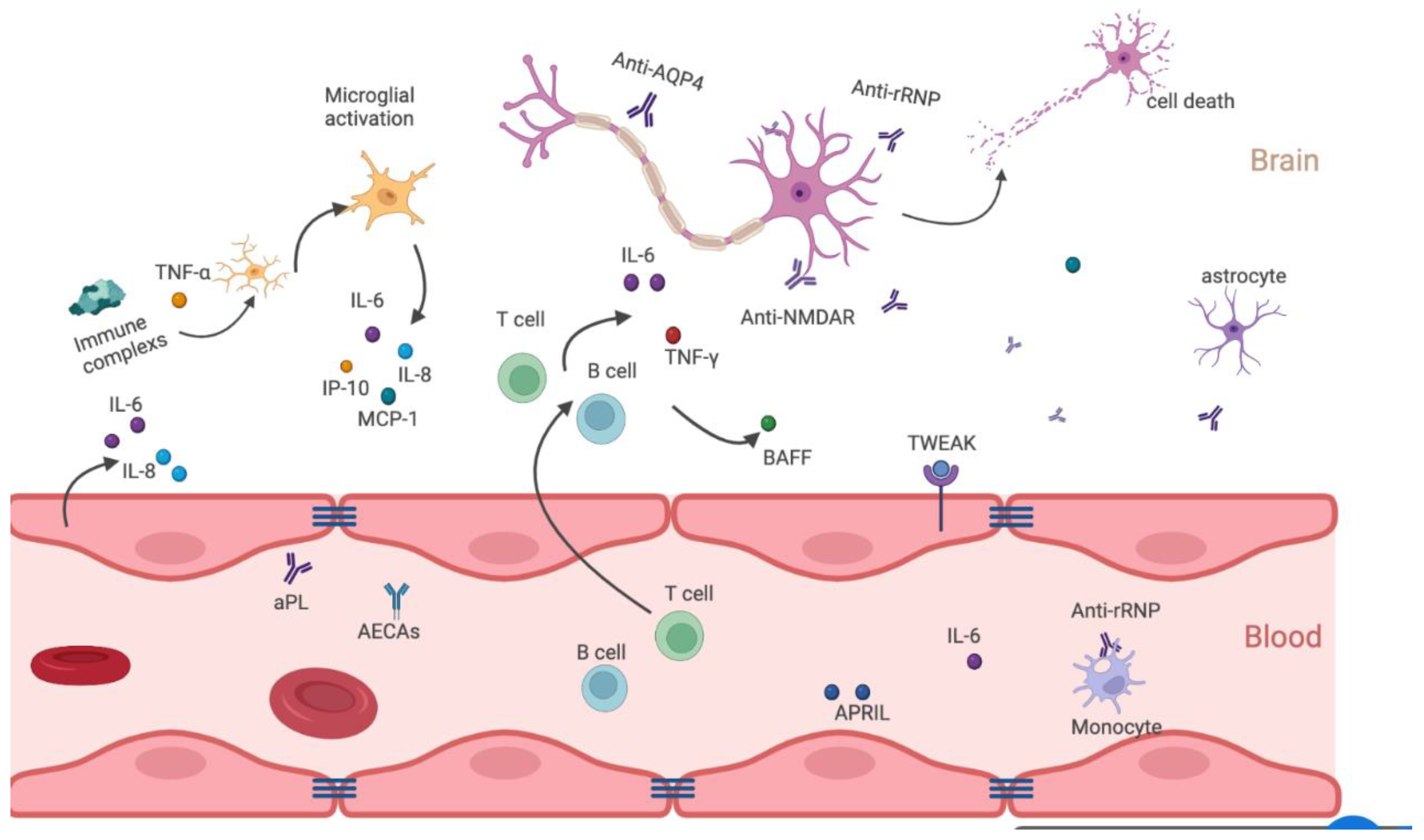
2.2. Neuroinflammatory Pathway
2.2.1. Enhanced Permeability of Brain Barrier
2.2.2. Autoantibody-Induced Inflammation
Anti-NMDAR Antibodies
Anti-RP Antibodies
AECAs
Anti-Ganglioside Antibodies
2.2.3. Cytokines-Mediated Inflammation
TWEAK/Fn14
IL-6
IFN-α
BAFF and APRIL
2.2.4. Brain-Resident and Infiltrating Cells
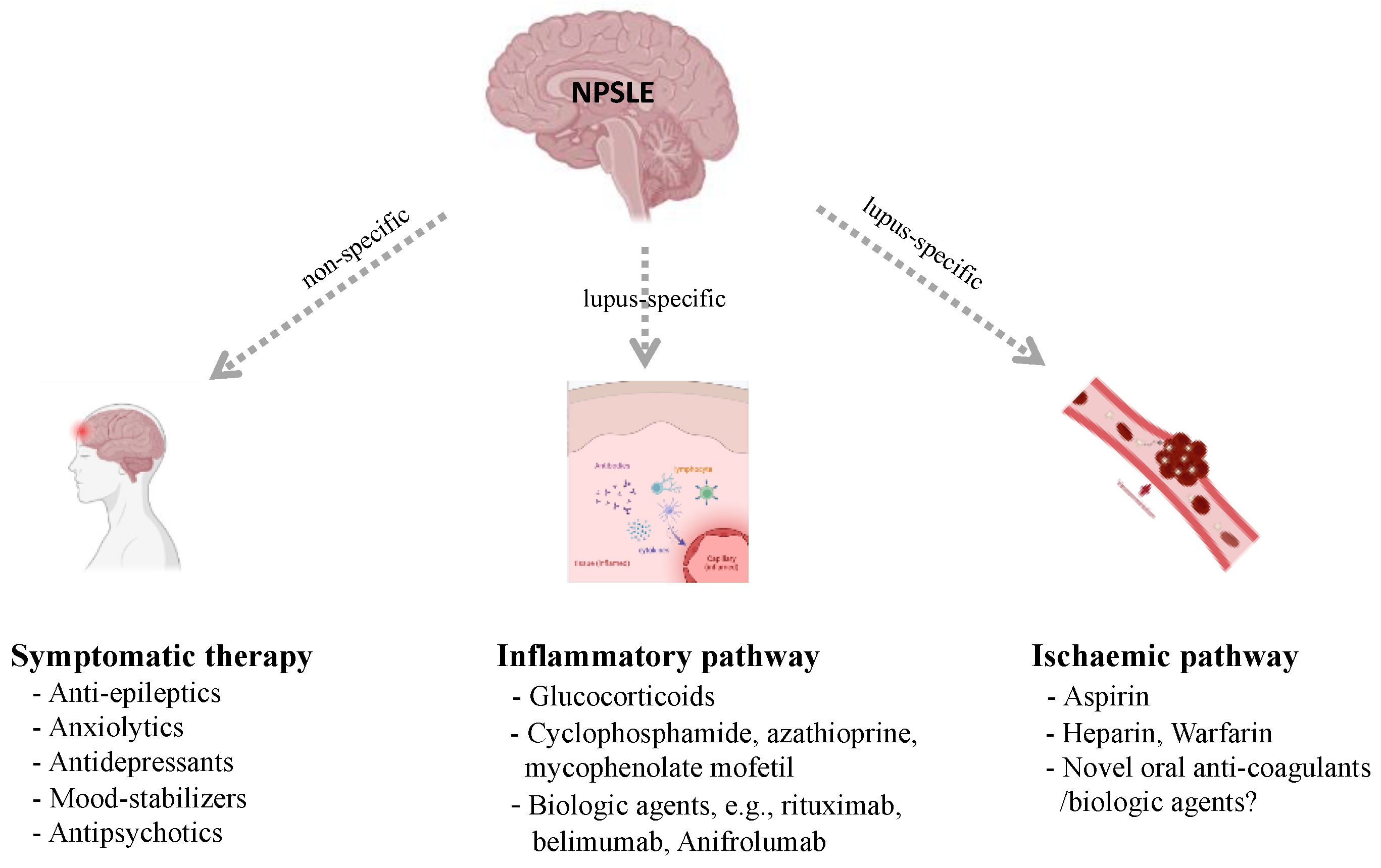
3. Current Management of NPSLE
3.1. Inflammatory Pathway Therapies
3.2. Ischemic Pathway Therapies
4. Promising Targeted Therapies
4.1. Complement Inhibitors
4.2. BBB-Targeted Therapies
4.3. MMPs Inhibitors
4.4. IFN-α/β Receptor Antagonists
4.5. BTK Inhibitors
4.6. S1P Receptor Modulator
4.7. ACE Inhibitors
4.8. CSF1R Inhibitors
4.9. Nogo-a/NgR1 Antagonists
4.10. JAK Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, M.; Wang, Y.; Zhao, J.; Wang, Q.; Wang, Z.; Tian, X.; Zeng, X. Chinese SLE Treatment and Research group (CSTAR) registry 2009–2019: Major clinical characteristics of Chinese patients with systemic lupus erythematosus. Rheumatol. Immunol. Res. 2021, 2, 43–47. [Google Scholar] [CrossRef]
- Wang, Z.; Li, M.; Ye, Z.; Li, C.; Li, Z.; Li, X.; Wu, L.; Liu, S.; Zuo, X.; Zhu, P.; et al. Long-term outcomes of patients with systemic lupus erythematosus: A Multicenter Cohort Study from CSTAR registry. Rheumatol. Immunol. Res. 2021, 2, 195–202. [Google Scholar] [CrossRef]
- Liang, M.H.; Corzillius, M.; Bae, S.C.; Lew, R.A.; Fortin, P.R.; Gordon, C.; Isenberg, D.; Alarcón, G.S.; Straaton, K.V.; Denburg, J.; et al. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999, 42, 599–608. [Google Scholar] [CrossRef]
- Bortoluzzi, A.; Piga, M.; Silvagni, E.; Chessa, E.; Mathieu, A.; Govoni, M. Peripheral nervous system involvement in systemic lupus erythematosus: A retrospective study on prevalence, associated factors and outcome. Lupus 2019, 28, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Hanly, J.G.; Li, Q.; Su, L.; Urowitz, M.B.; Gordon, C.; Bae, S.C.; Romero-Diaz, J.; Sanchez-Guerrero, J.; Bernatsky, S.; Clarke, A.E.; et al. Peripheral Nervous System Disease in Systemic Lupus Erythematosus: Results From an International Inception Cohort Study. Arthritis Rheumatol. 2020, 72, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.; Huang, M.W.; Putterman, C. Advances in the diagnosis, pathogenesis and treatment of neuropsychiatric systemic lupus erythematosus. Curr. Opin. Rheumatol. 2020, 32, 152–158. [Google Scholar] [CrossRef]
- Govoni, M.; Hanly, J.G. The management of neuropsychiatric lupus in the 21st century: Still so many unmet needs? Rheumatology 2020, 59 (Suppl. S5), v52–v62. [Google Scholar] [CrossRef]
- Schwartz, N.; Stock, A.D.; Putterman, C. Neuropsychiatric lupus: New mechanistic insights and future treatment directions. Nat. Rev. Rheumatol. 2019, 15, 137–152. [Google Scholar] [CrossRef]
- Ho, R.C.; Thiaghu, C.; Ong, H.; Lu, Y.; Ho, C.S.; Tam, W.W.; Zhang, M.W. A meta-analysis of serum and cerebrospinal fluid autoantibodies in neuropsychiatric systemic lupus erythematosus. Autoimmun. Rev. 2016, 15, 124–138. [Google Scholar] [CrossRef]
- Ben Salem, C. The pathogenesis of the antiphospholipid syndrome. N. Engl. J. Med. 2013, 368, 2334. [Google Scholar] [CrossRef]
- Mikdashi, J.; Handwerger, B. Predictors of neuropsychiatric damage in systemic lupus erythematosus: Data from the Maryland lupus cohort. Rheumatology 2004, 43, 1555–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, R.M.; Alarcón, G.S.; González, L.A.; Fernández, M.; Apte, M.; Vilá, L.M.; McGwin, G., Jr.; Reveille, J.D. Seizures in patients with systemic lupus erythematosus: Data from LUMINA, a multiethnic cohort (LUMINA LIV). Ann. Rheum. Dis. 2008, 67, 829–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomietto, P.; Annese, V.; D’Agostini, S.; Venturini, P.; La Torre, G.; De Vita, S.; Ferraccioli, G.F. General and specific factors associated with severity of cognitive impairment in systemic lupus erythematosus. Arthritis Rheum. 2007, 57, 1461–1472. [Google Scholar] [CrossRef]
