The Role of Rab Proteins in Neuronal Cells and in the Trafficking of Neurotrophin Receptors


{kind=link}
{kind=link}
Abstract
:1. Introduction
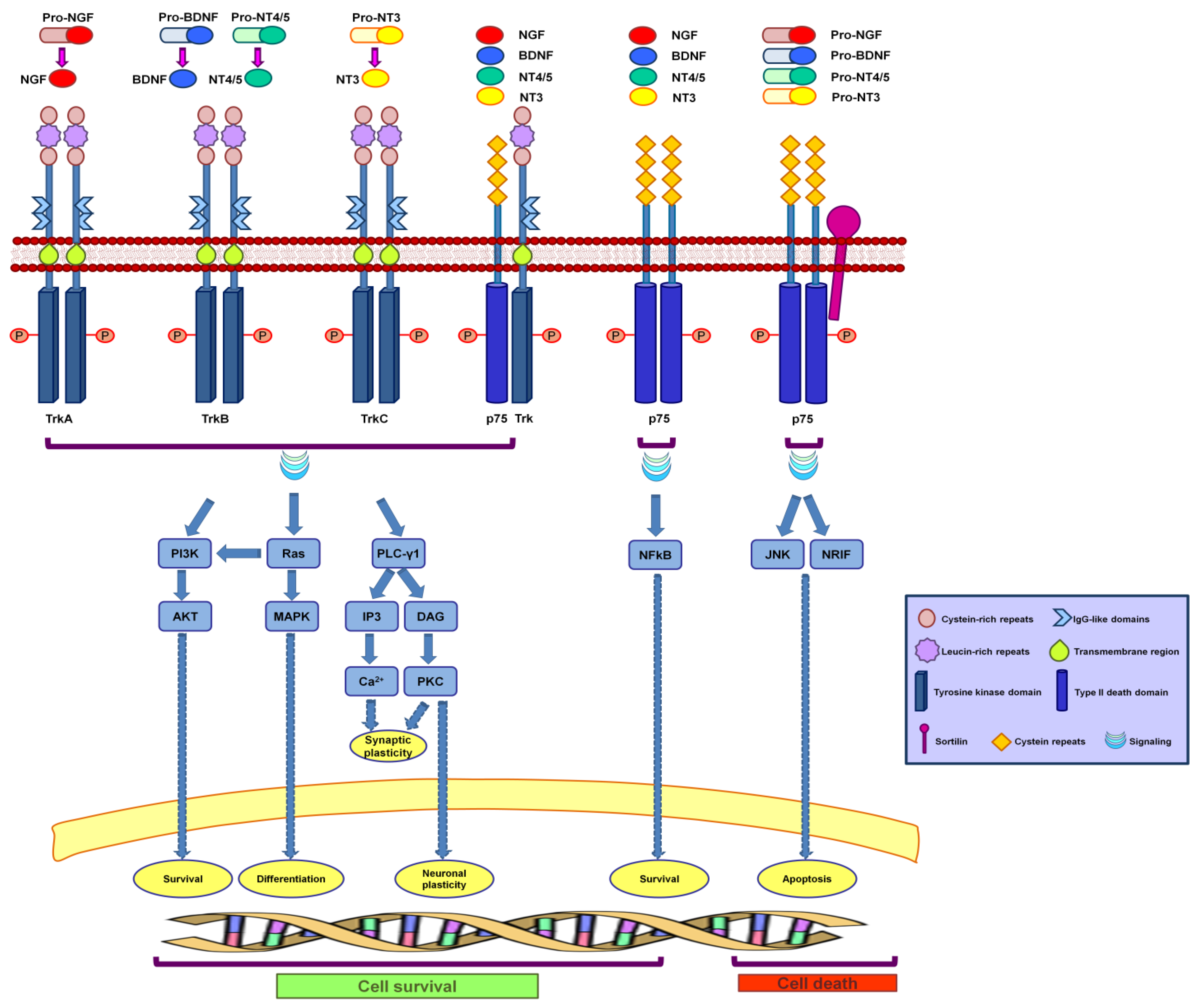
2. Neurotrophins and Their Receptors
2.1. Neurotrophins
2.2. Neurotrophin Receptors
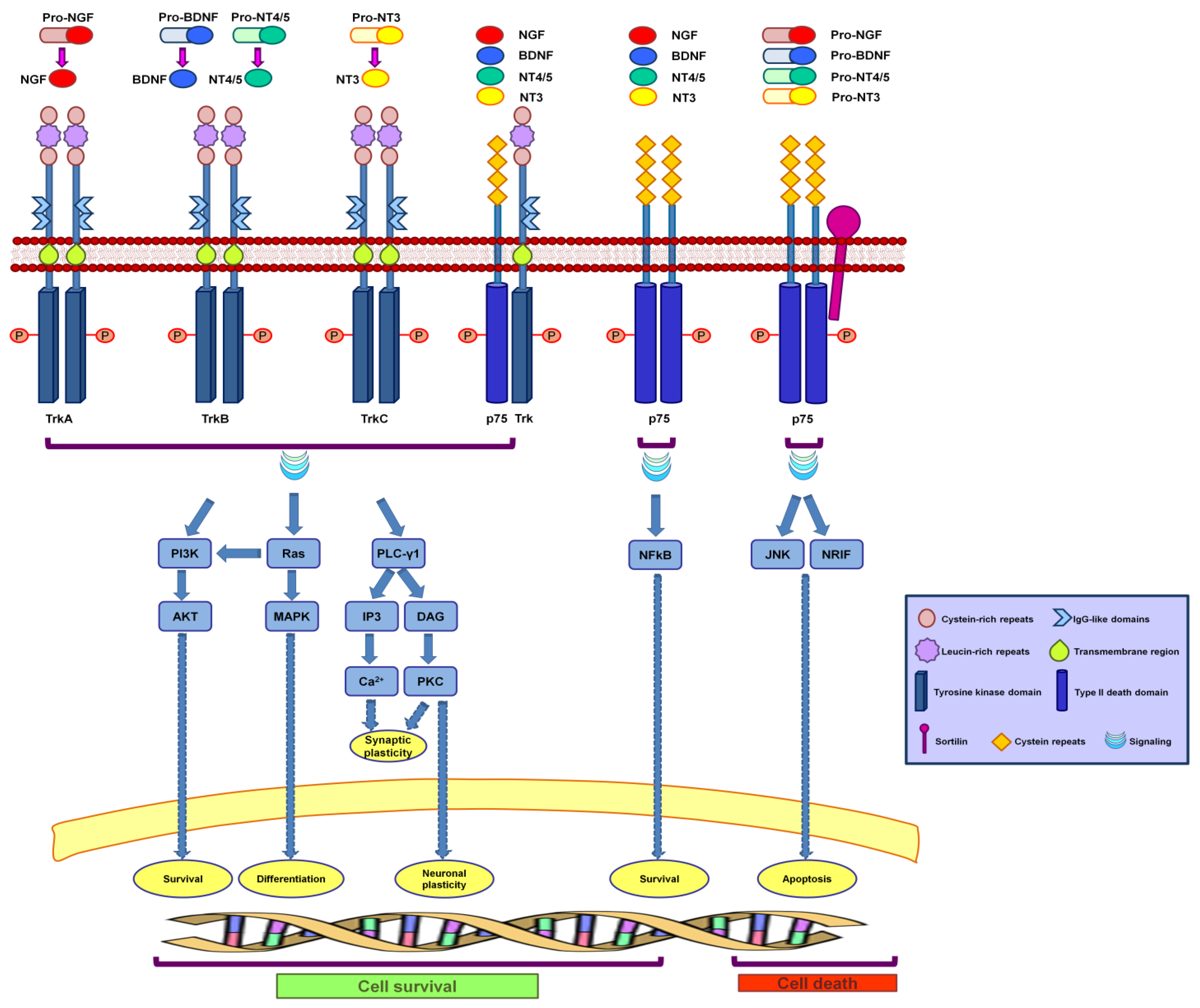
3. Neurotrophin Signaling
3.1. Neurotrophin Receptor Signaling
3.2. Neurotrophin Receptor Trafficking and the “Signaling Endosome” Hypothesis
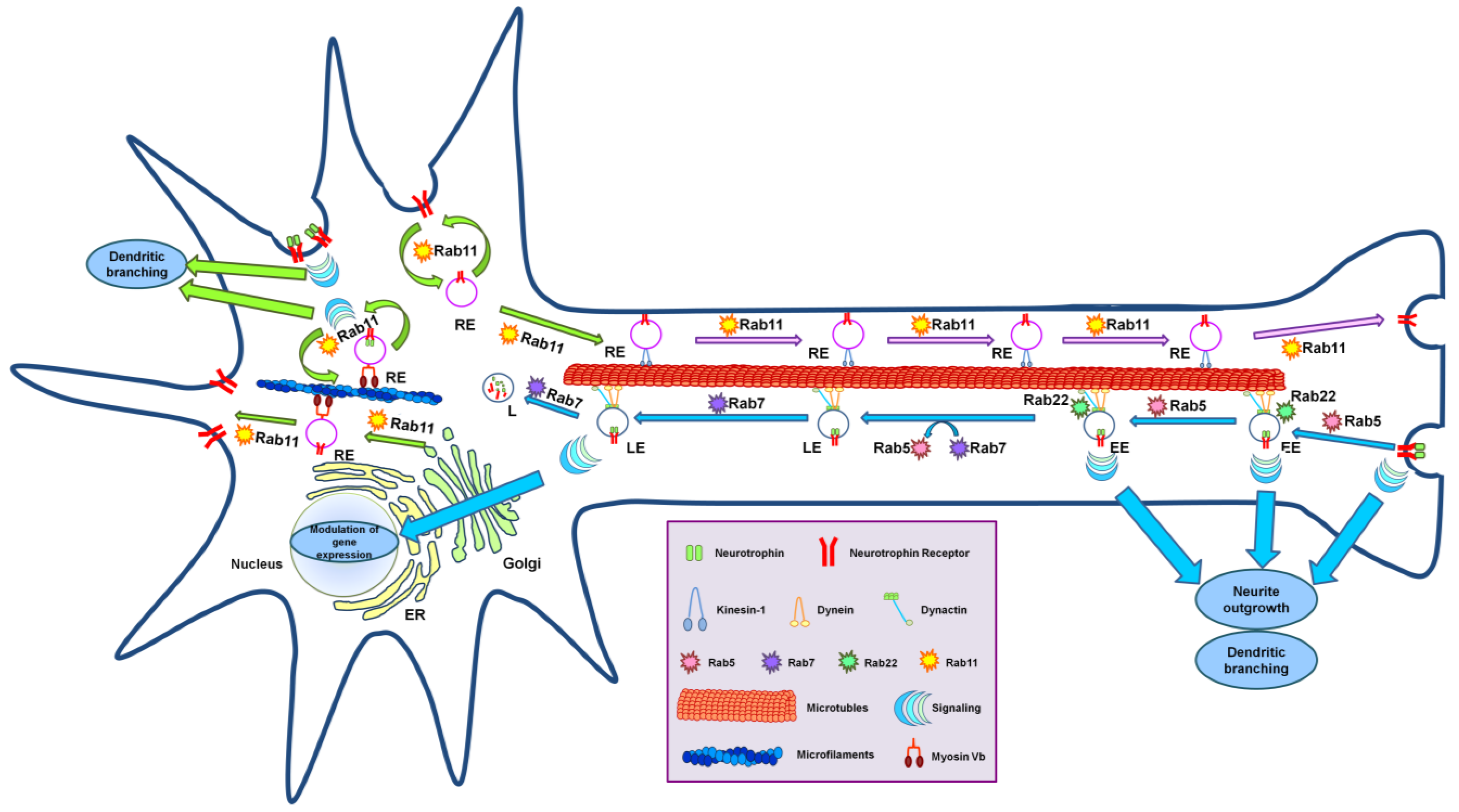
4. Neurotrophin Receptor Trafficking: The Role of Rab Proteins
4.1. Neuronal Trafficking and Axonal Transport
4.2. Rab Proteins and Neuronal Trafficking
4.3. Rab Proteins and Anterograde Axonal Transport
4.4. Rab Proteins and Anterograde Trafficking of Neurotrophin Receptors
4.5. Rab Proteins and Retrograde Axonal Transport of Neurotrophin Receptors
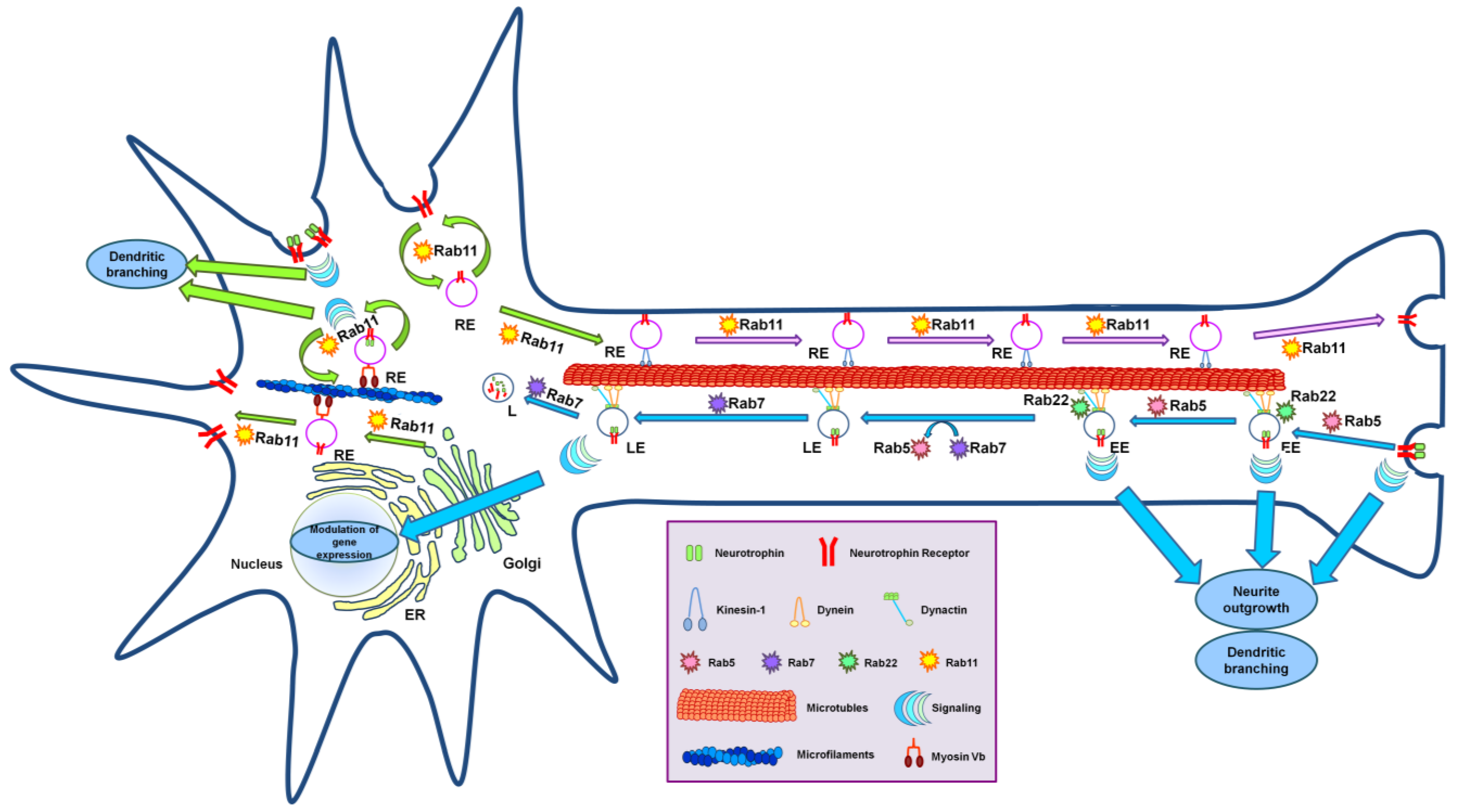
4.6. Rab Proteins and Microtubule Motors
5. Neurodegenerative and Age-Related Diseases: Consequences of Alterations of Neurotrophin Trafficking
5.1. Alzheimer’s Disease (AD)
5.2. Amyotrophic Lateral Sclerosis (ALS)
5.3. Charcot-Marie-Tooth Disease
5.4. Huntington’s Disease (HD)
5.5. Aging
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Levi-Montalcini, R.; Hamburger, V. Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J. Exp. Zool. 1951, 116, 321–361. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Levi-Montalcini, R.; Hamburger, V. A nerve growth-stimulating factor isolated from sarcom as 37 and 180. Proc. Natl. Acad. Sci. USA 1954, 40, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [PubMed]
- Maisonpierre, P.C.; Belluscio, L.; Squinto, S.; Ip, N.Y.; Furth, M.E.; Lindsay, R.M.; Yancopoulos, G.D. Neurotrophin-3: A neurotrophic factor related to NGF and BDNF. Science 1990, 247, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Hallböök, F.; Ibáñez, C.F.; Persson, H. Evolutionary studies of the nerve growth factor family reveal a novel member abundantly expressed in xenopus ovary. Neuron 1991, 6, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Ip, N.Y.; Ibáñez, C.F.; Nye, S.H.; McClain, J.; Jones, P.F.; Gies, D.R.; Belluscio, L.; Le Beau, M.M.; Espinosa, R.R.; Squinto, S.P.; et al. Mammalian neurotrophin-4: Structure, chromosomal localization, tissue distribution, and receptor specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 3060–3064. [Google Scholar] [CrossRef] [PubMed]
- Hennigan, A.; O’Callaghan, R.M.; Kelly, A.M. Neurotrophins and their receptors: Roles in plasticity, neurodegeneration and neuroprotection. Biochem. Soc. Trans. 2007, 35, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.L.; Mobley, W.C. Long-distance retrograde neurotrophic signaling. Curr. Opin. Neurobiol. 2005, 15, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.L.; Mobley, W.C. Signaling endosome hypothesis: A cellular mechanism for long distance communication. J. Neurobiol. 2004, 58, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Hutagalung, A.H.; Novick, P. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.; Lambright, D.G. Rab GEFs and GAPs. Curr. Opin. Cell Biol. 2010, 22, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. Rab GTPase regulation of membrane identity. Curr. Opin. Cell Biol. 2013, 25, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A. Review series: Rab GTPases and membrane identity: Causal or inconsequential? J. Cell Biol. 2013, 202, 191–199. [Google Scholar] [CrossRef]
- Bucci, C.; Chiariello, M. Signal transduction gRabs attention. Cell Signal 2006, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, A.L.; Bucci, C. Rab GTPases-cargo direct interactions: Fine modulators of intracellular trafficking. Histol. Histopathol. 2013, 28, 839–849. [Google Scholar] [PubMed]
- Poon, V.Y.; Choi, S.; Park, M. Growth factors in synaptic function. Front Synaptic Neurosci. 2013, 5. [Google Scholar] [CrossRef]
- Weisenhorn, D.M.; Roback, J.; Young, A.N.; Wainer, B.H. Cellular aspects of trophic actions in the nervous system. Int. Rev. Cytol. 1999, 189, 177–265. [Google Scholar] [PubMed]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Butte, M.J. Neurotrophic factor structures reveal clues to evolution, binding, specificity, and receptor activation. Cell Mol. Life Sci. 2001, 58, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed]
- Mowla, S.J.; Pareek, S.; Farhadi, H.F.; Petrecca, K.; Fawcett, J.P.; Seidah, N.G.; Morris, S.J.; Sossin, W.S.; Murphy, R.A. Differential sorting of nerve growth factor and brain-derived neurotrophic factor in hippocampal neurons. J. Neurosci. 1999, 19, 2069–2080. [Google Scholar] [PubMed]
- Dicou, E. Multiple biological activities for two peptides derived from the nerve growth factor precursor. Biochem. Biophys. Res. Commun. 2006, 347, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Dicou, E. Peptides other than the neurotrophins that can be cleaved from proneurotrophins: A neglected story. Arch. Physiol. Biochem. 2007, 113, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of cell survival by secreted proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Barker, P.A. Neurotrophin signaling through the p75 neurotrophin receptor. Prog. Neurobiol. 2002, 67, 203–233. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Clary, D.O.; Reichardt, L.F. An alternatively spliced form of the nerve growth factor receptor TrkA confers an enhanced response to neurotrophin 3. Proc. Natl. Acad. Sci. USA 1994, 91, 11133–11137. [Google Scholar] [CrossRef] [PubMed]
- Strohmaier, C.; Carter, B.D.; Urfer, R.; Barde, Y.A.; Dechant, G. A splice variant of the neurotrophin receptor TrkB with increased specificity for brain-derived neurotrophic factor. EMBO J. 1996, 15, 3332–3337. [Google Scholar] [PubMed]
- Eide, F.F.; Vining, E.R.; Eide, B.L.; Zang, K.; Wang, X.Y.; Reichardt, L.F. Naturally occurring truncated TrkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J. Neurosci. 1996, 16, 3123–3129. [Google Scholar] [PubMed]
- Sotthibundhu, A.; Sykes, A.M.; Fox, B.; Underwood, C.K.; Thangnipon, W.; Coulson, E.J. Beta-amyloid(1–42) induces neuronal death through the p75 neurotrophin receptor. J. Neurosci. 2008, 28, 3941–3946. [Google Scholar] [CrossRef] [PubMed]
- Hasebe, N.; Fujita, Y.; Ueno, M.; Yoshimura, K.; Fujino, Y.; Yamashita, T. Soluble β-amyloid precursor protein alpha binds to p75 neurotrophin receptor to promote neurite outgrowth. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Della-Bianca, V.; Rossi, F.; Armato, U.; Dal-Pra, I.; Costantini, C.; Perini, G.; Politi, V.; Della Valle, G. Neurotrophin p75 receptor is involved in neuronal damage by prion peptide-(106–126). J. Biol. Chem. 2001, 276, 38929–38933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Chen, Y.; Liu, Y.; Zhao, Y.; Liao, F.F.; Xu, H. App regulates NGF receptor trafficking and NGF-mediated neuronal differentiation and survival. PLoS One 2013, 8, e80571. [Google Scholar] [CrossRef] [PubMed]
