
Oxidative Stress and Endoplasmic Reticulum Stress in Rare Respiratory Diseases
 , , , and
, , , and
Abstract
:1. Introduction
2. What Is Oxidative Stress?
3. Clinical Relevance of Oxidative Stress
4. Endoplasmic Reticulum Stress
4.1. ATF6
4.2. IRE1
4.3. PERK
5. Endoplasmic Reticulum Stress and Oxidative Stress

{kind=link}
{kind=link}
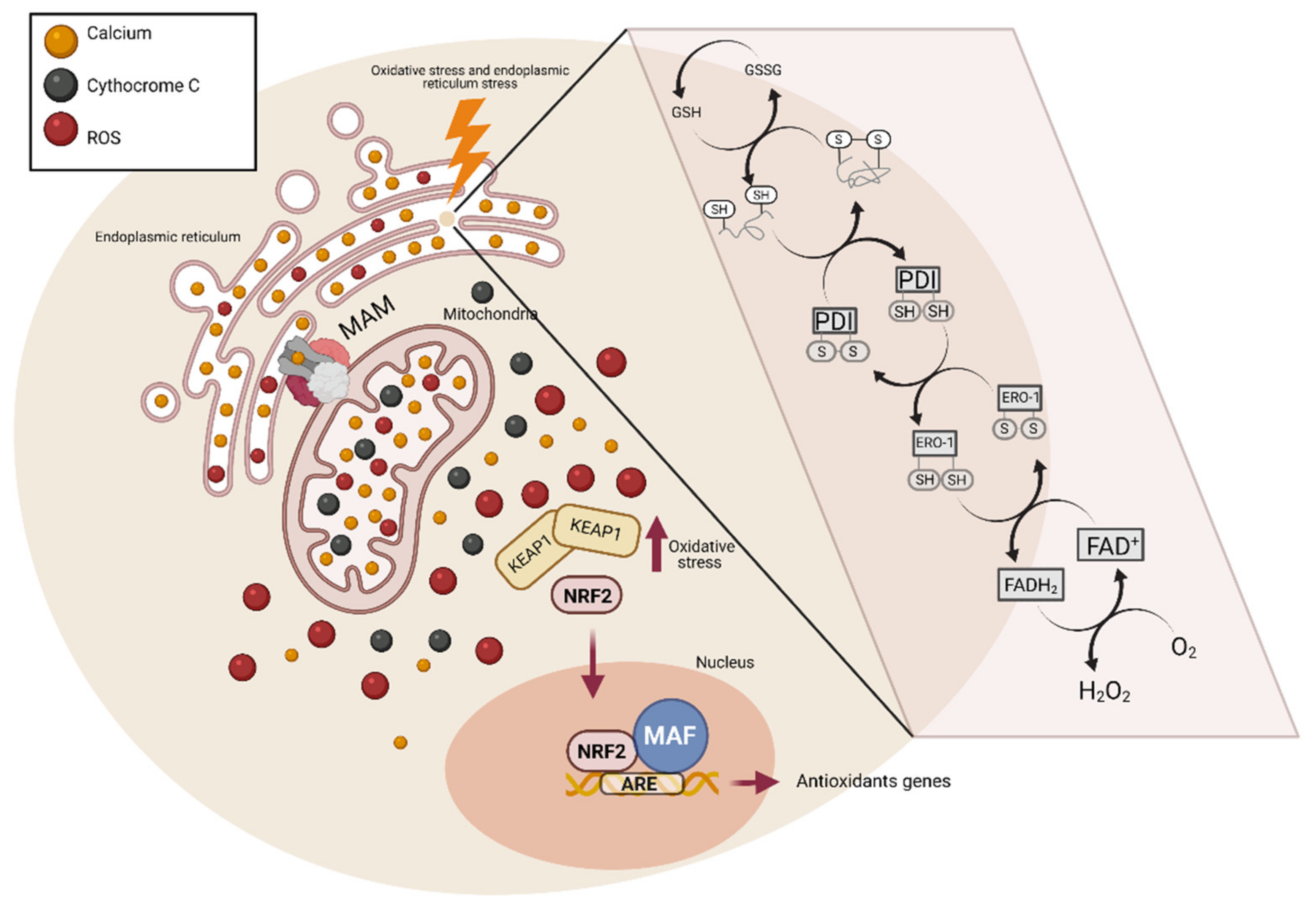{kind=link}
| Cell Type | Stimulus | ER Stress Measurement | OS Measurement | ER–OS Branch | Observation | Ref |
|---|---|---|---|---|---|---|
| PERK-/- and PERK+/+ MEFs | Phox-ER stress | CHOP and Chaperones ↑ | DiOC6 ↓ (Ψm) NAO ↑ (oxidized cardiolipin) | Cross-talk ER-mitochondria | PERK is a component of MAMs. | [73] |
| Bax-/- haemopoietic cells | Tunicamycin | CHOP ↑ BiP/GRP78 ↑ | Mitotracker Red ↓ (Ψm) → ↑mitochondrial O2− | Cross-talk ER-mitochondria | Mitochondrial mass, O2 consumption & ATP production ↓. | [77] |
| S. cerevisiae Hip deficient cells (erv29∆) | CPY (a misfolded mutant form of protein carboxypeptidase) | IRE1 ↑ | DHR-123 ↑ (General ROS) | Antioxidant genes | GSH suppress ROS and cell death but not ER stress. | [83] |
| CHO cells | Misfolded factor VIII expression | BiP ↑ eIF2α-P ↑ CHOP ↑ | DCF ↑ (peroxides), DHE ↑ (superoxide), MDA ↑, GSH ↓, GSSG ↑ HODE ↓ (hydroxioctadecaidienoic acid) Prot. Carbonyls ↓ | Antioxidant genes | BHA (butylated hydroxyanisole) antioxidant ↓apoptosis, ↓intracellular accumulation of misfolded proteins and ↑secretion of properly folded proteins. | [84] |
| PERK-/- and ATF4-/- fibroblast and C. elegans PERK -/- | Tunicamycin | Several genes | DCF ↑ (peroxides) | ER oxidative environment for disulfide bond forming | ATF4-/- cells are impaired in expressing genes involved in aa import, GSH synthesis, and OS resistance. PERK-/- cells accumulate endogenous peroxides during ER stress, whereas interference with the ER oxidase ERO1 abrogates such accumulation. eIF2α phosphorylation protects cells against metabolic consequences of ER oxidation by promoting the linked processed of sufficiency and resistance to OS. | [85] |
| Left ventricle cells form five-month-old Lee-Sung (Met-S) and Lanyu (MHO) obese minipig | High-fat diet | CHOP ↑ (Met S & MHO) PERK ↑ (Met S & MHO) IRE1α = (Met S & MHO) ATF6 (↑ Met S & = MHO) | TBARS (Thiobarbituric acid reactive substances) (↑ Met S & ↑ MHO) | [86] | ||
| Primary murine brain endothelial cells from 2 month-old BL/6 mice | T-BHP (Tert-butyl hydroperoxide) | XBP1-S ↓ CHOP ↑ | DCF ↑ (peroxides) MDA ↑ 4-HNE ↑ CAT = SOD = GPx = RH ↑ (Ψm) | Cross-talk ER-mitochondria | Down-regulation of Homer1 protects against t-BHP-induced endothelial injury. Down-regulation of Homer1 reduces t-BHP-induced OS. Down-regulation of Homer1 preserves Ca2+ homeostasis in mBECs. Down-regulation of Homer1 attenuates t-BHP-induced ER stress. | [87] |
| Human PBMCs cognitive impairment. PBMCs and brain cortex cells from a transgenic mouse with Alzheimer’s disease | Thapsigargin | GRP78 ↑ XBP1 ↑ | DCF ↑ (peroxides) Nrf2 ↑ GCLc | Antioxidant genes | [88] | |
| MIA PaCa-2 human pancreatic cells | Piperlongumine | ATF4 ↑ IRE1α ↑ XBP1 ↑ | OSGIN1 ↑ ABCB10 ↓ | [89] | ||
| Hepatopancreas from Litopenaeus vannamei | Ammonia nitrogen | BiP ↑ eIF2α = ATF4 ↑ IRE1 = XBP1-S ↑ | SOD ↓ MDA ↑ | [90] |
6. Endoplasmic Reticulum and Oxidative Stress in Rare Respiratory Diseases
6.1. Serpinopathies, Endoplasmic Reticulum, and Oxidative Stress
6.2. Interstitial Lung Diseases, Endoplasmic Reticulum, and Oxidative Stress
6.3. Cystic Fibrosis, Endoplasmic Reticulum, and Oxidative Stress
6.4. Primary Ciliary Dyskinesia and Oxidative Stress
7. Antioxidants Therapies in Rare Respiratory Diseases
7.1. Alpha-1 Antitrypsin Deficiency and Antioxidant Therapy
7.2. Idiopathic Pulmonary Fibrosis and Antioxidant Therapy
7.3. Cystic Fibrosis and Antioxidant Therapy
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Slimen, I.B.; Najar, T.; Ghram, A.; Dabbebi, H.; Ben Mrad, M.; Abdrabbah, M. Reactive Oxygen Species, Heat Stress and Oxidative-Induced Mitochondrial Damage. A Review. Int. J. Hyperth. 2014, 30, 513–523. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, A.; Sapey, E. Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms. J. Clin. Med. 2017, 6, 21. [Google Scholar] [CrossRef]
- Magallón, M.; Navarro-García, M.M.; Dasí, F. Oxidative Stress in COPD. J. Clin. Med. 2019, 8, 1953. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Blanco, I.; Lara, B. Déficit de alfa1-Antitripsina: Fisiopatología, Enfermedades Relacionadas, Diagnóstico y Tratamiento, 2nd ed.; Editorial Respira: Barcelona, Spain, 2016. [Google Scholar]
- Rahman, K. Studies on Free Radicals, Antioxidants, and Co-Factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, Oxidative Stress and the Biology of Ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive Oxygen Species as Intracellular Messengers during Cell Growth and Differentiation. Cell. Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Schröder, P.; Krutmann, J. Environmental Oxidative Stress—Environmental Sources of ROS. Handb. Environ. Chem. 2005, 2, 19–31. [Google Scholar] [CrossRef]
- Wang, X.; Hai, C. Novel Insights into Redox System and the Mechanism of Redox Regulation. Mol. Biol. Rep. 2016, 43, 607–628. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: Oxidants and Antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef]
- Ohnishi, S.; Ma, N.; Thanan, R.; Pinlaor, S.; Hammam, O.; Murata, M.; Kawanishi, S. DNA Damage in Inflammation-Related Carcinogenesis and Cancer Stem Cells. Oxid. Med. Cell. Longev. 2013, 2013, 387014. [Google Scholar] [CrossRef]