- Shoenfeld, Y.; Nahum, A.; Korczyn, A.D.; Dano, M.; Rabinowitz, R.; Beilin, O.; Pick, C.G.; Leider-Trejo, L.; Kalashnikova, L.; Blank, M.; et al. Neuronal-binding antibodies from patients with antiphospholipid syndrome induce cognitive deficits following intrathecal passive transfer. Lupus 2003, 12, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Caronti, B.; Calderaro, C.; Alessandri, C.; Conti, F.; Tinghino, R.; Pini, C.; Palladini, G.; Valesini, G. Serum anti-beta2-glycoprotein I antibodies from patients with antiphospholipid antibody syndrome bind central nervous system cells. J. Autoimmun. 1998, 11, 425–429. [Google Scholar] [CrossRef]
- Chapman, J.; Cohen-Armon, M.; Shoenfeld, Y.; Korczyn, A.D. Antiphospholipid antibodies permeabilize and depolarize brain synaptoneurosomes. Lupus 1999, 8, 127–133. [Google Scholar] [CrossRef]
- Katzav, A.; Ben-Ziv, T.; Blank, M.; Pick, C.G.; Shoenfeld, Y.; Chapman, J. Antibody-specific behavioral effects: Intracerebroventricular injection of antiphospholipid antibodies induces hyperactive behavior while anti-ribosomal-P antibodies induces depression and smell deficits in mice. J. Neuroimmunol. 2014, 272, 10–15. [Google Scholar] [CrossRef]
- Hanly, J.G.; Kozora, E.; Beyea, S.D.; Birnbaum, J. Review: Nervous System Disease in Systemic Lupus Erythematosus: Current Status and Future Directions. Arthritis Rheumatol. 2019, 71, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Magro-Checa, C.; Schaarenburg, R.A.; Beaart, H.J.; Huizinga, T.W.; Steup-Beekman, G.M.; Trouw, L.A. Complement levels and anti-C1q autoantibodies in patients with neuropsychiatric systemic lupus erythematosus. Lupus 2016, 25, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Kowal, C.; DeGiorgio, L.A.; Nakaoka, T.; Hetherington, H.; Huerta, P.T.; Diamond, B.; Volpe, B.T. Cognition and immunity; antibody impairs memory. Immunity 2004, 21, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Zehnder, M.; Toledo, E.M.; Segovia-Miranda, F.; Serrano, F.G.; Benito, M.J.; Metz, C.; Retamal, C.; Álvarez, A.; Massardo, L.; Inestrosa, N.C.; et al. Anti-ribosomal P protein autoantibodies from patients with neuropsychiatric lupus impair memory in mice. Arthritis Rheumatol. 2015, 67, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulos, D.; Fanouriakis, A.; Boumpas, D.T. Update on the pathogenesis of central nervous system lupus. Curr. Opin. Rheumatol. 2019, 31, 669–677. [Google Scholar] [CrossRef] [PubMed]
- de Boer, A.G.; Gaillard, P.J. Blood-brain barrier dysfunction and recovery. J. Neural. Transm. 2006, 113, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Lauvsnes, M.B.; Omdal, R. Systemic lupus erythematosus, the brain, and anti-NR2 antibodies. J. Neurol. 2012, 259, 622–629. [Google Scholar] [CrossRef] [Green Version]
- Duarte-Delgado, N.P.; Vásquez, G.; Ortiz-Reyes, B.L. Blood-brain barrier disruption and neuroinflammation as pathophysiological mechanisms of the diffuse manifestations of neuropsychiatric systemic lupus erythematosus. Autoimmun. Rev. 2019, 18, 426–432. [Google Scholar] [CrossRef]
- Tumani, H.; Huss, A.; Bachhuber, F. The cerebrospinal fluid and barriers—Anatomic and physiologic considerations. Handb. Clin. Neurol. 2017, 146, 21–32. [Google Scholar] [CrossRef]
- James, W.G.; Hutchinson, P.; Bullard, D.C.; Hickey, M.J. Cerebral leucocyte infiltration in lupus-prone MRL/MpJ-fas lpr mice—Roles of intercellular adhesion molecule-1 and P-selectin. Clin. Exp. Immunol. 2006, 144, 299–308. [Google Scholar] [CrossRef]
- Gelb, S.; Stock, A.D.; Anzi, S.; Putterman, C.; Ben-Zvi, A. Mechanisms of neuropsychiatric lupus: The relative roles of the blood-cerebrospinal fluid barrier versus blood-brain barrier. J. Autoimmun. 2018, 91, 34–44. [Google Scholar] [CrossRef]
- Ballok, D.A.; Ma, X.; Denburg, J.A.; Arsenault, L.; Sakic, B. Ibuprofen fails to prevent brain pathology in a model of neuropsychiatric lupus. J. Rheumatol. 2006, 33, 2199–2213. [Google Scholar]
- Verheggen, I.C.M.; Van Boxtel, M.P.J.; Verhey, F.R.J.; Jansen, J.F.A.; Backes, W.H. Interaction between blood-brain barrier and glymphatic system in solute clearance. Neurosci. Biobehav. Rev. 2018, 90, 26–33. [Google Scholar] [CrossRef]