- Skeldal, S.; Sykes, A.M.; Glerup, S.; Matusica, D.; Palstra, N.; Autio, H.; Boskovic, Z.; Madsen, P.; Castrén, E.; Nykjaer, A.; et al. Mapping of the interaction site between sortilin and the p75 neurotrophin receptor reveals a regulatory role for the sortilin intracellular domain in p75 neurotrophin receptor shedding and apoptosis. J. Biol. Chem. 2012, 287, 43798–43809. [Google Scholar] [CrossRef] [PubMed]
- Bibel, M.; Hoppe, E.; Barde, Y.A. Biochemical and functional interactions between the neurotrophin receptors Trk and p75NTR. EMBO J. 1999, 18, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Makkerh, J.P.; Ceni, C.; Auld, D.S.; Vaillancourt, F.; Dorval, G.; Barker, P.A. P75 neurotrophin receptor reduces ligand-induced Trk receptor ubiquitination and delays Trk receptor internalization and degradation. EMBO Rep. 2005, 6, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Ceni, C.; Kommaddi, R.P.; Thomas, R.; Vereker, E.; Liu, X.; McPherson, P.S.; Ritter, B.; Barker, P.A. The p75NTR intracellular domain generated by neurotrophin-induced receptor cleavage potentiates Trk signaling. J. Cell Sci. 2010, 123, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Volosin, M.; Cragnolini, A.B.; Hempstead, B.L.; Friedman, W.J. ProNGF induces PTEN via p75NTR to suppress Trk-mediated survival signaling in brain neurons. J. Neurosci. 2010, 30, 15608–15615. [Google Scholar] [CrossRef] [PubMed]
- Hamanoue, M.; Middleton, G.; Wyatt, S.; Jaffray, E.; Hay, R.T.; Davies, A.M. P75-mediated NF-kappaB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol. Cell Neurosci. 1999, 14, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Willnow, T.E.; Petersen, C.M.; Nykjaer, A. Vps10p-domain receptors regulators of neuronal viability and function. Nat. Rev. Neurosci. 2008, 9, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed]
- Segal, R.A.; Greenberg, M.E. Intracellular signaling pathways activated by neurotrophic factors. Annu. Rev. Neurosci. 1996, 19, 483–489. [Google Scholar] [CrossRef]
- Pawson, T.; Nash, P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000, 14, 1027–1047. [Google Scholar] [PubMed]
- Sciarretta, C.; Fritzsch, B.; Beisel, K.; Rocha-Sanchez, S.M.; Buniello, A.; Horn, J.M.; Minichiello, L. Plcγ-activated signaling is essential for TrkB mediated sensory neuron structural plasticity. BMC Dev. Biol. 2010, 10. [Google Scholar] [CrossRef]
- Iacaruso, M.F.; Galli, S.; Martí, M.; Villalta, J.I.; Estrin, D.A.; Jares-Erijman, E.A.; Pietrasanta, L.I. Structural model for p75(NTR)-TrkA intracellular domain interaction: A combined FRET and bioinformatics study. J. Mol. Biol. 2011, 414, 681–698. [Google Scholar] [CrossRef] [PubMed]
- Uren, R.T.; Turnley, A.M. Regulation of neurotrophin receptor (Trk) signaling: Suppressor of cytokine signaling 2 (SOCS2) is a new player. Front Mol. Neurosci. 2014, 7. [Google Scholar] [CrossRef]
- Sassone-Corsi, P.; Der, C.J.; Verma, I.M. Ras-induced neuronal differentiation of PC12 cells: Possible involvement of fos and jun. Mol. Cell Biol. 1989, 9, 3174–3183. [Google Scholar] [PubMed]
- Li, S.; Mattar, P.; Dixit, R.; Lawn, S.O.; Wilkinson, G.; Kinch, C.; Eisenstat, D.; Kurrasch, D.M.; Chan, J.A.; Schuurmans, C. Ras/ERK signaling controls proneural genetic programs in cortical development and gliomagenesis. J. Neurosci. 2014, 34, 2169–2190. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.; Ahn, S.; Davenport, C.M.; Blendy, J.A.; Ginty, D.D. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 1999, 286, 2358–2361. [Google Scholar] [CrossRef] [PubMed]
- Holgado-Madruga, M.; Moscatello, D.K.; Emlet, D.R.; Dieterich, R.; Wong, A.J. Grb2-associated binder-1 mediates phosphatidylinositol 3-kinase activation and the promotion of cell survival by nerve growth factor. Proc. Natl. Acad. Sci. USA 1997, 94, 12419–12424. [Google Scholar] [CrossRef] [PubMed]
- Marte, B.M.; Downward, J. Pkb/akt: Connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem. Sci. 1997, 22, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Lipinski, M.; Degterev, A. Diversity in the mechanisms of neuronal cell death. Neuron 2003, 40, 401–413. [Google Scholar] [CrossRef]
- Brunet, A.; Datta, S.R.; Greenberg, M.E. Transcription-dependent and -independent control of neuronal survival by the PI3K-AKT signaling pathway. Curr. Opin. Neurobiol. 2001, 11, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann-Zeh, A.; Rodriguez-Viciana, P.; Ulrich, E.; Gilbert, C.; Coffer, P.; Downward, J.; Evan, G. Suppression of c-Myc-induced apoptosis by Ras signaling through PI(3)K and PKB. Nature 1997, 385, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Canossa, M.; Griesbeck, O.; Berninger, B.; Campana, G.; Kolbeck, R.; Thoenen, H. Neurotrophin release by neurotrophins: Implications for activity-dependent neuronal plasticity. Proc. Natl. Acad. Sci. USA 1997, 94, 13279–13286. [Google Scholar] [CrossRef] [PubMed]
- Loeb, D.M.; Stephens, R.M.; Copeland, T.; Kaplan, D.R.; Greene, L.A. A trk nerve growth factor (NGF) receptor point mutation affecting interaction with phospholipase c-gamma 1 abolishes NGF-promoted peripherin induction but not neurite outgrowth. J. Biol. Chem. 1994, 269, 8901–8910. [Google Scholar] [PubMed]
- Wong, J.; Oblinger, M.M. Differential regulation of peripherin and neurofilament gene expression in regenerating rat DRG neurons. J. Neurosci. Res. 1990, 27, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Julien, J.P. Peripherin-mediated death of motor neurons rescued by overexpression of neurofilament NF-H proteins. J. Neurochem. 2003, 85, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Cogli, L.; Progida, C.; Thomas, C.L.; Spencer-Dene, B.; Donno, C.; Schiavo, G.; Bucci, C. Charcot-Marie-Tooth type 2b disease-causing rab7a mutant proteins show altered interaction with the neuronal intermediate filament peripherin. Acta Neuropathol. 2013, 25, 257–272. [Google Scholar] [CrossRef]
- Watson, F.L.; Porcionatto, M.A.; Bhattacharyya, A.; Stiles, C.D.; Segal, R.A. TrkA glycosylation regulates receptor localization and activity. J. Neurobiol. 1999, 39, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, P.P.; de Camilli, P. Endocytosis and signaling: An inseparable partnership. Cell 2001, 106, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Miaczynska, M.; Pelkmans, L.; Zerial, M. Not just a sink: Endosomes in control of signal transduction. Curr. Opin. Cell Biol. 2004, 16, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Grimes, M.L.; Zhou, J.; Beattie, E.C.; Yuen, E.C.; Hall, D.E.; Valletta, J.S.; Topp, K.S.; LaVail, J.H.; Bunnett, N.W.; Mobley, W.C. Endocytosis of activated TrkA: Evidence that nerve growth factor induces formation of signaling endosomes. J. Neurosci. 1996, 16, 7950–7964. [Google Scholar] [PubMed]