- Baierle, M.; Nascimento, S.N.; Moro, A.M.; Brucker, N.; Freitas, F.; Gauer, B.; Durgante, J.; Bordignon, S.; Zibetti, M.; Trentini, C.M.; et al. Relationship between Inflammation and Oxidative Stress and Cognitive Decline in the Institutionalized Elderly. Oxid. Med. Cell. Longev. 2015, 2015, 804198. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Salvioli, S.; Franceschi, C. Oxidative Stress and the Ageing Endocrine System. Nat. Rev. Endocrinol. 2013, 9, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Laddha, N.C.; Dwivedi, M.; Mansuri, M.S.; Singh, M.; Gani, A.R.; Yeola, A.P.; Panchal, V.N.; Khan, F.; Dave, D.J.; Patel, A.; et al. Role of Oxidative Stress and Autoimmunity in Onset and Progression of Vitiligo. Exp. Dermatol. 2014, 23, 352–353. [Google Scholar] [CrossRef] [PubMed]
- Assies, J.; Mocking, R.J.T.; Lok, A.; Ruhé, H.G.; Pouwer, F.; Schene, A.H. Effects of Oxidative Stress on Fatty Acid- and One-Carbon-Metabolism in Psychiatric and Cardiovascular Disease Comorbidity. Acta Psychiatr. Scand. 2014, 130, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive Oxygen Species (ROS) Homeostasis and Redox Regulation in Cellular Signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Sanz, A. Coenzyme Q Redox Signalling and Longevity. Free Radic. Biol. Med. 2021, 164, 187–205. [Google Scholar] [CrossRef]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA Damage and Disease: Induction, Repair and Significance. Mutat. Res. Rev. Mutat. Res. 2004, 567, 1–61. [Google Scholar] [CrossRef]
- Evans, M.D.; Cooke, M.S. Factors Contributing to the Outcome of Oxidative Damage to Nucleic Acids. BioEssays 2004, 26, 533–542. [Google Scholar] [CrossRef]
- Tsikas, D. Assessment of Lipid Peroxidation by Measuring Malondialdehyde (MDA) and Relatives in Biological Samples: Analytical and Biological Challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein Carbonyl Groups as Biomarkers of Oxidative Stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-Hydroxy-2′ -Deoxyguanosine (8-OHdG): A Critical Biomarker of Oxidative Stress and Carcinogenesis. J. Environ. Sci. Health Part C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 120–139. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, Oxidative Stress and Cell Death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling Functions of Reactive Oxygen Species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Bohme, E.; Schmidt, H.H.H.W. Nitric Oxide and Cytosolic Guanylate Cyclase: Components of an Intercellular Signalling System. Z. Kardiol. 1989, 78, 75–79. [Google Scholar]
- McIlwain, H. Extended Roles in the Brain for Second-Messenger Systems. Neuroscience 1977, 2, 357–372. [Google Scholar] [CrossRef]
- Forman, H.J.; Ursini, F.; Maiorino, M. An Overview of Mechanisms of Redox Signaling. J. Mol. Cell. Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide Dismutase. An Enzymic Function for Erythrocuprein (Hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Hawkins, B.J.; Madesh, M.; Kirkpatrick, C.J.; Fisher, A.B. Superoxide Flux in Endothelial Cells via the Chloride Channel-3 Mediates Intracellular Signaling. Mol. Biol. Cell 2007, 18, 2002–2012. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Revisiting the Reactions of Superoxide with Glutathione and Other Thiols. Arch. Biochem. Biophys. 2016, 595, 68–71. [Google Scholar] [CrossRef]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive Oxygen Intermediates as Apparently Widely Used Messengers in the Activation of the NF-Kappa B Transcription Factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Kaul, N.; Forman, H.J. Activation of NFκB by the Respiratory Burst of Macrophages. Free Radic. Biol. Med. 1996, 21, 401–405. [Google Scholar] [CrossRef]
- Kaul, N.; Gopalakrishna, R.; Gundimeda, U.; Choi, J.; Forman, H.J. Role of Protein Kinase C in Basal and Hydrogen Peroxide-Stimulated NF- ΚB Activation in the Murine Macrophage J774A.1 Cell Line. Arch. Biochem. Biophys. 1998, 350, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Hoyal, C.R.; Gozal, E.; Zhou, H.; Foldenauer, K.; Forman, H.J. Modulation of the Rat Alveolar Macrophage Respiratory Burst by Hydroperoxides Is Calcium Dependent. Arch. Biochem. Biophys. 1996, 326, 166–171. [Google Scholar] [CrossRef]
- Murphy, J.K.; Livingston, F.R.; Gozal, E.; Torres, M.; Forman, H.J. Stimulation of the Rat Alveolar Macrophage Respiratory Burst by Extracellular Adenine Nucleotides. Am. J. Respir. Cell Mol. Biol. 1993, 9, 505–510. [Google Scholar] [CrossRef]
- Hoyal, C.R.; Thomas, A.P.; Forman, H.J. Hydroperoxide-Induced Increases in Intracellular Calcium Due to Annexin VI Translocation and Inactivation of Plasma Membrane Ca 2+-ATPase. J. Biol. Chem. 1996, 271, 29205–29210. [Google Scholar] [CrossRef]
- Girón-Calle, J.; Srivatsa, K.; Forman, H.J. Priming of Alveolar Macrophage Respiratory Burst by H2O2 Is Prevented by Phosphatidylcholine-Specific Phospholipase C Inhibitor Tricyclodecan-9-Yl-Xanthate (D609). J. Pharmacol. Exp. Ther. 2002, 301, 87–94. [Google Scholar] [CrossRef]
- Torres, M.; Forman, H.J. Activation of Several MAP Kinases upon Stimulation of Rat Alveolar Macrophages: Role of the NADPH Oxidase. Arch. Biochem. Biophys. 1999, 366, 231–239. [Google Scholar] [CrossRef]
- Rinna, A.; Torres, M.; Forman, H.J. Stimulation of the Alveolar Macrophage Respiratory Burst by ADP Causes Selective Glutathionylation of Protein Tyrosine Phosphatase 1B. Free Radic. Biol. Med. 2006, 41, 86–91. [Google Scholar] [CrossRef]
- Denu, J.M.; Tanner, K.G. Specific and Reversible Inactivation of Protein Tyrosine Phosphatases by Hydrogen Peroxide: Evidence for a Sulfenic Acid Intermediate and Implications for Redox Regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef] [PubMed]
- Murad, F. Nitric Oxide: The Coming of the Second Messenger. Rambam Maimonides Med. J. 2011, 2, e0038. [Google Scholar] [CrossRef] [PubMed]