- Brøchner, C.B.; Holst, C.B.; Møllgård, K. Outer brain barriers in rat and human development. Front. Neurosci. 2015, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Aranow, C.; Diamond, B.; Mackay, M. Glutamate receptor biology and its clinical significance in neuropsychiatric systemic lupus erythematosus. Rheum. Dis. Clin. N. Am. 2010, 36, 187–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirohata, S.; Arinuma, Y.; Yanagida, T.; Yoshio, T. Blood-brain barrier damages and intrathecal synthesis of anti-N-methyl-D-aspartate receptor NR2 antibodies in diffuse psychiatric/neuropsychological syndromes in systemic lupus erythematosus. Arthritis Res. Ther. 2014, 16, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arinuma, Y.; Yanagida, T.; Hirohata, S. Association of cerebrospinal fluid anti-NR2 glutamate receptor antibodies with diffuse neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2008, 58, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Steup-Beekman, G.; Steens, S.; van Buchem, M.; Huizinga, T. Anti-NMDA receptor autoantibodies in patients with systemic lupus erythematosus and their first-degree relatives. Lupus 2007, 16, 329–334. [Google Scholar] [CrossRef]
- Diamond, B.; Volpe, B.T. A model for lupus brain disease. Immunol. Rev. 2012, 248, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Omdal, R.; Brokstad, K.; Waterloo, K.; Koldingsnes, W.; Jonsson, R.; Mellgren, S.I. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur. J. Neurol. 2005, 12, 392–398. [Google Scholar] [CrossRef]
- Lapteva, L.; Nowak, M.; Yarboro, C.H.; Takada, K.; Roebuck-Spencer, T.; Weickert, T.; Bleiberg, J.; Rosenstein, D.; Pao, M.; Patronas, N.; et al. Anti-N-methyl-D-aspartate receptor antibodies, cognitive dysfunction, and depression in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2505–2514. [Google Scholar] [CrossRef]
- Gao, H.X.; Campbell, S.R.; Cui, M.H.; Zong, P.; Hee-Hwang, J.; Gulinello, M.; Putterman, C. Depression is an early disease manifestation in lupus-prone MRL/lpr mice. J. Neuroimmunol. 2009, 207, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Brimberg, L.; Mader, S.; Fujieda, Y.; Arinuma, Y.; Kowal, C.; Volpe, B.T.; Diamond, B. Antibodies as Mediators of Brain Pathology. Trends Immunol. 2015, 36, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Y.; Zhao, Y.H.; Zhang, J.H.; Lei, H.W. Anti-N-Methyl-D-Aspartic Acid Receptor 2 (Anti-NR2) Antibody in Neuropsychiatric Lupus Serum Damages the Blood-Brain Barrier and Enters the Brain. Med. Sci. Monit. 2019, 25, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Karassa, F.B.; Afeltra, A.; Ambrozic, A.; Chang, D.M.; De Keyser, F.; Doria, A.; Galeazzi, M.; Hirohata, S.; Hoffman, I.E.; Inanc, M.; et al. Accuracy of anti-ribosomal P protein antibody testing for the diagnosis of neuropsychiatric systemic lupus erythematosus: An international meta-analysis. Arthritis Rheum. 2006, 54, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Husebye, E.S.; Sthoeger, Z.M.; Dayan, M.; Zinger, H.; Elbirt, D.; Levite, M.; Mozes, E. Autoantibodies to a NR2A peptide of the glutamate/NMDA receptor in sera of patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2005, 64, 1210–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isshi, K.; Hirohata, S. Association of anti-ribosomal P protein antibodies with neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 1996, 39, 1483–1490. [Google Scholar] [CrossRef]
- Choi, M.Y.; FitzPatrick, R.D.; Buhler, K.; Mahler, M.; Fritzler, M.J. A review and meta-analysis of anti-ribosomal P autoantibodies in systemic lupus erythematosus. Autoimmun. Rev. 2020, 19, 102463. [Google Scholar] [CrossRef]
- Gaber, W.; Ezzat, Y.; El Fayoumy, N.M.; Helmy, H.; Mohey, A.M. Detection of asymptomatic cranial neuropathies in patients with systemic lupus erythematosus and their relation to antiribosomal P antibody levels and disease activity. Clin. Rheumatol. 2014, 33, 1459–1466. [Google Scholar] [CrossRef]
- Arinuma, Y.; Kikuchi, H.; Hirohata, S. Anti-ribosomal P protein antibodies influence mortality of patients with diffuse psychiatric/neuropsychological syndromes in systemic lupus erythematous involving a severe form of the disease. Mod. Rheumatol. 2019, 29, 612–618. [Google Scholar] [CrossRef]
- Katzav, A.; Solodeev, I.; Brodsky, O.; Chapman, J.; Pick, C.G.; Blank, M.; Zhang, W.; Reichlin, M.; Shoenfeld, Y. Induction of autoimmune depression in mice by anti-ribosomal P antibodies via the limbic system. Arthritis Rheum. 2007, 56, 938–948. [Google Scholar] [CrossRef]