- Delcroix, J.D.; Valletta, J.S.; Wu, C.; Hunt, S.J.; Kowal, A.S.; Mobley, W.C. NGF signaling in sensory neurons: Evidence that early endosomes carry NGF retrograde signals. Neuron 2003, 39, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Grimes, M.L.; Beattie, E.; Mobley, W.C. A signaling organelle containing the nerve growth factor-activated receptor tyrosine kinase, trka. Proc. Natl. Acad. Sci. USA 1997, 94, 9909–9914. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Howe, C.L.; Cosgaya, J.M.; Steiner, P.; Hirling, H.; Chan, J.R.; Weis, J.; Kruttgen, A. Differential endocytic sorting of p75NTR and TrkA in response to NGF: A role for late endosomes in TrkA trafficking. Mol. Cell Neurosci. 2005, 28, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.; Paladino, S.; Mazzone, M.; Nitsch, L.; Gulisano, M.; Zurzolo, C. Functional interaction between p75NTR and TrkA: The endocytic trafficking of p75NTR is driven by TrkA and regulates TrkA-mediated signaling. Biochem. J. 2005, 385, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Boutilier, J.; Ceni, C.; Pagdala, P.C.; Forgie, A.; Neet, K.E.; Barker, P.A. Proneurotrophins require endocytosis and intracellular proteolysis to induce TrkA activation. J. Biol. Chem. 2008, 283, 12709–12716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Moheban, D.B.; Conway, B.R.; Bhattacharyya, A.; Segal, R.A. Cell surface trk receptors mediate NGF-induced survival while internalized receptors regulate NGF-induced differentiation. J. Neurosci. 2000, 20, 5671–5678. [Google Scholar] [PubMed]
- Kuruvilla, R.; Zweifel, L.S.; Glebova, N.O.; Lonze, B.E.; Valdez, G.; Ye, H.; Ginty, D.D. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell 2004, 118, 243–255. [Google Scholar] [CrossRef] [PubMed]
- York, R.D.; Molliver, D.C.; Grewal, S.S.; Stenberg, P.E.; McCleskey, E.W.; Stork, P.J. Role of phosphoinositide 3-kinase and endocytosis in nerve growth factor-induced extracellular signal-regulated kinase activation via Ras and Rap. Mol. Cell Biol. 2000, 20, 8069–8083. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.L.; Valletta, J.S.; Rusnak, A.S.; Mobley, W.C. NGF signaling from clathrin-coated vesicles: Evidence that signaling endosomes serve as a platform for the Ras-MAPK pathway. Neuron 2001, 32, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Hibbert, A.P.; Kramer, B.M.; Miller, F.D.; Kaplan, D. The localization, trafficking and retrograde transport of BDNF bound to p75NTR in sympathetic neurons. Mol. Cell Neurosci. 2006, 32, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Limpert, A.S.; Karlo, J.C.; Landreth, G.E. Nerve growth factor stimulates the concentration of TrkA within lipid rafts and extracellular signal-regulated kinase activation through c-Cbl-associated protein. Mol. Cell Biol. 2007, 27, 5686–5698. [Google Scholar] [CrossRef] [PubMed]
- Valdez, G.; Philippidou, P.; Rosenbaum, J.; Akmentin, W.; Shao, Y.; Halegoua, S. Trk-signaling endosomes are generated by Rac-dependent macroendocytosis. Proc. Natl. Acad. Sci. USA 2007, 104, 12270–12275. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Akmentin, W.; Toledo-Aral, J.J.; Rosenbaum, J.; Valdez, G.; Cabot, J.B.; Hilbush, B.S.; Halegoua, S. Pincher, a pinocytic chaperone for nerve growth factor/TrkA signaling endosomes. J. Cell Biol. 2002, 157, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Valdez, G.; Akmentin, W.; Philippidou, P.; Kuruvilla, R.; Ginty, D.D.; Halegoua, S. Pincher-mediated macroendocytosis underlies retrograde signaling by neurotrophin receptors. J. Neurosci. 2005, 25, 5236–5247. [Google Scholar] [PubMed]
- Philippidou, P.; Valdez, G.; Akmentin, W.; Bowers, W.J.; Federoff, H.J.; Halegoua, S. Trk retrograde signaling requires persistent, Pincher-directed endosomes. Proc. Natl. Acad. Sci. USA 2011, 108, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Geetha, T.; Wooten, M.W. TrkA receptor endolysosomal degradation is both ubiquitin and proteasome dependent. Traffic 2008, 9, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Yoo, Y.S. Nerve growth factor-induced neurite outgrowth is potentiated by stabilization of TrkA receptors. BMB Rep. 2011, 44, 182–186. [Google Scholar] [CrossRef]
- Takahashi, Y.; Shimokawa, N.; Esmaeili-Mahani, S.; Morita, A.; Masuda, H.; Iwasaki, T.; Tamura, J.; Haglund, K.; Koibuchi, N. Ligand-induced downregulation of TrkA is partly regulated through ubiquitination by CBL. FEBS Lett. 2011, 585, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.C.; Howe, C.L.; Wilde, A.; Brodsky, F.M.; Mobley, W.C. NGF signals through TrkA to increase clathrin at the plasma membrane and enhance clathrin-mediated membrane trafficking. J. Neurosci. 2000, 20, 7325–7333. [Google Scholar] [PubMed]
- Nomura, M.; Nagai, T.; Harada, Y.; Tani, T. Facilitated intracellular transport of TrkA by an interaction with Nerve Growth Factor. Dev. Neurobiol. 2011, 71, 634–649. [Google Scholar] [CrossRef] [PubMed]
- Ascaño, M.; Richmond, A.; Borden, P.; Kuruvilla, R. Axonal targeting of Trk receptors via transcytosis regulates sensitivity to neurotrophin responses. J. Neurosci. 2009, 29, 11674–11685. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; van Niekerk, E.; Willis, D.E.; Twiss, J.L. RNA transport and localized protein synthesis in neurological disorders and neural repair. Dev. Neurobiol. 2007, 67, 1166–1182. [Google Scholar] [PubMed]
- Goldstein, L.S. Molecular motors: From one motor many tails to one motor many tales. Trends Cell Biol. 2001, 11, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Takemura, R. Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci. 2005, 6, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Brown, A. Axonal transport of membranous and nonmembranous cargoes: A unified perspective. J. Cell. Biol. 2003, 160, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Verhey, K.J.; Hammond, J.W. Traffic control: Regulation of kinesin motors. Nat. Rev. Mol. Cell Biol. 2009, 10, 765–777. [Google Scholar] [CrossRef]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [PubMed]