- Reis, G.S.; Augusto, V.S.; Silveira, A.P.C.; Jordão, A.A.; Baddini-Martinez, J.; Poli Neto, O.; Rodrigues, A.J.; Evora, P.R.B. Oxidative-Stress Biomarkers in Patients with Pulmonary Hypertension. Pulm. Circ. 2013, 3, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Torres-Durán, M.; Lopez-Campos, J.L.; Barrecheguren, M.; Miravitlles, M.; Martinez-Delgado, B.; Castillo, S.; Escribano, A.; Baloira, A.; Navarro-Garcia, M.M.; Pellicer, D.; et al. Alpha-1 Antitrypsin Deficiency: Outstanding Questions and Future Directions. Orphanet J. Rare Dis. 2018, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, J.; Prywes, R. The Luminal Domain of ATF6 Senses Endoplasmic Reticulum (ER) Stress and Causes Translocation of ATF6 from the Er to the Golgi. J. Biol. Chem. 2002, 277, 13045–13052. [Google Scholar] [CrossRef]
- Credle, J.J.; Finer-Moore, J.S.; Papa, F.R.; Stroud, R.M.; Walter, P. On the Mechanism of Sensing Unfolded Protein in the Endoplasmic Reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 18773–18784. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic Interaction of BiP and ER Stress Transducers in the Unfolded-Protein Response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Kimata, Y.; Oikawa, D.; Shimizu, Y.; Ishiwata-Kimata, Y.; Kohno, K. A Role for BiP as an Adjustor for the Endoplasmic Reticulum Stress-Sensing Protein Ire1. J. Cell Biol. 2004, 167, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the Cis-Acting Endoplasmic Reticulum Stress Response Element Responsible for Transcriptional Induction of Mammalian Glucose- Regulated Proteins: Involvement of Basic Leucine Zipper Transcription Factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. Of Membrane-Bound ATF6 by the Same Proteases That Process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. Endoplasmic Reticulum Stress-Induced Formation of Transcription Factor Complex ERSF Including NF-Y (CBF) and Activating Transcription Factors 6α and 6β That Activates the Mammalian Unfolded Protein Response. Mol. Cell. Biol. 2001, 21, 1239–1248. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A Time-Dependent Phase Shift in the Mammalian Unfolded Protein Response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef]
- Cox, J.S.; Walter, P. A Novel Mechanism for Regulating Activity of a Transcription Factor That Controls the Unfolded Protein Response. Cell 1996, 87, 391–404. [Google Scholar] [CrossRef]
- Morl, K.; Ma, W.; Gething, M.J.; Sambrook, J. A Transmembrane Protein with a Cdc2+ CDC28-Related Kinase Activity Is Required for Signaling from the ER to the Nucleus. Cell 1993, 74, 743–756. [Google Scholar] [CrossRef]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional Induction of Genes Encoding Endoplasmic Reticulum Resident Proteins Requires a Transmembrane Protein Kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 MRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 Couples Endoplasmic Reticulum Load to Secretory Capacity by Processing the XBP-1 MRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein Translation and Folding Are Coupled by an Endoplasmic- Reticulum-Resident Kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized Redox State of Glutathione in the Endoplasmic Reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Van der Vlies, D.; Makkinje, M.; Jansens, A.; Braakman, I.; Verkleij, A.J.; Wirtz, K.W.A.; Post, J.A. Oxidation of ER Resident Proteins upon Oxidative Stress: Effects of Altering Cellular Redox/Antioxidant Status and Implications for Protein Maturation. Antioxid. Redox Signal. 2003, 5, 381–387. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. Oxidative Protein Folding in Eukaryotes: Mechanisms and Consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic Reticulum and the Unfolded Protein Response. Dynamics and Metabolic Integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid Synthesis in a Membrane Fraction Associated with Mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Bravo, R.; Gutierrez, T.; Paredes, F.; Gatica, D.; Rodriguez, A.E.; Pedrozo, Z.; Chiong, M.; Parra, V.; Quest, A.F.G.; Rothermel, B.A.; et al. Endoplasmic Reticulum: ER Stress Regulates Mitochondrial Bioenergetics. Int. J. Biochem. Cell Biol. 2012, 44, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.; Walker, A.N.; Kaur, J.; Thomas, J.L.; Powell, S.A.; Pandey, A.V.; Whittal, R.M.; Burak, W.E.; Petruzzelli, G.; Bose, H.S. Endoplasmic Reticulum Stress Enhances Mitochondrial Metabolic Activity in Mammalian Adrenals and Gonads. Mol. Cell. Biol. 2016, 36, 3058–3074. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, Y.C. Endoplasmic Reticulum Stress and the Related Signaling Networks in Severe Asthma. Allergy Asthma Immunol. Res. 2015, 7, 106–117. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-Mediated Coupling of Endoplasmic Reticulum and Mitochondrial Ca2+ Channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK Is Required at the ER-Mitochondrial Contact Sites to Convey Apoptosis after ROS-Based ER Stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-Mitochondrial Coupling Promotes Mitochondrial Respiration and Bioenergetics during Early Phases of ER Stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Monaco, G.; Bultynck, G.; Missiaen, L.; De Smedt, H.; Parys, J.B. The IP3 Receptor-Mitochondria Connection in Apoptosis and Autophagy. Biochim. Biophys. Acta 2011, 1813, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Eno, C.O.; Altman, B.J.; Zhu, Y.; Zhao, G.; Olberding, K.E.; Rathmell, J.C.; Li, C. ER Stress Modulates Cellular Metabolism. Biochem. J. 2011, 435, 285–296. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 Positively and C-Fos and Fra1 Negatively Regulate the Human Antioxidant Response Element-Mediated Expression of NAD(P)H:Quinone Oxidoreductase1 Gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 Represses Nuclear Activation of Antioxidant Responsive Elements by Nrf2 through Binding to the Amino-Terminal Neh2 Domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. PERK-Dependent Activation of Nrf2 Contributes to Redox Homeostasis and Cell Survival Following Endoplasmic Reticulum Stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of Misfolded Proteins Prevents ER-Derived Oxidative Stress and Cell Death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants Reduce Endoplasmic Reticulum Stress and Improve Protein Secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Li, S.J.; Liu, C.H.; Chu, H.P.; Mersmann, H.J.; Ding, S.T.; Chu, C.H.; Wang, C.Y.; Chen, C.Y. The High-Fat Diet Induces Myocardial Fibrosis in the Metabolically Healthy Obese Minipigs—The Role of ER Stress and Oxidative Stress. Clin. Nutr. 2017, 36, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.Y.; Zhang, Y.H.; Xie, G.Q.; Liu, C.X.; Zhou, R.; Shi, W. Down-Regulation of Homer1 Attenuates t-BHP-Induced Oxidative Stress through Regulating Calcium Homeostasis and ER Stress in Brain Endothelial Cells. Biochem. Biophys. Res. Commun. 