- Matus, S.; Burgos, P.V.; Bravo-Zehnder, M.; Kraft, R.; Porras, O.H.; Farías, P.; Barros, L.F.; Torrealba, F.; Massardo, L.; Jacobelli, S.; et al. Antiribosomal-P autoantibodies from psychiatric lupus target a novel neuronal surface protein causing calcium influx and apoptosis. J. Exp. Med. 2007, 204, 3221–3234. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Park, Y.B.; Lee, W.K.; Lee, K.H.; Lee, S.K. Clinical associations of anti-endothelial cell antibodies in patients with systemic lupus erythematosus. Rheumatol. Int. 2000, 20, 1–7. [Google Scholar] [CrossRef]
- Conti, F.; Alessandri, C.; Bompane, D.; Bombardieri, M.; Spinelli, F.R.; Rusconi, A.C.; Valesini, G. Autoantibody profile in systemic lupus erythematosus with psychiatric manifestations: A role for anti-endothelial-cell antibodies. Arthritis Res. Ther. 2004, 6, R366–R372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubarski, K.; Mania, A.; Michalak, S.; Osztynowicz, K.; Mazur-Melewska, K.; Figlerowicz, M. The Clinical Spectrum of Autoimmune-Mediated Neurological Diseases in Paediatric Population. Brain Sci. 2022, 12, 584. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, A.; Silvagni, E.; Furini, F.; Piga, M.; Govoni, M. Peripheral nervous system involvement in systemic lupus erythematosus: A review of the evidence. Clin. Exp. Rheumatol. 2019, 37, 146–155. [Google Scholar]
- Labrador-Horrillo, M.; Martinez-Valle, F.; Gallardo, E.; Rojas-Garcia, R.; Ordi-Ros, J.; Vilardell, M. Anti-ganglioside antibodies in patients with systemic lupus erythematosus and neurological manifestations. Lupus 2012, 21, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Loftis, J.M.; Huckans, M.; Morasco, B.J. Neuroimmune mechanisms of cytokine-induced depression: Current theories and novel treatment strategies. Neurobiol. Dis. 2010, 37, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Yepes, M. TWEAK and Fn14 in the Neurovascular Unit. Front. Immunol. 2013, 4, 367. [Google Scholar] [CrossRef] [Green Version]
- Stock, A.D.; Wen, J.; Putterman, C. Neuropsychiatric Lupus, the Blood Brain Barrier, and the TWEAK/Fn14 Pathway. Front. Immunol. 2013, 4, 484. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Xia, Y.; Stock, A.; Michaelson, J.S.; Burkly, L.C.; Gulinello, M.; Putterman, C. Neuropsychiatric disease in murine lupus is dependent on the TWEAK/Fn14 pathway. J. Autoimmun. 2013, 43, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Chen, C.H.; Stock, A.; Doerner, J.; Gulinello, M.; Putterman, C. Intracerebroventricular administration of TNF-like weak inducer of apoptosis induces depression-like behavior and cognitive dysfunction in non-autoimmune mice. Brain Behav. Immun. 2016, 54, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Hirohata, S.; Kanai, Y.; Mitsuo, A.; Tokano, Y.; Hashimoto, H. Accuracy of cerebrospinal fluid IL-6 testing for diagnosis of lupus psychosis. A multicenter retrospective study. Clin. Rheumatol. 2009, 28, 1319–1323. [Google Scholar] [CrossRef]
- Katsumata, Y.; Harigai, M.; Kawaguchi, Y.; Fukasawa, C.; Soejima, M.; Takagi, K.; Tanaka, M.; Ichida, H.; Tochimoto, A.; Kanno, T.; et al. Diagnostic reliability of cerebral spinal fluid tests for acute confusional state (delirium) in patients with systemic lupus erythematosus: Interleukin 6 (IL-6), IL-8, interferon-alpha, IgG index, and Q-albumin. J. Rheumatol. 2007, 34, 2010–2017. [Google Scholar] [PubMed]
- Trysberg, E.; Nylen, K.; Rosengren, L.E.; Tarkowski, A. Neuronal and astrocytic damage in systemic lupus erythematosus patients with central nervous system involvement. Arthritis Rheum. 2003, 48, 2881–2887. [Google Scholar] [CrossRef] [PubMed]
- Dellalibera-Joviliano, R.; Dos Reis, M.L.; Cunha Fde, Q.; Donadi, E.A. Kinins and cytokines in plasma and cerebrospinal fluid of patients with neuropsychiatric lupus. J. Rheumatol. 2003, 30, 485–492. [Google Scholar] [PubMed]
- Bialas, A.R.; Presumey, J.; Das, A.; van der Poel, C.E.; Lapchak, P.H.; Mesin, L.; Victora, G.; Tsokos, G.C.; Mawrin, C.; Herbst, R.; et al. Retraction Note: Microglia-dependent synapse loss in type I interferon-mediated lupus. Nature 2020, 578, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santer, D.M.; Yoshio, T.; Minota, S.; Möller, T.; Elkon, K.B. Potent induction of IFN-alpha and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus. J. Immunol. 2009, 182, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Kobayashi, A.; Yamanaka, H. Cytokines and chemokines in neuropsychiatric syndromes of systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 2010, 268436. [Google Scholar] [CrossRef] [Green Version]