- King, S.J.; Schroer, T.A. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat. Cell Biol. 2000, 2, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yao, X.; Fischer, L.; Abenza, J.F.; Peñalva, M.A.; Xiang, X. The p25 subunit of the dynactin complex is required for dynein-early endosome interaction. J. Cell Biol. 2011, 193, 1245–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavo, G.; Greensmith, L.; Hafezparast, M.; Fisher, E.M. Cytoplasmic dynein heavy chain: The servant of many masters. Trends Neurosci. 2013, 36, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Hammer, J.A., III; Wagner, W. Functions of class V myosins in neurons. J. Biol. Chem. 2013, 288, 28428–28434. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Chylinski, T.M.; Pimenta, A.; Ortiz, D.; Shea, T.B. Neurofilament transport is dependent on actin and myosin. J. Neurosci. 2004, 24, 9486–9496. [Google Scholar] [CrossRef] [PubMed]
- Alami, N.H.; Jung, P.; Brown, A. Myosin Va increases the efficiency of neurofilament transport by decreasing the duration of long-term pauses. J. Neurosci. 2009, 29, 6625–6634. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Sepp, K.J.; Hollenbeck, P.J. Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J. Neurosci. 2010, 30, 8984–8992. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.L., Jr.; Mao, T.; Arnold, D.B. A role for myosin VI in the localization of axonal proteins. PLoS Biol. 2011, 9. [Google Scholar] [CrossRef]
- Rudolf, R.; Bittins, C.M.; Gerdes, H.H. The role of myosin V in exocytosis and synaptic plasticity. J. Neurochem. 2011, 116, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Lalli, G.; Gschmeissner, S.; Schiavo, G. Myosin Va and microtubule-based motors are required for fast axonal retrograde transport of tetanus toxin in motor neurons. J. Cell Sci. 2003, 116, 4639–4650. [Google Scholar] [CrossRef] [PubMed]
- Bittins, C.M.; Eichler, T.W.; Hammer, J.A., III; Gerdes, H.H. Dominant-negative myosin Va impairs retrograde but not anterograde axonal transport of large dense core vesicles. Cell Mol. Neurobiol. 2010, 30, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.L.; Tang, B.L. Rab GTPases and their roles in brain neurons and glia. Brain Res. Rev. 2008, 58, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Star, E.N.; Newton, A.J.; Murthy, V.N. Real-time imaging of Rab3a and Rab5a reveals differential roles in presynaptic function. J. Physiol. 2005, 569, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Szodorai, A.; Kuan, Y.H.; Hunzelmann, S.; Engel, U.; Sakane, A.; Sasaki, T.; Takai, Y.; Kirsch, J.; Müller, U.; Beyreuther, K.; et al. App anterograde transport requires Rab3a GTPase activity for assembly of the transport vesicle. J. Neurosci. 2009, 29, 14534–14544. [Google Scholar] [PubMed]
- Li, J.Y.; Jahn, R.; Dahlström, A. Rab3a, a small GTP-binding protein, undergoes fast anterograde transport but not retrograde transport in neurons. Eur. J. Cell Biol. 1995, 67, 297–307. [Google Scholar] [PubMed]
- Huber, L.A.; Dupree, P.; Dotti, C.G. A deficiency of the small GTPase Rab8 inhibits membrane traffic in developing neurons. Mol. Cell Biol. 1995, 15, 918–924. [Google Scholar] [PubMed]
- Hattula, K.; Furuhjelm, J.; Tikkanen, J.; Tanhuanpää, K.; Laakkonen, P.; Peränen, J. Characterization of the Rab8-specific membrane traffic route linked to protrusion formation. J. Cell Sci. 2006, 119, 4866–4877. [Google Scholar] [CrossRef] [PubMed]
- Sakane, A.; Honda, K.; Sasaki, T. Rab13 regulates neurite outgrowth in PC12 cells through its effector protein, jrab/mical-l2. Mol. Cell Biol. 2010, 30, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Pfenninger, K.H. Plasma membrane expansion: A neuron’s herculean task. Nat. Rev. Neurosci. 2009, 10, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.Y.; Lei, W.L.; Xu, X.H.; Ju, X.C.; Liu, Y.; Luo, Z.G. Jip1 mediates anterograde transport of Rab10 cargos during neuronal polarization. J. Neurosci. 2014, 34, 1710–1723. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.H.; Deng, C.Y.; Liu, Y.; He, M.; Peng, J.; Wang, T.; Yuan, L.; Zheng, Z.S.; Blackshear, P.J.; Luo, Z.G. MARCKS regulates membrane targeting of Rab10 vesicles to promote axon development. Cell Res. 2014, 24, 576–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, X.H.; Chen, Q.; Wang, T.; Deng, C.Y.; Song, B.L.; Du, J.L.; Luo, Z.G. Myosin Vb controls biogenesis of post-golgi Rab10 carriers during axon development. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Schuck, S.; Gerl, M.J.; Ang, A.; Manninen, A.; Keller, P.; Mellman, I.; Simons, K. Rab10 is involved in basolateral transport in polarized madin-darby canine kidney cells. Traffic 2007, 8, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, Y.; Xu, X.H.; Deng, C.Y.; Wu, K.Y.; Zhu, J.; Fu, X.Q.; He, M.; Luo, Z.G. Lgl1 activation of Rab10 promotes axonal membrane trafficking underlying neuronal polarization. Dev. Cell 2011, 21, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Duan, S.; Sun, T.; Wang, J.; Zhao, L.; Geng, Z.; Yan, J.; Sun, H.J.; Chen, Z.Y. JIP3 mediates TrkB axonal anterograde transport and enhances BDNF signaling by directly bridging TrkB with kinesin-1. J. Neurosci. 2011, 31, 10602–10614. [Google Scholar] [CrossRef] [PubMed]
- Vaegter, C.B.; Jansen, P.; Fjorback, A.W.; Glerup, S.; Skeldal, S.; Kjolby, M.; Richner, M.; Erdmann, B.; Nyengaard, J.R.; Tessarollo, L.; et al. Sortilin associates with Trk receptors to enhance anterograde transport and neurotrophin signaling. Nat. Neurosci. 2011, 14, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Kimura, T.; Nakamuta, S.; Taya, S.; Funahashi, Y.; Hattori, A.; Shimada, A.; Ménager, C.; Kawabata, S.; Fujii, K.; et al. Anterograde transport of TrkB in axons is mediated by direct interaction with SLP1 and Rab27. Dev. Cell 2009, 16, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Torii, S.; Yokota-Hashimoto, H.; Takeuchi, T.; Izumi, T. Involvement of Rab27b in the regulated secretion of pituitary hormones. Endocrinology 2002, 143, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Progida, C.; Cogli, L.; Piro, F.; de Luca, A.; Bakke, O.; Bucci, C. Rab7b controls trafficking from endosomes to the TGN. J. Cell Sci. 2010, 123, 1480–1491. [Google Scholar] [CrossRef] [PubMed]
- Progida, C.; Nielsen, M.S.; Koster, G.; Bucci, C.; Bakke, O. Dynamics of Rab7b-dependent transport of sorting receptors. Traffic 2012, 13, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, O.; Reinsch, S.; Urbé, S.; Zerial, M.; Parton, R.G. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 1996, 135, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Shirane, M.; Nakayama, K.I. Protrudin induces neurite formation by directional membrane trafficking. Science 2006, 314, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Eva, R.; Dassie, E.; Caswell, P.T.; Dick, G.; ffrench-Constant, C.; Norman, J.C.; Fawcett, J.W. Rab11 and its effector Rab Coupling Protein contribute to the trafficking of beta 1 integrins during axon growth in adult dorsal root ganglion neurons and PC12 cells. J. Neurosci. 2010, 30, 11654–11669. [Google Scholar] [CrossRef] [PubMed]