2016, 477, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.I.; Costa, R.O.; Ferreira, I.L.; Santana, I.; Caldeira, G.L.; Padovano, C.; Fonseca, A.C.; Baldeiras, I.; Cunha, C.; Letra, L.; et al. Oxidative Stress Involving Changes in Nrf2 and ER Stress in Early Stages of Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1428–1441. [Google Scholar] [CrossRef]
- Dhillon, H.; Mamidi, S.; Mcclean, P.; Reindl, K.M. Transcriptome Analysis of Piperlongumine-Treated Human Pancreatic Cancer Cells Reveals Involvement of Oxidative Stress and Endoplasmic Reticulum Stress Pathways. J. Med. Food 2016, 19, 578–585. [Google Scholar] [CrossRef]
- Liang, Z.; Liu, R.; Zhao, D.; Wang, L.; Sun, M.; Wang, M.; Song, L. Ammonia Exposure Induces Oxidative Stress, Endoplasmic Reticulum Stress and Apoptosis in Hepatopancreas of Pacific White Shrimp (Litopenaeus Vannamei). Fish Shellfish Immunol. 2016, 54, 523–528. [Google Scholar] [CrossRef]
- Silverman, G.A.; Bird, P.I.; Carrell, R.W.; Church, F.C.; Coughlin, P.B.; Gettins, P.G.W.; Irving, J.A.; Lomas, D.A.; Luke, C.J.; Moyer, R.W.; et al. The Serpins Are an Expanding Superfamily of Structurally Similar but Functionally Diverse Proteins. J. Biol. Chem. 2001, 276, 33293–33296. [Google Scholar] [CrossRef]
- Silverman, G.A.; Whisstock, J.C.; Bottomley, S.P.; Huntington, J.A.; Kaiserman, D.; Luke, C.J.; Pak, S.C.; Reichhart, J.-M.; Bird, P.I. Serpins Flex Their Muscle. J. Biol. Chem. 2010, 285, 24299–24305. [Google Scholar] [CrossRef]
- Stein, P.E.; Carrell, R.W. What Do Dysfunctional Serpins Tell Us about Molecular Mobility and Disease? Nat. Struct. Biol. 1995, 2, 96–113. [Google Scholar] [CrossRef] [PubMed]
- Janoff, A. Inhibition of Human Granulocyte Elastase by Serum Alpha-1-Antitrypsin. Am. Rev. Respir. Dis. 1972, 105, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, A.; Snapp, E.L.; Tan, L.; Miranda, E.; Marciniak, S.J.; Lomas, D.A. Endoplasmic Reticulum Polymers Impair Luminal Protein Mobility and Sensitize to Cellular Stress in Alpha1-Antitrypsin Deficiency. Hepatology 2013, 57, 2049–2060. [Google Scholar] [CrossRef]
- Hidvegi, T.; Schmidt, B.Z.; Hale, P.; Perlmutter, D.H. Accumulation of Mutant A1-Antitrypsin Z in the Endoplasmic Reticulum Activities Caspases-4 and -12, NFκB, and BAP31 but Not the Unfolded Protein Response. J. Biol. Chem. 2005, 280, 39002–39015. [Google Scholar] [CrossRef] [PubMed]
- Papp, E.; Száiraz, P.; Korcsmáiros, T.; Csermely, P.; Papp, E.; Száiraz, P.; Korcsmáiros, T.; Csermely, P. Changes of Endoplasmic Reticulum Chaperone Complexes, Redox State, and Impaired Protein Disulfide Reductase Activity in Misfolding Ai-antitrypsin Transgenic Mice. FASEB J. 2006, 20, 1018–1020. [Google Scholar] [CrossRef]
- Carroll, T.P.; Greene, C.M.; O’Connor, C.A.; Nolan, Á.M.; O’Neill, S.J.; McElvaney, N.G. Evidence for Unfolded Protein Response Activation in Monocytes from Individuals with α-1 Antitrypsin Deficiency. J. Immunol. 2010, 184, 4538–4546. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Greene, C.M.; Carroll, T.P.; McElvaney, N.G.; O’Neill, S.J. Selenoprotein S/SEPS1 Modifies Endoplasmic Reticulum Stress in Z Variant A1-Antitrypsin Deficiency. J. Biol. Chem. 2009, 284, 16891–16897. [Google Scholar] [CrossRef]
- Lawless, M.W.; Greene, C.M.; Mulgrew, A.; Taggart, C.C.; O’Neill, S.J.; McElvaney, N.G. Activation of Endoplasmic Reticulum-Specific Stress Responses Associated with the Conformational Disease Z A1-Antitrypsin Deficiency. J. Immunol. 2004, 172, 5722–5726. [Google Scholar] [CrossRef]
- Raeymaekers, L.; Larivière, E. Vesicularization of the Endoplasmic Reticulum Is a Fast Response to Plasma Membrane Injury. Biochem. Biophys. Res. Commun. 2011, 414, 246–251. [Google Scholar] [CrossRef]
- Subramanian, K.; Meyer, T. Calcium-Induced Restructuring of Nuclear Envelope and Endoplasmic Reticulum Calcium Stores. Cell 1997, 89, 963–971. [Google Scholar] [CrossRef]
- Escribano, A.; Amor, M.; Pastor, S.; Castillo, S.; Sanz, F.; Codoñer-Franch, P.; Dasí, F. Decreased Glutathione and Low Catalase Activity Contribute to Oxidative Stress in Children with α-1 Antitrypsin Deficiency. Thorax 2015, 70, 82–83. [Google Scholar] [CrossRef]
- Sitia, R.; Molteni, S.N. Stress, Protein (Mis)Folding, and Signaling: The Redox Connection. Sci. STKE 2004, 2004, e27. [Google Scholar] [CrossRef]
- Nardai, G.; Stadler, K.; Papp, E.; Korcsmáros, T.; Jakus, J.; Csermely, P. Diabetic Changes in the Redox Status of the Microsomal Protein Folding Machinery. Biochem. Biophys. Res. Commun. 2005, 334, 787–795. [Google Scholar] [CrossRef]
- Marcus, N.Y.; Blomenkamp, K.; Ahmad, M.; Teckman, J.H. Oxidative Stress Contributes to Liver Damage in a Murine Model of Alpha-1-Antitrypsin Deficiency. Exp. Biol. Med. 2012, 237, 1163–1172. [Google Scholar] [CrossRef]
- Topic, A.; Nagorni-Obradovic, L.; Francuski, D.; Ljujic, M.; Malic, Z.; Radojkovic, D. Oxidative Stress and Polymorphism of Xenobiotic-Metabolizing Enzymes in Two Patients with Severe Alpha-1-Antitrypsin Deficiency. Biochem. Genet. 2016, 54, 746–752. [Google Scholar] [CrossRef] [PubMed]
- GSTP1 Glutathione S-Transferase Pi 1 [Homo Sapiens (Human)]-Gene-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene?Cmd=DetailsSearch&Term=2950 (accessed on 28 January 2020).