- George-Chandy, A.; Trysberg, E.; Eriksson, K. Raised intrathecal levels of APRIL and BAFF in patients with systemic lupus erythematosus: Relationship to neuropsychiatric symptoms. Arthritis Res. Ther. 2008, 10, R97. [Google Scholar] [CrossRef] [Green Version]
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Doerner, J.; Weidenheim, K.; Xia, Y.; Stock, A.; Michaelson, J.S.; Baruch, K.; Deczkowska, A.; Gulinello, M.; Schwartz, M.; et al. TNF-like weak inducer of apoptosis promotes blood brain barrier disruption and increases neuronal cell death in MRL/lpr mice. J. Autoimmun. 2015, 60, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Crupi, R.; Cambiaghi, M.; Spatz, L.; Hen, R.; Thorn, M.; Friedman, E.; Vita, G.; Battaglia, F. Reduced adult neurogenesis and altered emotional behaviors in autoimmune-prone B-cell activating factor transgenic mice. Biol. Psychiatry 2010, 67, 558–566. [Google Scholar] [CrossRef]
- Cunningham, M.A.; Wirth, J.R.; Freeman, L.R.; Boger, H.A.; Granholm, A.C.; Gilkeson, G.S. Estrogen receptor alpha deficiency protects against development of cognitive impairment in murine lupus. J. Neuroinflamm. 2014, 11, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragoso-Loyo, H.; Richaud-Patin, Y.; Orozco-Narváez, A.; Dávila-Maldonado, L.; Atisha-Fregoso, Y.; Llorente, L.; Sánchez-Guerrero, J. Interleukin-6 and chemokines in the neuropsychiatric manifestations of systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Nestor, J.; Arinuma, Y.; Huerta, T.S.; Kowal, C.; Nasiri, E.; Kello, N.; Fujieda, Y.; Bialas, A.; Hammond, T.; Sriram, U.; et al. Lupus antibodies induce behavioral changes mediated by microglia and blocked by ACE inhibitors. J. Exp. Med. 2018, 215, 2554–2566. [Google Scholar] [CrossRef] [Green Version]
- Stock, A.D.; Der, E.; Gelb, S.; Huang, M.; Weidenheim, K.; Ben-Zvi, A.; Putterman, C. Tertiary lymphoid structures in the choroid plexus in neuropsychiatric lupus. JCI Insight 2019, 4, e124203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zhao, Y.; Zhang, Z.; Huang, C.; Liu, Y.; Gu, J.; Zhang, X.; Xu, H.; Li, X.; Wu, L.; et al. 2020 Chinese guidelines for the diagnosis and treatment of systemic lupus erythematosus. Rheumatol. Immunol. Res. 2020, 1, 5–23. [Google Scholar] [CrossRef]
- Barile-Fabris, L.; Ariza-Andraca, R.; Olguín-Ortega, L.; Jara, L.J.; Fraga-Mouret, A.; Miranda-Limón, J.M.; Fuentes de la Mata, J.; Clark, P.; Vargas, F.; Alocer-Varela, J. Controlled clinical trial of IV cyclophosphamide versus IV methylprednisolone in severe neurological manifestations in systemic lupus erythematosus. Ann. Rheum. Dis. 2005, 64, 620–625. [Google Scholar] [CrossRef]
- Denburg, S.D.; Carbotte, R.M.; Denburg, J.A. Corticosteroids and neuropsychological functioning in patients with systemic lupus erythematosus. Arthritis Rheum. 1994, 37, 1311–1320. [Google Scholar] [CrossRef]
- Ruiz-Arruza, I.; Lozano, J.; Cabezas-Rodriguez, I.; Medina, J.A.; Ugarte, A.; Erdozain, J.G.; Ruiz-Irastorza, G. Restrictive Use of Oral Glucocorticoids in Systemic Lupus Erythematosus and Prevention of Damage Without Worsening Long-Term Disease Control: An Observational Study. Arthritis Care Res. 2018, 70, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Bolanos, S.H.; Khan, D.A.; Hanczyc, M.; Bauer, M.S.; Dhanani, N.; Brown, E.S. Assessment of mood states in patients receiving long-term corticosteroid therapy and in controls with patient-rated and clinician-rated scales. Ann. Allergy Asthma Immunol. 2004, 92, 500–505. [Google Scholar] [CrossRef]
- Lewis, D.A.; Smith, R.E. Steroid-induced psychiatric syndromes. A report of 14 cases and a review of the literature. J. Affect. Disord. 1983, 5, 319–332. [Google Scholar] [CrossRef]
- Mok, C.C.; Lau, C.S.; Wong, R.W. Treatment of lupus psychosis with oral cyclophosphamide followed by azathioprine maintenance: An open-label study. Am. J. Med. 2003, 115, 59–62. [Google Scholar] [CrossRef]