- Lazo, O.M.; Gonzalez, A.; Ascano, M.; Kuruvilla, R.; Couve, A.; Bronfman, F.C. BDNF regulates Rab11-mediated recycling endosome dynamics to induce dendritic branching. J. Neurosci. 2013, 33, 6112–6122. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Wang, J.; Sui, W.H.; Chen, B.; Zhang, X.Y.; Yan, J.; Geng, Z.; Chen, Z.Y. BDNF-dependent recycling facilitates TrkB translocation to postsynaptic density during LTP via a Rab11-dependent pathway. J. Neurosci. 2013, 33, 9214–9230. [Google Scholar]
- Ehlers, M.D.; Kaplan, D.R.; Price, D.L.; Koliatsos, V.E. NGF-stimulated retrograde transport of TrkA in the mammalian nervous system. J. Cell Biol. 1995, 130, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Watson, F.L.; Heerssen, H.M.; Moheban, D.B.; Lin, M.Z.; Sauvageot, C.M.; Bhattacharyya, A.; Pomeroy, S.L.; Segal, R.A. Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J. Neurosci. 1999, 19, 7889–7900. [Google Scholar] [PubMed]
- Deinhardt, K.; Salinas, S.; Verastegui, C.; Watson, R.; Worth, D.; Hanrahan, S.; Bucci, C.; Schiavo, G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 2006, 52, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.A.; Hampton, C.; El-Sabeawy, F.; Sabo, S.L.; McAllister, A.K. The dynamic distribution of TrkB receptors before, during, and after synapse formation between cortical neurons. J. Neurosci. 2006, 26, 11487–11500. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Parton, R.G.; Mather, I.H.; Stunnenberg, H.; Simons, K.; Hoflack, B.; Zerial, M. The small GTPase Rab5 functions as a regulatory factor in the early endocytic pathway. Cell 1992, 70, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Vitelli, R.; Santillo, M.; Lattero, D.; Chiariello, M.; Bifulco, M.; Bruni, C.; Bucci, C. Role of the small GTPase Rab7 in the late endocytic pathway. J. Biol. Chem. 1997, 272, 4391–4397. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: A key to lysosome biogenesis. Mol. Biol. Cell. 2000, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lai, C.; Mobley, W.C. Nerve growth factor activates persistent Rap1 signaling in endosomes. J. Neurosci. 2001, 21, 5406–5416. [Google Scholar]
- Deinhardt, K.; Reversi, A.; Berninghausen, O.; Hopkins, C.R.; Schiavo, G. Neurotrophins redirect p75NTR from a clathrin-independent to a clathrin-dependent endocytic pathway coupled to axonal transport. Traffic 2007, 8, 1736–1749. [Google Scholar] [CrossRef] [PubMed]
- Christoforidis, S.; Miaczynska, M.; Ashman, K.; Wilm, M.; Zhao, L.; Yip, S.C.; Waterfield, M.D.; Backer, J.M.; Zerial, M. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat. Cell Biol. 1999, 1, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lamb, D.; Chou, M.M.; Liu, Y.J.; Li, G. Nerve growth factor-mediated neurite outgrowth via regulation of Rab5. Mol. Biol. Cell 2007, 18, 1375–13854. [Google Scholar] [PubMed]
- Haas, A.K.; Fuchs, E.; Kopajtich, R.; Barr, F.A. A GTPase-activating protein controls Rab5 function in endocytic trafficking. Nat. Cell Biol. 2005, 7, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, H.; Lippé, R.; McBride, H.M.; Rubino, M.; Woodman, P.; Stenmark, H.; Rybin, V.; Wilm, M.; Ashman, K.; Mann, M.; et al. A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell 1997, 90, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Delprato, A.; Merithew, E.; Lambright, D.G. Structure, exchange determinants, and family-wide Rab specificity of the tandem helical bundle and VPS9 domains of RABEX-5. Cell 2004, 118, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Kauppi, M.; Simonsen, A.; Bremnes, B.; Vieira, A.; Callaghan, J.; Stenmark, H.; Olkkonen, V.M. The small GTPase Rab22 interacts with EEA1 and controls endosomal membrane trafficking. J. Cell Sci. 2002, 115, 899–911. [Google Scholar] [PubMed]
- Magadán, J.G.; Barbieri, M.A.; Mesa, R.; Stahl, P.D.; Mayorga, L.S. Rab22a regulates the sorting of transferrin to recycling endosomes. Mol. Cell Biol. 2006, 26, 2595–2614. [Google Scholar] [CrossRef] [PubMed]
- Burgo, A.; Sotirakis, E.; Simmler, M.C.; Verraes, A.; Chamot, C.; Simpson, J.C.; Lanzetti, L.; Proux-Gillardeaux, V.; Galli, T. Role of VARP, a Rab21 exchange factor and TI-VAMP/VAMP7 partner, in neurite growth. EMBO Rep. 2009, 10, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Fukuda, M. Rabex-5 determines the neurite localization of its downstream rab proteins in hippocampal neurons. Commun. Integr. Biol. 2013, 6. [Google Scholar] [CrossRef]
- Simpson, J.C.; Griffiths, G.; Wessling-Resnick, M.; Fransen, J.A.; Bennett, H.; Jones, A.T. A role for the small GTPase Rab21 in the early endocytic pathway. J. Cell Sci. 2004, 117, 6297–6311. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liang, Z.; Li, G. Rab22 controls NGF signaling and neurite outgrowth in PC12 cells. Mol. Biol. Cell 2011, 22, 3853–3860. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.; Bucci, C.; Vieira, O.; Schroer, T.; Grinstein, S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: Role of Rab7 and RILP. Mol. Cell Biol. 2003, 23, 6494–6506. [Google Scholar] [CrossRef] [PubMed]
- Vieira, O.V.; Bucci, C.; Harrison, R.E.; Trimble, W.S.; Lanzetti, L.; Gruenberg, J.; Schreiber, A.D.; Stahl, P.D.; Grinstein, S. Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol. Cell Biol. 2003, 23, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Bucci, C.; Weis, J.; Kruttgen, A. The small GTPase Rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor TrkA. J. Neurosci. 2005, 25, 10930–10940. [Google Scholar] [CrossRef] [PubMed]
- Cogli, L.; Progida, C.; Lecci, R.; Bramato, R.; Krüttgen, A.; Bucci, C. CMT2b-associated Rab7 mutants inhibit neurite outgrowth. Acta Neuropathol. 2010, 120, 491–501. [Google Scholar] [PubMed]
- Bronfman, F.C.; Escudero, C.A.; Weis, J.; Kruttgen, A. Endosomal transport of neurotrophins: Roles in signaling and neurodegenerative diseases. Dev. Neurobiol. 2007, 67, 1183–1203. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.J.; Blasier, K.R.; Jeffery, E.D.; Ross, M.W.; Pullikuth, A.K.; Suo, D.; Park, J.; Smiley, W.R.; Lo, K.W.; Shabanowitz, J.; et al. Trk activation of the ERK1/2 kinase pathway stimulates intermediate chain phosphorylation and recruits cytoplasmic dynein to signaling endosomes for retrograde axonal transport. J. Neurosci. 2012, 32, 15495–15510. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.W.; St Hillaire, C.; Zweifel, L.S.; Glebova, N.O.; Philippidou, P.; Halegoua, S.; Ginty, D.D. Recruitment of actin modifiers to TrkA endosomes governs retrograde NGF signaling and survival. Cell 2011, 146, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Holzbaur, E.L. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 2014, 30, 71–85. [Google Scholar] [CrossRef] [PubMed]