- Escribano, A.; Pastor, S.; Reula, A.; Castillo, S.; Vicente, S.; Sanz, F.; Casas, F.; Torres, M.; Fernández-Fabrellas, E.; Codoñer-Franch, P.; et al. Accelerated Telomere Attrition in Children and Teenagers with A1-Antitrypsin Deficiency. Eur. Respir. J. 2016, 48, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Berlett, B.S.; Moskovitz, J.; Mosoni, L.; Stadtman, E.R. Methionine Residues May Protect Proteins from Critical Oxidative Damage. Mech. Ageing Dev. 1999, 107, 323–332. [Google Scholar] [CrossRef]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of Either Methionine 351 or Methionine 358 in A1-Antitrypsin Causes Loss of Anti-Neutrophil Elastase Activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [CrossRef]
- Feng, Y.; Xu, J.; Zhou, Q.; Wang, R.; Liu, N.; Wu, Y.; Yuan, H.; Che, H. Alpha-1 Antitrypsin Prevents the Development of Preeclampsia through Suppression of Oxidative Stress. Front. Physiol. 2016, 7, 176. [Google Scholar] [CrossRef]
- Barratt, S.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [PubMed]
- Kliment, C.R.; Oury, T.D. Oxidative Stress, Extracellular Matrix Targets, and Idiopathic Pulmonary Fibrosis. Free Radic. Biol. Med. 2010, 49, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.M.; Seeger, W.; et al. Epithelial Endoplasmic Reticulum Stress and Apoptosis in Sporadic Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic Reticulum Stress in Alveolar Epithelial Cells Is Prominent in IPF: Association with Altered Surfactant Protein Processing and Herpesvirus Infection. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2008, 294, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Baek, H.A.; Kim, D.S.; Park, H.S.; Jang, K.Y.; Kang, M.J.; Lee, D.G.; Moon, W.S.; Chae, H.J.; Chung, M.J. Involvement of Endoplasmic Reticulum Stress in Myofibroblastic Differentiation of Lung Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2012, 46, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.J.; Larson-Casey, J.L.; He, C.; Murthy, S.; Brent Carter, A. 3 Asbestos-Induced Disruption of Calcium Homeostasis Induces Endoplasmic Reticulum Stress in Macrophages. J. Biol. Chem. 2014, 289, 33391–33403. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, Y.; Zhang, Z.; He, L.; Zhu, J.; Zhang, M.; He, X.; Cheng, Z.; Ao, Q.; Cao, Y.; et al. Chop Deficiency Protects Mice against Bleomycin-Induced Pulmonary Fibrosis by Attenuating M2 Macrophage Production. Mol. Ther. 2016, 24, 915–925. [Google Scholar] [CrossRef]
- Burman, A.; Tanjore, H.; Blackwell, T.S. Endoplasmic Reticulum Stress in Pulmonary Fibrosis. Matrix Biol. 2018, 68–69, 355–365. [Google Scholar] [CrossRef]
- Kamp, D.W.; Liu, G.; Cheresh, P.; Kim, S.J.; Mueller, A.; Lam, A.P.; Trejo, H.; Williams, D.; Tulasiram, S.; Baker, M.; et al. Asbestos-Induced Alveolar Epithelial Cell Apoptosis: The Role of Endoplasmic Reticulum Stress Response. Am. J. Respir. Cell Mol. Biol. 2013, 49, 892–901. [Google Scholar] [CrossRef]
- Zhong, Q.; Zhou, B.; Ann, D.K.; Minoo, P.; Liu, Y.; Banfalvi, A.; Krishnaveni, M.S.; Dubourd, M.; Demaio, L.; Willis, B.C.; et al. Role of Endoplasmic Reticulum Stress in Epithelial-Mesenchymal Transition of Alveolar Epithelial Cells: Effects of Misfolded Surfactant Protein. Am. J. Respir. Cell Mol. Biol. 2011, 45, 498–509. [Google Scholar] [CrossRef]
- Tanjore, H.; Cheng, D.S.; Degryse, A.L.; Zoz, D.F.; Abdolrasulnia, R.; Lawson, W.E.; Blackwell, T.S. Alveolar Epithelial Cells Undergo Epithelial-to-Mesenchymal Transition in Response to Endoplasmic Reticulum Stress. J. Biol. Chem. 2011, 286, 30972–30980. [Google Scholar] [CrossRef] [PubMed]
- Bargagli, E.; Penza, F.; Vagaggini, C.; Magi, B.; Perari, M.G.; Rottoli, P. Analysis of Carbonylated Proteins in Bronchoalveolar Lavage of Patients with Diffuse Lung Diseases. Lung 2007, 185, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Rottoli, P.; Magi, B.; Cianti, R.; Bargagli, E.; Vagaggini, C.; Nikiforakis, N.; Pallini, V.; Bini, L. Carbonylated Proteins in Bronchoalveolar Lavage of Patients with Sarcoidosis, Pulmonary Fibrosis Associated with Systemic Sclerosis and Idiopathic Pulmonary Fibrosis. Proteomics 2005, 5, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Malli, F.; Bardaka, F.; Tsilioni, I.; Karetsi, E.; Gourgoulianis, K.I.; Daniil, Z. 8-Isoprostane Levels in Serum and Bronchoalveolar Lavage in Idiopathic Pulmonary Fibrosis and Sarcoidosis. Food Chem. Toxicol. 2013, 61, 160–163. [Google Scholar] [CrossRef]
- Daniil, Z.D.; Papageorgiou, E.; Koutsokera, A.; Kostikas, K.; Kiropoulos, T.; Papaioannou, A.I.; Gourgoulianis, K.I. Serum Levels of Oxidative Stress as a Marker of Disease Severity in Idiopathic Pulmonary Fibrosis. Pulm. Pharmacol. Ther. 2008, 21, 26–31. [Google Scholar] [CrossRef]
- Psathakis, K.; Mermigkis, D.; Papatheodorou, G.; Loukides, S.; Panagou, P.; Polychronopoulos, V.; Siafakas, N.M.; Bouros, D. Exhaled Markers of Oxidative Stress in Idiopathic Pulmonary Fibrosis. Eur. J. Clin. Investig. 2006, 36, 362–367. [Google Scholar] [CrossRef]
- Cantin, A.M.; North, S.L.; Fells, G.A.; Hubbard, R.C.; Crystal, R.G. Oxidant-Mediated Epithelial Cell Injury in Idiopathic Pulmonary Fibrosis. J. Clin. Investig. 1987, 79, 1665–1673. [Google Scholar] [CrossRef]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH Oxidase-4 Mediates Myofibroblast Activation and Fibrogenic Responses to Lung Injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef]
- Veith, C.; Boots, A.W.; Idris, M.; Van Schooten, F.J.; Van Der Vliet, A. Redox Imbalance in Idiopathic Pulmonary Fibrosis: A Role for Oxidant Cross-Talk between NADPH Oxidase Enzymes and Mitochondria. Antioxid. Redox Signal. 2019, 31, 1092–1115. [Google Scholar] [CrossRef]
- Zeidler, P.C.; Hubbs, A.; Battelli, L.; Castranova, V. Role of Inducible Nitric Oxide Synthase-Derived Nitric Oxide in Silica-Induced Pulmonary Inflammation and Fibrosis. J. Toxicol. Environ. Health Part A 2004, 67, 1001–1026. [Google Scholar] [CrossRef]
- Saleh, D.; Barnes, P.J.; Giaid, A. Increased Production of the Potent Oxidant Peroxynitrite in the Lungs of Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 1763–1769. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hubbard, R.C.; Crystal, R.G. Glutathione Deficiency in the Epithelial Lining Fluid of the Lower Respiratory Tract in Idiopathic Pulmonary Fibrosis. Am. Rev. Respir. Dis. 1989, 139, 370–372. [Google Scholar] [CrossRef]
- Mazur, W.; Lindholm, P.; Vuorinen, K.; MyllÄrniemi, M.; Salmenkivi, K.; Kinnula, V.L. Cell-Specific Elevation of NRF2 and Sulfiredoxin-1 as Markers of Oxidative Stress in the Lungs of Idiopathic Pulmonary Fibrosis and Non-Specific Interstitial Pneumonia. APMIS 2010, 118, 703–712. [Google Scholar] [CrossRef]