- Dale, R.C.; Brilot, F.; Duffy, L.V.; Twilt, M.; Waldman, A.T.; Narula, S.; Muscal, E.; Deiva, K.; Andersen, E.; Eyre, M.R.; et al. Utility and safety of rituximab in pediatric autoimmune and inflammatory CNS disease. Neurology 2014, 83, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narváez, J.; Ríos-Rodriguez, V.; de la Fuente, D.; Estrada, P.; López-Vives, L.; Gómez-Vaquero, C.; Nolla, J.M. Rituximab therapy in refractory neuropsychiatric lupus: Current clinical evidence. Semin. Arthritis Rheum. 2011, 41, 364–372. [Google Scholar] [CrossRef]
- Pranzatelli, M.R.; Tate, E.D.; Travelstead, A.L.; Barbosa, J.; Bergamini, R.A.; Civitello, L.; Franz, D.N.; Greffe, B.S.; Hanson, R.D.; Hurwitz, C.A.; et al. Rituximab (anti-CD20) adjunctive therapy for opsoclonus-myoclonus syndrome. J. Pediatr. Hematol. Oncol. 2006, 28, 585–593. [Google Scholar] [CrossRef]
- Jacob, A.; Weinshenker, B.G.; Violich, I.; McLinskey, N.; Krupp, L.; Fox, R.J.; Wingerchuk, D.M.; Boggild, M.; Constantinescu, C.S.; Miller, A.; et al. Treatment of neuromyelitis optica with rituximab: Retrospective analysis of 25 patients. Arch. Neurol. 2008, 65, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Manzi, S.; Sánchez-Guerrero, J.; Merrill, J.T.; Furie, R.; Gladman, D.; Navarra, S.V.; Ginzler, E.M.; D’Cruz, D.P.; Doria, A.; Cooper, S.; et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: Combined results from two phase III trials. Ann. Rheum. Dis. 2012, 71, 1833–1838. [Google Scholar] [CrossRef]
- de Amorim, L.C.; Maia, F.M.; Rodrigues, C.E. Stroke in systemic lupus erythematosus and antiphospholipid syndrome: Risk factors, clinical manifestations, neuroimaging, and treatment. Lupus 2017, 26, 529–536. [Google Scholar] [CrossRef]
- Vadgama, T.S.; Smith, A.; Bertolaccini, M.L. Treatment in thrombotic antiphospholipid syndrome: A review. Lupus 2019, 28, 1181–1188. [Google Scholar] [CrossRef]
- Finazzi, G.; Marchioli, R.; Brancaccio, V.; Schinco, P.; Wisloff, F.; Musial, J.; Baudo, F.; Berrettini, M.; Testa, S.; D’Angelo, A.; et al. A randomized clinical trial of high-intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS). J. Thromb. Haemost. 2005, 3, 848–853. [Google Scholar] [CrossRef]
- Crowther, M.A.; Ginsberg, J.S.; Julian, J.; Denburg, J.; Hirsh, J.; Douketis, J.; Laskin, C.; Fortin, P.; Anderson, D.; Kearon, C.; et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N. Engl. J. Med. 2003, 349, 1133–1138. [Google Scholar] [CrossRef]
- Dobrowolski, C.; Erkan, D. Treatment of antiphospholipid syndrome beyond anticoagulation. Clin. Immunol. 2019, 206, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Raschi, E.; Testoni, C.; Tincani, A.; Balestrieri, G.; Molteni, R.; Khamashta, M.A.; Tremoli, E.; Camera, M. Statins prevent endothelial cell activation induced by antiphospholipid (anti-beta2-glycoprotein I) antibodies: Effect on the proadhesive and proinflammatory phenotype. Arthritis Rheum. 2001, 44, 2870–2878. [Google Scholar] [CrossRef]
- Jung, H.; Bobba, R.; Su, J.; Shariati-Sarabi, Z.; Gladman, D.D.; Urowitz, M.; Lou, W.; Fortin, P.R. The protective effect of antimalarial drugs on thrombovascular events in systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 863–868. [Google Scholar] [CrossRef]
- Jacob, A.; Hack, B.; Chiang, E.; Garcia, J.G.; Quigg, R.J.; Alexander, J.J. C5a alters blood-brain barrier integrity in experimental lupus. FASEB J. 2010, 24, 1682–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, S.D.; Parikh, N.U.; Woodruff, T.M.; Jarvis, J.N.; Lopez, M.; Hennon, T.; Cunningham, P.; Quigg, R.J.; Schwartz, S.A.; Alexander, J.J. C5a alters blood-brain barrier integrity in a human in vitro model of systemic lupus erythematosus. Immunology 2015, 146, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Di Minno, M.N.D.; Emmi, G.; Ambrosino, P.; Scalera, A.; Tufano, A.; Cafaro, G.; Peluso, R.; Bettiol, A.; Di Scala, G.; Silvestri, E.; et al. Subclinical atherosclerosis in asymptomatic carriers of persistent antiphospholipid antibodies positivity: A cross-sectional study. Int. J. Cardiol. 2019, 274, 1–6. [Google Scholar] [CrossRef]
- De Luca, C.; Virtuoso, A.; Maggio, N.; Papa, M. Neuro-Coagulopathy: Blood Coagulation Factors in Central Nervous System Diseases. Int. J. Mol. Sci. 2017, 18, 2128. [Google Scholar] [CrossRef]