- García-Arencibia, M.; Hochfeld, W.E.; Toh, P.P.; Rubinsztein, D.C. Autophagy, a guardian against neurodegeneration. Semin. Cell Dev. Biol. 2010, 21, 691–698. [Google Scholar] [PubMed]
- Hansen, T.E.; Johansen, T. Following autophagy step by step. BMC Biol. 2011, 9. [Google Scholar] [CrossRef]
- Hansen, K.; Wagner, B.; Hamel, W.; Schweizer, M.; Haag, F.; Westphal, M.; Lamszus, K. Autophagic cell death induced by TrkA receptor activation in human glioblastoma cells. J. Neurochem. 2007, 103, 259–275. [Google Scholar] [PubMed]
- Florez-McClure, M.L.; Linseman, D.A.; Chu, C.T.; Barker, P.A.; Bouchard, R.J.; Le, S.S.; Laessig, T.A.; Heidenreich, K. The p75 neurotrophin receptor can induce autophagy and death of cerebellar Purkinje neurons. J. Neurosci. 2004, 24, 4498–4509. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Stauber, T.; del Nery, E.; Vernos, I.; Pepperkok, R.; Nilsson, T. Regulation of microtubule-dependent recycling at the trans-Golgi network by Rab6a and Rab6a’. Mol. Biol. Cell 2005, 16, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Huang, X.; Tanaka, Y.; Hirokawa, N. KIF16b/Rab14 molecular motor complex is critical for early embryonic development by transporting FGF receptor. Dev. Cell 2011, 20, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Schonteich, E.; Wilson, G.M.; Burden, J.; Hopkins, C.R.; Anderson, K.; Goldenring, J.R.; Prekeris, R. The RIP11/Rab11-FIP5 and kinesin ii complex regulates endocytic protein recycling. J. Cell Sci. 2008, 121, 3824–3833. [Google Scholar] [CrossRef] [PubMed]
- Horgan, C.P.; Hanscom, S.R.; Jolly, R.S.; Futter, C.E.; McCaffrey, M.W. Rab11-FIP3 links the Rab11 GTPase and cytoplasmic dynein to mediate transport to the endosomal-recycling compartment. J. Cell Sci. 2010, 123, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Perlson, E.; Maday, S.; Fu, M.M.; Moughamian, A.J.; Holzbaur, E.L. Retrograde axonal transport: Pathways to cell death? Trends Neurosci. 2010, 33, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chan, C.C.; Cherry, S.; Hiesinger, P.R. Membrane trafficking in neuronal maintenance and degeneration. Cell Mol. Life Sci. 2013, 70, 2919–2934. [Google Scholar] [CrossRef] [PubMed]
- Schindowski, K.; Belarbi, K.; Buée, L. Neurotrophic factors in Alzheimer's disease: Role of axonal transport. Genes Brain Behav. 2008, 7, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Annaert, W. Membrane trafficking pathways in Alzheimer’s disease. Traffic 2012, 13, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Buggia-Prévot, V.; Fernandez, C.G.; Riordan, S.; Vetrivel, K.S.; Roseman, J.; Waters, J.; Bindokas, V.P.; Vassar, R.; Thinakaran, G. Axonal BACE1 dynamics and targeting in hippocampal neurons: A role for Rab11 gtpase. Mol. Neurodegener. 2014, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Poon, W.W.; Blurton-Jones, M.; Tu, C.H.; Feinberg, L.M.; Chabrier, M.A.; Harris, J.W.; Jeon, N.L.; Cotman, C.W. β-amyloid impairs axonal BDNF retrograde trafficking. Neurobiol. Aging 2011, 32, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Poon, W.W.; Carlos, A.J.; Aguilar, B.L.; Berchtold, N.C.; Kawano, C.K.; Zograbyan, V.; Yaopruke, T.; Shelanski, M.; Cotman, C.W. β-amyloid (a β) oligomers impair brain-derived neurotrophic factor retrograde trafficking by down-regulating ubiquitin c-terminal hydrolase, uch-l1. J. Biol. Chem. 2013, 288, 16937–16948. [Google Scholar] [CrossRef] [PubMed]
- Shelton, S.B.; Johnson, G.V. Tau and HMW tau phosphorylation and compartmentalization in apoptotic neuronal PC12 cells. J. Neurosci. Res. 2001, 66, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.; Heldman, E.; Gurwitz, D.; Haring, R.; Karton, Y.; Meshulam, H.; Pittel, Z.; Marciano, D.; Brandeis, R.; Sadot, E.; et al. Review M1 agonists for the treatment of Alzheimer’s disease. Novel properties and clinical update. Ann. N.Y. Acad. Sci. 1996, 777, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Holsinger, R.M.; Schnarr, J.; Henry, P.; Castelo, V.T.; Fahnestock, M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: Decreased levels in alzheimer’s disease. Brain Res. Mol. Brain Res. 2000, 76, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D. Down regulation of Trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J. Neurochem. 2006, 97, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Almenar-Queralt, A.; LeBlanc, J.F.; Roberts, E.A.; Goldstein, L.S. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 2001, 414, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Udayar, V.; Buggia-Prévot, V.; Guerreiro, R.L.; Siegel, G.; Rambabu, N.; Soohoo, A.L.; Ponnusamy, M.; Siegenthaler, B.; Bali, J.; Simons, M.; et al. A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of β-amyloid production. Cell Rep. 2013, 5, 1536–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginsberg, S.D.; Mufson, E.J.; Alldred, M.J.; Counts, S.E.; Wuu, J.; Nixon, R.A.; Che, S. Upregulation of select Rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J. Chem. Neuroanat. 2011, 42, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Mufson, E.J.; Counts, S.E.; Wuu, J.; Alldred, M.J.; Nixon, R.A.; Che, S. Regional selectivity of Rab5 and Rab7 protein upregulation in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 631–639. [Google Scholar] [PubMed]
- Gunawardena, S.; Yang, G.; Goldstein, L.S. Presenilin controls kinesin-1 and dynein function during APP-vesicle transport in vivo. Hum. Mol. Genet 2013, 22, 3828–3843. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. Gamma-secretase is a membrane protein complex comprised of Presenilin, Nicastrin, APH-1, and PEN-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef] [PubMed]
- Dumanchin, C.; Czech, C.; Campion, D.; Cuif, M.H.; Poyot, T.; Martin, C.; Charbonnier, F.; Goud, B.; Pradier, L.; Frebourg, T. Presenilins interact with Rab11, a small GTPase involved in the regulation of vesicular transport. Hum. Mol. Genet 1999, 8, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Zwart, R.; Baas, F. Rab6 membrane association is dependent of presenilin 1 and cellular phosphorylation events. Brain Res. Mol. Brain Res. 2004, 122, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Funk, K.E.; Kuret, J. Lysosomal fusion dysfunction as a unifying hypothesis for Alzheimer’s disease pathology. Int. J. Alzheimers Dis. 2012, 2012, 752894:1–752894:10. [Google Scholar]