- Markart, P.; Luboeinski, T.; Korfei, M.; Schmidt, R.; Wygrecka, M.; Mahavadi, P.; Mayer, K.; Wilhelm, J.; Seeger, W.; Guenther, A.; et al. Alveolar Oxidative Stress Is Associated with Elevated Levels of Nonenzymatic Low-Molecular-Weight Antioxidants in Patients with Different Forms of Chronic Fibrosing Interstitial Lung Diseases. Antioxid. Redox Signal. 2009, 11, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.M.; Cho, H.Y.; Kleeberger, S.R. Oxidative Stress and Antioxidants in the Pathogenesis of Pulmonary Fibrosis: A Potential Role for Nrf2. Antioxid. Redox Signal. 2008, 10, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Fois, A.G.; Paliogiannis, P.; Sotgia, S.; Mangoni, A.A.; Zinellu, E.; Pirina, P.; Carru, C.; Zinellu, A. Evaluation of Oxidative Stress Biomarkers in Idiopathic Pulmonary Fibrosis and Therapeutic Applications: A Systematic Review. Respir. Res. 2018, 19, 51. [Google Scholar] [CrossRef] [PubMed]
- Scotet, V.; Gutierrez, H.; Farrell, P.M. Newborn Screening for CF across the Globe-Where Is Itworthwhile? Int. J. Neonatal Screen. 2020, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Battistoni, A.; Gambari, R.; Pompella, A.; Bragonzi, A.; Pilolli, F.; Iuliano, L.; Piroddi, M.; Dechecchi, M.C.; Cabrini, G. Oxidative Stress and Antioxidant Therapy in Cystic Fibrosis. Biochim. Biophys. Acta 2012, 1822, 690–713. [Google Scholar] [CrossRef] [PubMed]
- Fanen, P.; Wohlhuter-Haddad, A.; Hinzpeter, A. Genetics of Cystic Fibrosis: CFTR Mutation Classifications toward Genotype-Based CF Therapies. Int. J. Biochem. Cell Biol. 2014, 52, 94–102. [Google Scholar] [CrossRef]
- Bareil, C.; Bergougnoux, A. CFTR Gene Variants, Epidemiology and Molecular Pathology. Arch. Pediatr. 2020, 27, eS8–eS12. [Google Scholar] [CrossRef]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15.e1. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective Intracellular Transport and Processing of CFTR Is the Molecular Basis of Most Cystic Fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Younger, J.M.; Ren, H.Y.; Chen, L.; Fan, C.Y.; Fields, A.; Patterson, C.; Cyr, D.M. A Foldable CFTRΔF508 Biogenic Intermediate Accumulates upon Inhibition of the Hsc70-CHIP E3 Ubiquitin Ligase. J. Cell Biol. 2004, 167, 1075–1085. [Google Scholar] [CrossRef]
- Gilbert, A.; Jadot, M.; Leontieva, E.; Wattiaux-De Coninck, S.; Wattiaux, R. ΔF508 CFTR Localizes in the Endoplasmic Reticulum—Golgi Intermediate Compartment in Cystic Fibrosis Cells. Exp. Cell Res. 1998, 242, 144–152. [Google Scholar] [CrossRef]
- Kerbiriou, M.; Le Drévo, M.A.; Férec, C.; Trouvé, P. Coupling Cystic Fibrosis to Endoplasmic Reticulum Stress: Differential Role of Grp78 and ATF6. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 1236–1249. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, J.; Arenzana, N.; Tirasophon, W.; Kaufman, R.J.; Prywes, R. Activation of ATF6 and an ATF6 DNA Binding Site by the Endoplasmic Reticulum Stress Response. J. Biol. Chem. 2000, 275, 27013–27020. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Tang, A.C.; Saferali, A.; He, G.; Sandford, A.J.; Strug, L.; Turvey, S.E. Endoplasmic Reticulum Stress and Chemokine Production in Cystic Fibrosis Airway Cells: Regulation by STAT3 Modulation. J. Infect. Dis. 2017, 215, 293–302. [Google Scholar] [CrossRef]
- Stutts, M.J.; Knowles, M.R.; Gatzy, J.T.; Boucher, R.C. Oxygen Consumption and Ouabain Binding Sites in Cystic Fibrosis Nasal Epithelium. Pediatr. Res. 1986, 20, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Worlitzsch, D.; Tarran, R.; Ulrich, M.; Schwab, U.; Cekici, A.; Meyer, K.C.; Birrer, P.; Bellon, G.; Berger, J.; Weiss, T.; et al. Effects of Reduced Mucus Oxygen Concentration in Airway Pseudomonas Infections of Cystic Fibrosis Patients. J. Clin. Investig. 2002, 109, 317–325. [Google Scholar] [CrossRef]
- Colarusso, C.; Terlizzi, M.; Molino, A.; Pinto, A.; Sorrentino, R. Role of the Inflammasome in Chronic Obstructive Pulmonary Disease (COPD). Oncotarget 2017, 8, 81813–81824. [Google Scholar] [CrossRef]
- Kelly-Aubert, M.; Trudel, S.; Fritsch, J.; Nguyen-Khoa, T.; Baudouin-Legros, M.; Moriceau, S.; Jeanson, L.; Djouadi, F.; Matar, C.; Conti, M.; et al. GSH Monoethyl Ester Rescues Mitochondrial Defects in Cystic Fibrosis Models. Hum. Mol. Genet. 2011, 20, 2745–2759. [Google Scholar] [CrossRef] [PubMed]
- Laval, J.; Ralhan, A.; Hartl, D. Neutrophils in Cystic Fibrosis. Biol. Chem. 2016, 397, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.; Brennan, S.; Senthilmohan, R.; Gangell, C.L.; Chapman, A.L.; Sly, P.D.; Kettle, A.J. Identifying Peroxidases and Their Oxidants in the Early Pathology of Cystic Fibrosis. Free Radic. Biol. Med. 2010, 49, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Chan, T.; Osberg, I.; Senthilmohan, R.; Chapman, A.L.P.; Mocatta, T.J.; Wagener, J.S. Myeloperoxidase and Protein Oxidation in the Airways of Young Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2004, 170, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H. Mechanisms and Function of DUOX in Epithelia of the Lung. Antioxid. Redox Signal. 2009, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Roum, J.H.; Buhl, R.; McElvaney, N.G.; Borok, Z.; Crystal, R.G. Systemic Deficiency of Glutathione in Cystic Fibrosis. J. Appl. Physiol. 1993, 75, 2419–2424. [Google Scholar] [CrossRef]
- Linsdell, P.; Hanrahan, J.W. Glutathione Permeability of CFTR. Am. J. Physiol. Cell Physiol. 1998, 275, C323–C326. [Google Scholar] [CrossRef]
- Grasemann, H.; Michler, E.; Wallot, M.; Ratjen, F. Decreased Concentration of Exhaled Nitric Oxide (NO) in Patients with Cystic Fibrosis. Pediatr. Pulmonol. 1997, 24, 173–177. [Google Scholar] [CrossRef]
- Grasemann, H.; Al-Saleh, S.; Scott, J.A.; Shehnaz, D.; Mehl, A.; Amin, R.; Rafii, M.; Pencharz, P.; Belik, J.; Ratjen, F. Asymmetric Dimethylarginine Contributes to Airway Nitric Oxide Deficiency in Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 1363–1368. [Google Scholar] [CrossRef]
- Chen, J.; Kinter, M.; Shank, S.; Cotton, C.; Kelley, T.J.; Ziady, A.G. Dysfunction of Nrf-2 in CF Epithelia Leads to Excess Intracellular H2O2 and Inflammatory Cytokine Production. PLoS ONE 2008, 3, e3367. [Google Scholar] [CrossRef]
- Causer, A.J.; Shute, J.K.; Cummings, M.H.; Shepherd, A.I.; Gruet, M.; Costello, J.T.; Bailey, S.; Lindley, M.; Pearson, C.; Connett, G.; et al. Circulating Biomarkers of Antioxidant Status and Oxidative Stress in People with Cystic Fibrosis: A Systematic Review and Meta-Analysis. Redox Biol. 2020, 32, 101436. [Google Scholar] [CrossRef]