- Chehaibi, K.; le Maire, L.; Bradoni, S.; Escola, J.C.; Blanco-Vaca, F.; Slimane, M.N. Effect of PPAR-β/δ agonist GW0742 treatment in the acute phase response and blood-brain barrier permeability following brain injury. Transl. Res. 2017, 182, 27–48. [Google Scholar] [CrossRef]
- Niego, B.; Lee, N.; Larsson, P.; De Silva, T.M.; Au, A.E.; McCutcheon, F.; Medcalf, R.L. Selective inhibition of brain endothelial Rho-kinase-2 provides optimal protection of an in vitro blood-brain barrier from tissue-type plasminogen activator and plasmin. PLoS ONE 2017, 12, e0177332. [Google Scholar] [CrossRef]
- Lu, X.Y.; Zhu, C.Q.; Qian, J.; Chen, X.X.; Ye, S.; Gu, Y.Y. Intrathecal cytokine and chemokine profiling in neuropsychiatric lupus or lupus complicated with central nervous system infection. Lupus 2010, 19, 689–695. [Google Scholar] [CrossRef]
- Klein-Gitelman, M.; Brunner, H.I. The impact and implications of neuropsychiatric systemic lupus erythematosus in adolescents. Curr. Rheumatol. Rep. 2009, 11, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Ainiala, H.; Hietaharju, A.; Dastidar, P.; Loukkola, J.; Lehtimäki, T.; Peltola, J.; Korpela, M.; Heinonen, T.; Nikkari, S.T. Increased serum matrix metalloproteinase 9 levels in systemic lupus erythematosus patients with neuropsychiatric manifestations and brain magnetic resonance imaging abnormalities. Arthritis Rheum. 2004, 50, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Trysberg, E.; Blennow, K.; Zachrisson, O.; Tarkowski, A. Intrathecal levels of matrix metalloproteinases in systemic lupus erythematosus with central nervous system engagement. Arthritis Res. Ther. 2004, 6, R551–R556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S. Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Mok, C.C. The dawn of a new era of therapies in systemic lupus erythematosus. Rheumatol. Immunol. Res. 2020, 1, 31–37. [Google Scholar] [CrossRef]
- Hendriks, R.W. Drug discovery: New Btk inhibitor holds promise. Nat. Chem. Biol. 2011, 7, 4–5. [Google Scholar] [CrossRef]
- Jongstra-Bilen, J.; Puig Cano, A.; Hasija, M.; Xiao, H.; Smith, C.I.; Cybulsky, M.I. Dual functions of Bruton’s tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J. Immunol. 2008, 181, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Gabhann-Dromgoole, J.N.; Hams, E.; Smith, S.; Wynne, C.; Byrne, J.C.; Brennan, K.; Spence, S.; Kissenpfennig, A.; Johnston, J.A.; Fallon, P.G.; et al. Btk regulates macrophage polarization in response to lipopolysaccharide. PLoS ONE 2014, 9, e85834. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, S.A.; Wen, J.; Doerner, J.; Stock, A.; Cuda, C.M.; Makinde, H.M.; Perlman, H.; Bosanac, T.; Webb, D.; Nabozny, G.; et al. Highly selective inhibition of Bruton’s tyrosine kinase attenuates skin and brain disease in murine lupus. Arthritis Res. Ther. 2018, 20, 10. [Google Scholar] [CrossRef] [Green Version]
- Werth, V.P.; Merrill, J.T. A double-blind, randomized, placebo-controlled, phase II trial of baricitinib for systemic lupus erythematosus: How to optimize lupus trials to examine effects on cutaneous lupus erythematosus. Br. J. Dermatol. 2019, 180, 964–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Mike, E.V.; Makinde, H.M.; Der, E.; Stock, A.; Gulinello, M.; Gadhvi, G.T.; Winter, D.R.; Cuda, C.M.; Putterman, C. Neuropsychiatric Systemic Lupus Erythematosus Is Dependent on Sphingosine-1-Phosphate Signaling. Front. Immunol. 2018, 9, 2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, D.; Tian, T.; Yao, S.; Cao, K.; Zhu, X.; Zhang, M.; Wen, S.; Li, L.; Shi, M.; Zhou, H. FTY720 attenuates behavioral deficits in a murine model of systemic lupus erythematosus. Brain Behav. Immun. 2018, 70, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Chitu, V.; Stanley, E.R. Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol. 2006, 18, 39–48. [Google Scholar] [CrossRef]
- Chalmers, S.A.; Wen, J.; Shum, J.; Doerner, J.; Herlitz, L.; Putterman, C. CSF-1R inhibition attenuates renal and neuropsychiatric disease in murine lupus. Clin. Immunol. 2017, 185, 100–108. [Google Scholar] [CrossRef]
- Chi, O.Z.; Hunter, C.; Liu, X.; Weiss, H.R. Effects of exogenous excitatory amino acid neurotransmitters on blood-brain barrier disruption in focal cerebral ischemia. Neurochem. Res. 2009, 34, 1249–1254. [Google Scholar] [CrossRef]