- Hamano, T.; Mutoh, T.; Tabira, T.; Araki, W.; Kuriyama, M.; Mihara, T.; Yano, S.; Yamamoto, H. Abnormal intracellular trafficking of high affinity nerve growth factor receptor, Trk, in stable transfectants expressing presenilin 1 protein. Brain Res. Mol. Brain Res. 2005, 137, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Millecamps, S.; Julien, J.P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ström, A.L.; Fukada, K.; Lee, S.; Hayward, L.J.; Zhu, H. Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J. Biol. Chem. 2007, 282, 16691–16699. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, O.T.; Aytan, N.; Carreras, I.; Choi, J.K.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. 7,8-dihydroxyflavone improves motor performance and enhances lower motor neuronal survival in a mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 2014, 566, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Lowry, K.S.; Murray, S.S.; McLean, C.A.; Talman, P.; Mathers, S.; Lopes, E.C.; Cheema, S.S. A potential role for the p75 low-affinity neurotrophin receptor in spinal motor neuron degeneration in murine and human amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2001, 2, 127–134. [Google Scholar] [CrossRef]
- Ibáñez, C.F.; Simi, A. P75 neurotrophin receptor signaling in nervous system injury and degeneration: Paradox and opportunity. Trends Neurosci. 2012, 35, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, R.; Golfi, P.; Heimann, P.; Shaw, C.; Troakes, C.; Schmitt-John, T.; Bartsch, J.W. Endosomal accumulation of APP in wobbler motor neurons reflects impaired vesicle trafficking: Implications for human motor neuron disease. BMC Neurosci. 2011, 12. [Google Scholar] [CrossRef]
- Koistinen, H.; Prinjha, R.; Soden, P.; Harper, A.; Banner, S.J.; Pradat, P.F.; Loeffler, J.P.; Dingwall, C. Elevated levels of amyloid precursor protein in muscle of patients with amyotrophic lateral sclerosis and a mouse model of the disease. Muscle Nerve 2006, 34, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Shim, H.; Lai, C.; Xie, C.; Lin, X.; Yang, W.J.; Chandran, J. ALS2/alsin knockout mice and motor neuron diseases. Neurodegener. Dis. 2008, 5, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Otomo, A.; Hadano, S.; Okada, T.; Mizumura, H.; Kunita, R.; Nishijima, H.; Showguchi-Miyata, J.; Yanagisawa, Y.; Kohiki, E.; Suga, E.; et al. ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum. Mol. Genet 2003, 12, 1671–1687. [Google Scholar] [CrossRef] [PubMed]
- Topp, J.D.; Gray, N.W.; Gerard, R.D.; Horazdovsky, B.F. Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J. Biol. Chem. 2004, 279, 24612–24623. [Google Scholar] [CrossRef] [PubMed]
- Devon, R.S.; Orban, P.C.; Gerrow, K.; Barbieri, M.A.; Schwab, C.; Cao, L.P.; Helm, J.R.; Bissada, N.; Cruz-Aguado, R.; Davidson, T.L.; et al. ALS2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 9595–9600. [Google Scholar] [CrossRef] [PubMed]
- Levine, T.P.; Daniels, R.D.; Gatta, A.T.; Wong, L.H.; Hayes, M.J. The product of c9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013, 29, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9orf72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef] [PubMed]
- Cogli, L.; Piro, F.; Bucci, C. Rab7 and the CMT2b disease. Biochem. Soc. Trans. 2009, 37, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; de Luca, M. Molecular basis of Charcot-Marie-Tooth type 2b disease. Biochem. Soc. Trans. 2012, 40, 1368–1372. [Google Scholar] [CrossRef] [PubMed]
- Gentil, B.J.; Cooper, L. Molecular basis of axonal dysfunction and traffic impairments in CMT. Brain Res. Bull. 2012, 88, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Meggouh, F.; Bienfait, H.M.; Weterman, M.A.; de Visser, M.; Baas, F. Charcot-Marie-Tooth disease due to a de novo mutation of the rab7 gene. Neurology 2006, 67, 1476–1478. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, K.; de Jonghe, P.; Coen, K.; Verpoorten, N.; Auer-Grumbach, M.; Kwon, J.M.; FitzPatrick, D.; Schmedding, E.; de Vriendt, E.; Jacobs, A.; et al. Mutations in the small GTP-ase late endosomal protein Rab7 cause Charcot-Marie-Tooth type 2b neuropathy. Am. J. Hum. Genet. 2003, 72, 722–727. [Google Scholar] [PubMed]
- Houlden, H.; King, R.H.; Muddle, J.R.; Warner, T.T.; Reilly, M.M.; Orrell, R.W.; Ginsberg, L. A novel Rab7 mutation associated with ulcero-mutilating neuropathy. Ann. Neurol. 2004, 56, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Han, C.; Liu, W.; Wang, P.; Zhang, X. A novel Rab7 mutation in a chinese family with Charcot-Marie-Tooth type 2b disease. Gene 2014, 534, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, J.; Torii, T.; Kusakawa, S.; Sanbe, A.; Nakamura, K.; Takashima, S.; Hamasaki, H.; Kawaguchi, S.; Miyamoto, Y.; Tanoue, A. The mood stabilizer valproic acid improves defective neurite formation caused by Charcot-Marie-Tooth disease-associated mutant Rab7 through the JNK signaling pathway. J. Neurosci. Res. 2010, 88, 3189–3197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Fishel Ben Kenan, R.; Osakada, Y.; Xu, W.; Sinit, R.S.; Chen, L.; Zhao, X.; Chen, J.Y.; Cui, B.; Wu, C. Defective axonal transport of Rab7 GTPase results in dysregulated trophic signaling. J. Neurosci. 2013, 33, 7451–7462. [Google Scholar] [CrossRef] [PubMed]
- del Toro, D.; Alberch, J.; Lázaro-Diéguez, F.; Martín-Ibáñez, R.; Xifró, X.; Egea, G.; Canals, J.M. Mutant Huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing Optineurin/Rab8 complex from the Golgi apparatus. Mol. Biol. Cell 2009, 20, 1478–1492. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Severin, F.; Lommer, B.; Shevchenko, A.; Zerial, M. Huntingtin-hap40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. J. Cell Biol. 2006, 172, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sapp, E.; Chase, K.; Comer-Tierney, L.A.; Masso, N.; Alexander, J.; Reeves, P.; Kegel, K.B.; Valencia, A.; Esteves, M.; et al. Disruption of Rab11 activity in a knock-in mouse model of Huntington’s disease. Neurobiol. Dis. 2009, 36, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Sepers, M.D.; Raymond, L.A. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov. Today 2014, 19, 990–996. [Google Scholar] [PubMed]
- Her, L.S.; Goldstein, L.S. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant Huntingtin. J. Neurosci. 2008, 28, 13662–13672. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [PubMed]