- Collins, C.E.; Quaggiotto, P.; Wood, L.; O’Loughlin, E.V.; Henry, R.L.; Garg, M.L. Elevated Plasma Levels of F2(α) Isoprostane in Cystic Fibrosis. Lipids 1999, 34, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Back, E.I.; Frindt, C.; Nohr, D.; Frank, J.; Ziebach, R.; Stern, M.; Ranke, M.; Biesalski, H.K. Antioxidant Deficiency in Cystic Fibrosis: When Is the Right Time to Take Action? Am. J. Clin. Nutr. 2004, 80, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Kharitonov, S.A.; Corradi, M.; van Rensen, L.; Geddes, D.M.; Hodson, M.E.; Barnes, P.J.; Montuschi, P.; Ciabattoni, G. Exhaled 8-Isoprostane as a New Non-Invasive Biomarker of Oxidative Stress in Cystic Fibrosis. Thorax 2000, 55, 205–209. [Google Scholar] [CrossRef]
- Iuliano, L.; Monticolo, R.; Straface, G.; Zullo, S.; Galli, F.; Boaz, M.; Quattrucci, S. Association of Cholesterol Oxidation and Abnormalities in Fatty Acid Metabolism in Cystic Fibrosis. Am. J. Clin. Nutr. 2009, 90, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.S.; Davis, S.D.; Omran, H.; Shoemark, A. Primary Ciliary Dyskinesia in the Genomics Age. Lancet Respir. Med. 2020, 8, 202–216. [Google Scholar] [CrossRef]
- Baz-Redón, N.; Rovira-Amigo, S.; Paramonov, I.; Castillo-Corullón, S.; Cols Roig, M.; Antolín, M.; García Arumí, E.; Torrent-Vernetta, A.; de Mir Messa, I.; Gartner, S.; et al. Implementation of a Gene Panel for Genetic Diagnosis of Primary Ciliary Dyskinesia. Arch. Bronconeumol. 2020. [Google Scholar] [CrossRef]
- Cockx, M.; Gouwy, M.; Van Damme, J.; Struyf, S. Chemoattractants and Cytokines in Primary Ciliary Dyskinesia and Cystic Fibrosis: Key Players in Chronic Respiratory Diseases. Cell. Mol. Immunol. 2018, 15, 312–323. [Google Scholar] [CrossRef]
- Güney, E.; Emiralioğlu, N.; Cinel, G.; Yalçın, E.; Doğru, D.; Kiper, N.; Uğur Özçelik, H. Nasal Nitric Oxide Levels in Primary Ciliary Dyskinesia, Cystic Fibrosis and Healthy Children. Turk. J. Pediatr. 2019, 61, 20–25. [Google Scholar] [CrossRef]
- Lucas, J.S.; Barbato, A.; Collins, S.A.; Goutaki, M.; Behan, L.; Caudri, D.; Dell, S.; Eber, E.; Escudier, E.; Hirst, R.A.; et al. European Respiratory Society Guidelines for the Diagnosis of Primary Ciliary Dyskinesia. Eur. Respir. J. 2017, 49, 1601090. [Google Scholar] [CrossRef]
- Collins, S.A.; Gove, K.; Walker, W.; Lucas, J.S.A. Nasal Nitric Oxide Screening for Primary Ciliary Dyskinesia: Systematic Review and Meta-Analysis. Eur. Respir. J. 2014, 44, 1589–1599. [Google Scholar] [CrossRef]
- Zihlif, N.; Paraskakis, E.; Tripoli, C.; Lex, C.; Bush, A. Makers of Airway Inflammation in Primary Ciliary Dyskinesia Studied Using Exhaled Breath Condensate. Pediatr. Pulmonol. 2006, 41, 509–514. [Google Scholar] [CrossRef]
- Reula, A.; Pellicer, D.; Castillo, S.; Banyuls, L.; Magallón, M.; Navarro, M.M.; Escribano, A.; Armengot, M.; Dasí, F. Caracterización del perfil oxidativo en células epiteliales nasales de pacientes con discinesia ciliar primaria desarrollo de un nuevo algoritmo diagnóstico. In Proceedings of the 52º Congreso SEPAR, Santiago de Compostela, Spain, 13–16 June 2019; p. 476. [Google Scholar]
- Janciauskiene, S. The Beneficial Effects of Antioxidants in Health and Diseases. Chronic Obstr. Pulm. Dis. J. COPD Found. 2020, 7, 182–202. [Google Scholar] [CrossRef] [PubMed]
- Otoupalova, E.; Smith, S.; Cheng, G.; Thannickal, V.J. Oxidative Stress in Pulmonary Fibrosis. Compr. Physiol. 2020, 10, 509–547. [Google Scholar] [CrossRef] [PubMed]
- Ciofu, O.; Smith, S.; Lykkesfeldt, J. Antioxidant Supplementation for Lung Disease in Cystic Fibrosis. Cochrane Database Syst. Rev. 2019, 2019, CD007020. [Google Scholar] [CrossRef]
- Tonelli, A.R.; Brantly, M.L. Augmentation Therapy in Alpha-1 Antitrypsin Deficiency: Advances and Controversies. Ther. Adv. Respir. Dis. 2010, 4, 289–312. [Google Scholar] [CrossRef]
- Chapman, K.R.; Burdon, J.G.W.; Piitulainen, E.; Sandhaus, R.A.; Seersholm, N.; Stocks, J.M.; Stoel, B.C.; Huang, L.; Yao, Z.; Edelman, J.M.; et al. Intravenous Augmentation Treatment and Lung Density in Severe A1 Antitrypsin Deficiency (RAPID): A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2015, 386, 360–368. [Google Scholar] [CrossRef]
- Barnes, P.J. Oxidative Stress-Based Therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef] [PubMed]
- Bals, R. Alpha-1-Antitrypsin Deficiency. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 629–633. [Google Scholar] [CrossRef]
- Ronzoni, R.; Berardelli, R.; Medicina, D.; Sitia, R.; Gooptu, B.; Fra, A.M. Aberrant Disulphide Bonding Contributes to the ER Retention of Alpha1-Antitrypsin Deficiency Variants. Hum. Mol. Genet. 2016, 25, 642–650. [Google Scholar] [CrossRef]
- Alam, S.; Li, Z.; Janciauskiene, S.; Mahadeva, R. Oxidation of Z α 1 -Antitrypsin by Cigarette Smoke Induces Polymerization. Am. J. Respir. Cell Mol. Biol. 2011, 45, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Shahzeidi, S.; Sarnstrand, B.; Jeffery, P.K.; McAnulty, R.J.; Laurent, G.J. Oral N-Acetylcysteine Reduces Bleomycin-Induced Collagen Deposition in the Lungs of Mice. Eur. Respir. J. 1991, 4, 845–852. [Google Scholar] [PubMed]
- Hagiwara, S.I.; Ishii, Y.; Kitamura, S. Aerosolized Administration of N-Acetylcysteine Attenuates Lung Fibrosis Induced by Bleomycin in Mice. Am. J. Respir. Crit. Care Med. 2000, 162, 225–231. [Google Scholar] [CrossRef]
- Berend, N. Inhibition of Bleomycin Lung Toxicityby N-Acetyl Cysteine in the Rat. Pathology 1985, 17, 108–110. [Google Scholar] [CrossRef]
- Sun, T.; Liu, J.; Zhao, D.W. Efficacy of N-Acetylcysteine in Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Medicine 2016, 95, e3629. [Google Scholar] [CrossRef]
- Oldham, J.M.; Ma, S.F.; Martinez, F.J.; Anstrom, K.J.; Raghu, G.; Schwartz, D.A.; Valenzi, E.; Witt, L.; Lee, C.; Vij, R.; et al. TOLLIP, MUC5B, and the Response to N-Acetylcysteine among Individuals with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1475–1482. [Google Scholar] [CrossRef]
- Tanaka, K.I.; Ishihara, T.; Azuma, A.; Kudoh, S.; Ebina, M.; Nukiwa, T.; Sugiyama, Y.; Tasaka, Y.; Namba, T.; Ishihara, T.; et al. Therapeutic Effect of Lecithinized Superoxide Dismutase on Bleomycin-Induced Pulmonary Fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2010, 298, 348–360. [Google Scholar] [CrossRef]
- Kamio, K.; Azuma, A.; Ohta, K.; Sugiyama, Y.; Nukiwa, T.; Kudoh, S.; Mizushima, T. Double-Blind Controlled Trial of Lecithinized Superoxide Dismutase in Patients with Idiopathic Interstitial Pneumonia—Short Term Evaluation of Safety and Tolerability. BMC Pulm. Med. 2014, 14, 86. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of Persistent Fibrosis in Aging by Targeting Nox4-Nrf2 Redox Imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef]
- Sato, N.; Takasaka, N.; Yoshida, M.; Tsubouchi, K.; Minagawa, S.; Araya, J.; Saito, N.; Fujita, Y.; Kurita, Y.; Kobayashi, K.; et al. Metformin Attenuates Lung Fibrosis Development via NOX4 Suppression. Respir. Res. 2016, 17, 1–12. [Google Scholar] [CrossRef]