- Fukuyama, T.; Tschernig, T.; Qi, Y.; Volmer, D.A.; Bäumer, W. Aggression behaviour induced by oral administration of the Janus-kinase inhibitor tofacitinib, but not oclacitinib, under stressful conditions. Eur. J. Pharmacol. 2015, 764, 278–282. [Google Scholar] [CrossRef]
- Hasni, S.A.; Gupta, S.; Davis, M.; Poncio, E.; Temesgen-Oyelakin, Y.; Carlucci, P.M.; Wang, X.; Naqi, M.; Playford, M.P.; Goel, R.R.; et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat. Commun. 2021, 12, 3391. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Promising Targeted Therapies | Underlying Mechanisms and Clinical Findings | Experimental Arrangement | Potential Drugs |
|---|---|---|---|
| Complement inhibitors | Complement signaling promotes the loss of BBB integrity. Blocking the complement cascades relieved the symptoms of NPSLE. Complement deposits were present in most of patients with NPSLE. | Human brain autopsies | Eculizumab |
| BBB-targeted therapy | BBB disruption is essential in the neuronal damage process. Restoration of normal BBB function may reduce the development of neuropsychiatric manifestations | Human and mouse cells; C57 BL/6J mice, respectively | GW0742, a peroxisome proliferator-activated receptor β/δ agonist; KD025, a rho kinase inhibitor. |
| MMPs inhibitors | There is an association between CSF/serum levels of MMP-9, psychiatric NPSLE, and markers for neuronal/astrocytic damage. MMP-9 may contribute to the pathogenesis of psychiatric NPSLE by stimulating T-cell migration | - | - |
| IFN-α/β receptor antagonists | IFN receptor inhibition decreased microglia-related synaptic loss and attenuated anxiety-like behavior and cognitive deficits in animal models. | 564Igi lupus-prone mice | Anifrolumab |
| BTK inhibitors | Use of BI-BTK-1 (an inhibitor of BTK) in MRL/lpr mice, decreased the infiltration of macrophages, T cells, and B cells in the choroid plexus, and improved cognitive function. | MRL/lpr mice | Ibrutinib; Evobrutinib |
| S1P receptor modulator | S1P receptor modulators decreased proinflammatory cytokine secretion by microglia and significantly improved spatial memory and depression-like behavior. Fingolimod (a S1P receptor modulator) treatment attenuated neuropsychiatric manifestations, reversed the entry of immune components, and decreased BBB leakage. Fingolimod-treated microglia revealed down- regulated of multiple immune-mediated pathways, including NF-kB signaling and the IFN response with the negative regulation of type I IFN-mediated signaling. | MRL/lpr mice | Fingolimod |
| ACE inhibitors | ACE inhibitors treatment suppressed microglial activation and promoted cognitive status. | BALB/c mice | Captopril; Perindopril |
| CSF1R inhibitors | CSF1R is essential in both macrophage and microglia function. Inhibition of CSF1R signaling in MRL/lpr mice reduced the brain expression of proinflammatory cytokines and attenuated depression performance. | MRL/lpr mice | GW2580, a small CSF-1R kinase inhibitor; depletion of microglia |
| Nogo-a/NgR1 antagonists | Nogo-a/NgR1 in the CSF is significantly increased in NPSLE. Nogo-a/NgR1 antagonists improved cognitive function, decreased the expression of pro-inflammatory components, and reduced axonal degeneration and demyelination. | MRL/lpr mice | Nogo-66 |
| JAK inhibitors | JAK inhibitors penetrate the BBB and reduce the production of several cytokines, including type I IFNs. | - | Tofacitinib |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Wang, Z.; Zhang, S.; Wu, Y.; Zhang, L.; Zhao, J.; Wang, Q.; Tian, X.; Li, M.; Zeng, X. Progress in the Pathogenesis and Treatment of Neuropsychiatric Systemic Lupus Erythematosus. J. Clin. Med. 2022, 11, 4955. https://doi.org/10.3390/jcm11174955
Wang M, Wang Z, Zhang S, Wu Y, Zhang L, Zhao J, Wang Q, Tian X, Li M, Zeng X. Progress in the Pathogenesis and Treatment of Neuropsychiatric Systemic Lupus Erythematosus. Journal of Clinical Medicine. 2022; 11(17):4955. https://doi.org/10.3390/jcm11174955
Chicago/Turabian StyleWang, Minhui, Ziqian Wang, Shangzhu Zhang, Yang Wu, Li Zhang, Jiuliang Zhao, Qian Wang, Xinping Tian, Mengtao Li, and Xiaofeng Zeng. 2022. "Progress in the Pathogenesis and Treatment of Neuropsychiatric Systemic Lupus Erythematosus" Journal of Clinical Medicine 11, no. 17: 4955. https://doi.org/10.3390/jcm11174955