- McGuire, J.R.; Rong, J.; Li, S.H.; Li, X.J. Interaction of Huntingtin-associated protein-1 with kinesin light chain: Implications in intracellular trafficking in neurons. J. Biol. Chem. 2006, 281, 3552–3559. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; de Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [PubMed]
- Colin, E.; Zala, D.; Liot, G.; Rangone, H.; Borrell-Pagès, M.; Li, X.J.; Saudou, F.; Humbert, S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008, 27, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J. Neurosci. 2013, 33, 6298–6309. [Google Scholar] [CrossRef] [PubMed]
- Sahlender, D.A.; Roberts, R.C.; Arden, S.D.; Spudich, G.; Taylor, M.J.; Luzio, J.P.; Kendrick-Jones, J.; Buss, F. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J. Cell Biol. 2005, 169, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Imarisio, S.; Sarkar, S.; O’Kane, C.J.; Rubinsztein, D.C. Rab5 modulates aggregation and toxicity of mutant Huntingtin through macroautophagy in cell and fly models of Huntington disease. J. Cell Sci. 2008, 121, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sapp, E.; Valencia, A.; Kegel, K.B.; Qin, Z.H.; Alexander, J.; Masso, N.; Reeves, P.; Ritch, J.J.; Zeitlin, S.; et al. A function of Huntingtin in guanine nucleotide exchange on Rab11. Neuroreport 2008, 19, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Standley, C.; Sapp, E.; Valencia, A.; Qin, Z.H.; Kegel, K.B.; Yoder, J.; Comer-Tierney, L.A.; Esteves, M.; Chase, K.; et al. Mutant Huntingtin impairs vesicle formation from recycling endosomes by interfering with Rab11 activity. Mol. Cell Biol. 2009, 29, 6106–6116. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; DiFiglia, M. The recycling endosome and its role in neurological disorders. Prog. Neurobiol. 2012, 97, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Power, D.; Srinivasan, S.; Gunawardena, S. In-vivo evidence for the disruption of Rab11 vesicle transport by loss of Huntingtin. Neuroreport 2012, 23, 970–977. [Google Scholar] [CrossRef]
- Li, X.; Valencia, A.; Sapp, E.; Masso, N.; Alexander, J.; Reeves, P.; Kegel, K.B.; Aronin, N.; Difiglia, M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J. Neurosci. 2010, 30, 4552–4561. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Valencia, A.; McClory, H.; Sapp, E.; Kegel, K.B.; Difiglia, M. Deficient Rab11 activity underlies glucose hypometabolism in primary neurons of Huntington’s disease mice. Biochem. Biophys. Res. Commun. 2012, 421, 727–730. [Google Scholar] [CrossRef]
- Steinert, J.R.; Campesan, S.; Richards, P.; Kyriacou, C.P.; Forsythe, I.D.; Giorgini, F. Rab11 rescues synaptic dysfunction and behavioral deficits in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 2012, 21, 2912–2922. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.M.; Nirschl, J.J.; Holzbaur, E.L. LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev. Cell 2014, 29, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Bergman, E.; Fundin, B.T.; Ulfhake, B. Effects of aging and axotomy on the expression of neurotrophin receptors in primary sensory neurons. J. Comp. Neurol. 1999, 410, 368–386. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Guidotti, G.; Racagni, G.; Riva, M.A. Reduced neuroplasticity in aged rats: A role for the neurotrophin brain-derived neurotrophic factor. Neurobiol. Aging 2013, 34, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Ulfhake, B.; Bergman, E.; Edstrom, E.; Fundin, B.T.; Johnson, H.; Kullberg, S.; Ming, Y. Regulation of neurotrophin signaling in aging sensory and motoneurons: Dissipation of target support? Mol. Neurobiol. 2000, 21, 109–135. [Google Scholar] [CrossRef]
- Wu, C.; Cui, B.; He, L.; Chen, L.; Mobley, W.C. The coming of age of axonal neurotrophin signaling endosomes. J. Proteomics 2009, 72, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.I.; Aronow, B.J.; Wise, T.M.; Williams, S.S.; Couget, J.A.; Howley, P.M. Transcriptome signature of irreversible senescence in human papillomavirus-positive cervical cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 7093–7098. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Kawai, Y.; Endoh, M.; Hossain, M.N.; Nakabayashi, K.; Ayusawa, D. Expression of Rab27b is up-regulated in senescent human cells. Mech. Ageing Dev. 2006, 127, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Saetre, P.; Jazin, E.; Emilsson, L. Age-related changes in gene expression are accelerated in Alzheimer’s disease. Synapse 2011, 65, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Afshordel, S.; Wood, W.G.; Igbavboa, U.; Muller, W.E.; Eckert, G.P. Impaired geranylgeranyltransferase-i regulation reduces membrane-associated Rho protein levels in aged mouse brain. J. Neurochem. 2014, 129, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Okabayashi, S.; Ono, F. Dynein dysfunction disrupts intracellular vesicle trafficking bidirectionally and perturbs synaptic vesicle docking via endocytic disturbances a potential mechanism underlying age-dependent impairment of cognitive function. Am. J. Pathol. 2012, 180, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Bahr, B.A.; Wisniewski, M.L.; Butler, D. Positive lysosomal modulation as a unique strategy to treat age-related protein accumulation diseases. Rejuvenation Res. 2012, 15, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Sann, S.B.; Crane, M.M.; Lu, H.; Jin, Y. Rabx-5 regulates Rab-5 early endosomal compartments and synaptic vesicles in c. Elegans. PLoS One 2012, 7, e37930. [Google Scholar] [CrossRef] [PubMed]
- Wavre-Shapton, S.T.; Tolmachova, T.; Lopes da Silva, M.; Futter, C.E.; Seabra, M.C. Conditional ablation of the choroideremia gene causes age-related changes in mouse retinal pigment epithelium. PLoS One 2013, 8, e57769. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.; Giehl, K.; Nyengaard, J.R.; Teng, K.; Lioubinski, O.; Sjoegaard, S.S.; Breiderhoff, T.; Gotthardt, M.; Lin, F.; Eilers, A.; et al. Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat. Neurosci. 2007, 10, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bucci, C.; Alifano, P.; Cogli, L. The Role of Rab Proteins in Neuronal Cells and in the Trafficking of Neurotrophin Receptors. Membranes 2014, 4, 642-677. https://doi.org/10.3390/membranes4040642
Bucci C, Alifano P, Cogli L. The Role of Rab Proteins in Neuronal Cells and in the Trafficking of Neurotrophin Receptors. Membranes. 2014; 4(4):642-677. https://doi.org/10.3390/membranes4040642
Chicago/Turabian StyleBucci, Cecilia, Pietro Alifano, and Laura Cogli. 2014. "The Role of Rab Proteins in Neuronal Cells and in the Trafficking of Neurotrophin Receptors" Membranes 4, no. 4: 642-677. https://doi.org/10.3390/membranes4040642