- Tsubouchi, K.; Araya, J.; Minagawa, S.; Hara, H.; Ichikawa, A.; Saito, N.; Kadota, T.; Sato, N.; Yoshida, M.; Kurita, Y.; et al. Azithromycin Attenuates Myofibroblast Differentiation and Lung Fibrosis Development through Proteasomal Degradation of NOX4. Autophagy 2017, 13, 1420–1434. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, W.A.; Willems, S.; Vos, R.; Vanaudenaerde, B.M.; De Vleeschauwer, S.I.; Rinaldi, M.; Vanhooren, H.M.; Geudens, N.; Verleden, S.E.; Demedts, M.G.; et al. Azithromycin Reduces Pulmonary Fibrosis in a Bleomycin Mouse Model. Exp. Lung Res. 2010, 36, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Ichikado, K.; Yasuda, Y.; Anan, K.; Suga, M. Azithromycin for Idiopathic Acute Exacerbation of Idiopathic Pulmonary Fibrosis: A Retrospective Single-Center Study. BMC Pulm. Med. 2017, 17, 94. [Google Scholar] [CrossRef]
- Xu, Y.; Tai, W.; Qu, X.; Wu, W.; Li, Z.K.; Deng, S.; Vongphouttha, C.; Dong, Z. Rapamycin Protects against Paraquat-Induced Pulmonary Fibrosis: Activation of Nrf2 Signaling Pathway. Biochem. Biophys. Res. Commun. 2017, 490, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Ma, Z.; Shi, S.; Hu, Y.; Ma, T.; Rong, G.; Yang, J. Sulforaphane Prevents Bleomycin-Induced Pulmonary Fibrosis in Mice by Inhibiting Oxidative Stress via Nuclear Factor Erythroid 2-Related Factor-2 Activation. Mol. Med. Rep. 2017, 15, 4005–4014. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, F.; Kang, L.; Wang, Z.; Wang, Y. Pirfenidone Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice by Regulating Nrf2/Bach1 Equilibrium. BMC Pulm. Med. 2017, 17, 63. [Google Scholar] [CrossRef]
- Tang, H.; Gao, L.; Mao, J.; He, H.; Liu, J.; Cai, X.; Lin, H.; Wu, T. Salidroside Protects against Bleomycin-Induced Pulmonary Fibrosis: Activation of Nrf2-Antioxidant Signaling, and Inhibition of NF-ΚB and TGF-Β1/Smad-2/-3 Pathways. Cell Stress Chaperones 2016, 21, 239–249. [Google Scholar] [CrossRef]
- Liu, M.; Xu, H.; Zhang, L.; Zhang, C.; Yang, L.; Ma, E.; Liu, L.; Li, Y. Salvianolic Acid B Inhibits Myofibroblast Transdifferentiation in Experimental Pulmonary Fibrosis via the Up-Regulation of Nrf2. Biochem. Biophys. Res. Commun. 2018, 495, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Qin, H.Y.; Cao, L.F.; Chen, Y.H.; Tan, Z.X.; Zhang, C.; Xu, D.X. Phenylbutyric Acid Inhibits Epithelial-Mesenchymal Transition during Bleomycin-Induced Lung Fibrosis. Toxicol. Lett. 2015, 232, 213–220. [Google Scholar] [CrossRef]
- Griese, M.; Kappler, M.; Eismann, C.; Ballmann, M.; Junge, S.; Rietschel, E.; Van Koningsbruggen-Rietschel, S.; Staab, D.; Rolinck-Werninghaus, C.; Mellies, U.; et al. Inhalation Treatment with Glutathione in Patients with Cystic Fibrosis: A Randomized Clinical Trial. Am. J. Respir. Crit. Care Med. 2013, 188, 83–89. [Google Scholar] [CrossRef]
- Kariya, C.; Leitner, H.; Min, E.; Van Heeckeren, C.; Van Heeckeren, A.; Day, B.J. A Role for CFTR in the Elevation of Glutathione Levels in the Lung by Oral Glutathione Administration. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2007, 292, 1590–1597. [Google Scholar] [CrossRef]
- Conrad, C.; Lymp, J.; Thompson, V.; Dunn, C.; Davies, Z.; Chatfield, B.; Nichols, D.; Clancy, J.; Vender, R.; Egan, M.E.; et al. Long-Term Treatment with Oral N-Acetylcysteine: Affects Lung Function but Not Sputum Inflammation in Cystic Fibrosis Subjects. A Phase II Randomized Placebo-Controlled Trial. J. Cyst. Fibros. 2015, 14, 219–227. [Google Scholar] [CrossRef]
- Niki, E. Evidence for Beneficial Effects of Vitamin E. Korean J. Intern. Med. 2015, 30, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Khan, U.; Jain, R.; Graff, G.; Daines, C.L.; Dunitz, J.M.; Borowitz, D.; Orenstein, D.M.; Abdulhamid, I.; Noe, J.; et al. Effects of an Antioxidant-Enriched Multivitamin in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 198, 639–647. [Google Scholar] [CrossRef]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes Mellitus and Exocrine Pancreatic Dysfunction in Perk-/- Mice Reveals a Role for Translational Control in Secretory Cell Survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Zhang, K.; Wong, H.N.; Song, B.; Miller, C.N.; Scheuner, D.; Kaufman, R.J. The Unfolded Protein Response Sensor IRE1α Is Required at 2 Distinct Steps in B Cell Lymphopoiesis. J. Clin. Investig. 2005, 115, 268–281. [Google Scholar] [CrossRef]
- Lee, A.H.; Chu, G.C.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Is Required for Biogenesis of Cellular Secretory Machinery of Exocrine Glands. EMBO J. 2005, 24, 4368–4380. [Google Scholar] [CrossRef]
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An Essential Role in Liver Development for Transcription Factor XBP-1. Genes Dev. 2000, 14, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Yang, C.C.; Chan, D.C.; Wu, C.T.; Chen, L.P.; Huang, J.W.; Hung, K.Y.; Chiang, C.K. Chemical Chaperon 4-Phenylbutyrate Protects against the Endoplasmic Reticulum Stress-Mediated Renal Fibrosis in Vivo and in Vitro. Oncotarget 2016, 7, 22116–22127. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Ishitsuka, Y.; Hayasaka, M.; Yamada, Y.; Miyata, K.; Endo, M.; Kondo, Y.; Moriuchi, H.; Irikura, M.; Tanaka, K.I.; et al. The Exacerbating Roles of CCAAT/Enhancer-Binding Protein Homologous Protein (CHOP) in the Development of Bleomycin-Induced Pulmonary Fibrosis and the Preventive Effects of Tauroursodeoxycholic Acid (TUDCA) against Pulmonary Fibrosis in Mice. Pharmacol. Res. 2015, 99, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.; Sreenivasaiah, P.K.; Kim, J.O.; Lee, M.Y.; Kang, W.S.; Kim, Y.S.; Ahn, Y.; Park, W.J.; Cho, C.; Kim, D.H. Tauroursodeoxycholic Acid (TUDCA) Attenuates Pressure Overload-Induced Cardiac Remodeling by Reducing Endoplasmic Reticulum Stress. PLoS ONE 2017, 12, e0176071. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magallón, M.; Pastor, S.; Carrión, A.E.; Bañuls, L.; Pellicer, D.; Castillo, S.; Bondía, S.; Navarro-García, M.M.; González, C.; Dasí, F. Oxidative Stress and Endoplasmic Reticulum Stress in Rare Respiratory Diseases. J. Clin. Med. 2021, 10, 1268. https://doi.org/10.3390/jcm10061268
Magallón M, Pastor S, Carrión AE, Bañuls L, Pellicer D, Castillo S, Bondía S, Navarro-García MM, González C, Dasí F. Oxidative Stress and Endoplasmic Reticulum Stress in Rare Respiratory Diseases. Journal of Clinical Medicine. 2021; 10(6):1268. https://doi.org/10.3390/jcm10061268
Chicago/Turabian StyleMagallón, María, Sara Pastor, Ana Esther Carrión, Lucía Bañuls, Daniel Pellicer, Silvia Castillo, Sergio Bondía, María Mercedes Navarro-García, Cruz González, and Francisco Dasí. 2021. "Oxidative Stress and Endoplasmic Reticulum Stress in Rare Respiratory Diseases" Journal of Clinical Medicine 10, no. 6: 1268. https://doi.org/10.3390/jcm10061268