

Functional Characterization of Four Known Cav2.1 Variants Associated with Neurodevelopmental Disorders

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Biology
2.2. Xenopus Oocyte Preparation and Injection
2.3. Electrophysiology
2.4. Expression in HEK Cells
2.5. Homology Modeling of Human Cav2.1
2.6. Computational Modeling
2.7. Data Analysis
3. Results and Discussion
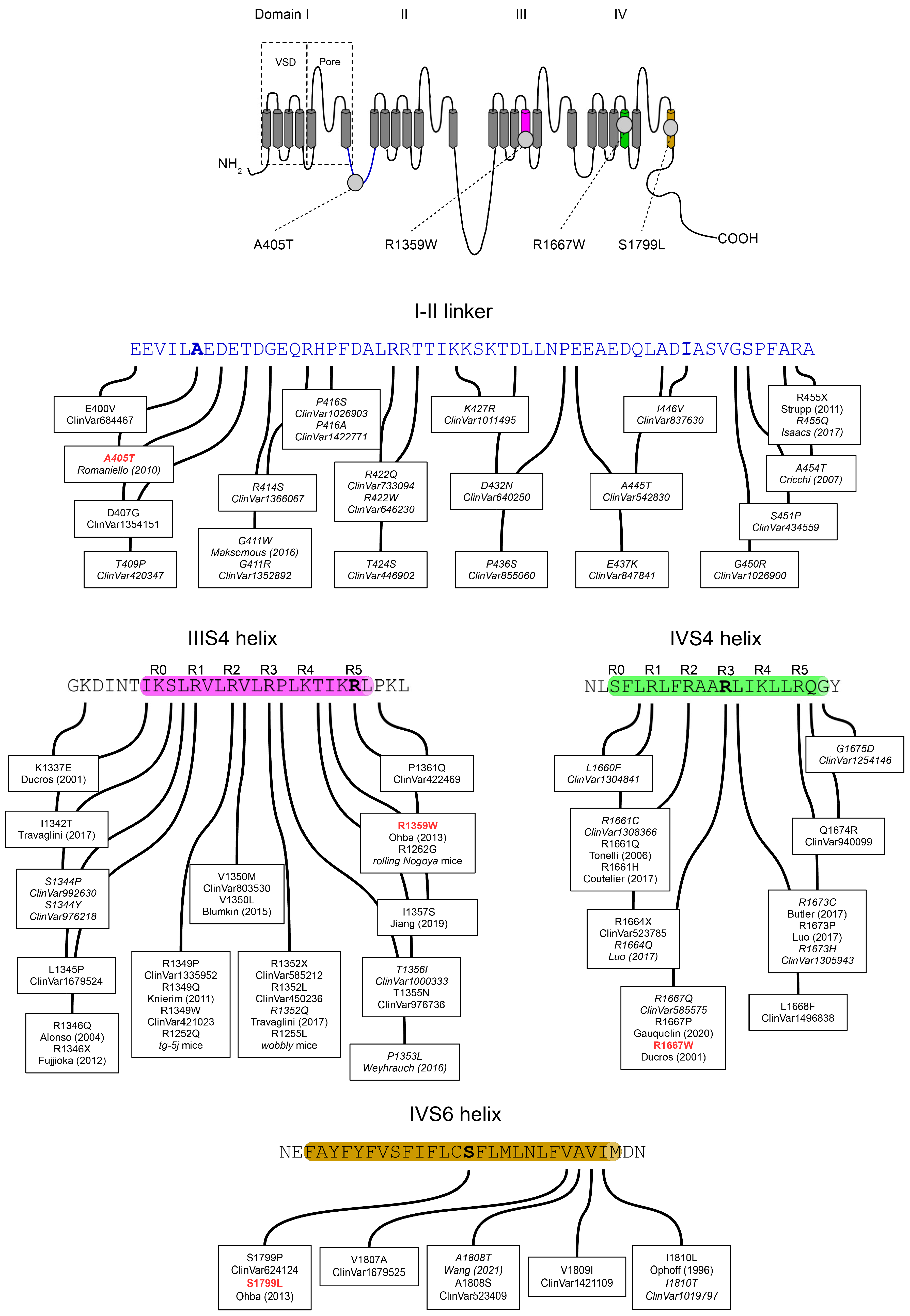
3.1. Clinical Features Associated with A405T, R1359W, R1667W and S1799L Variants
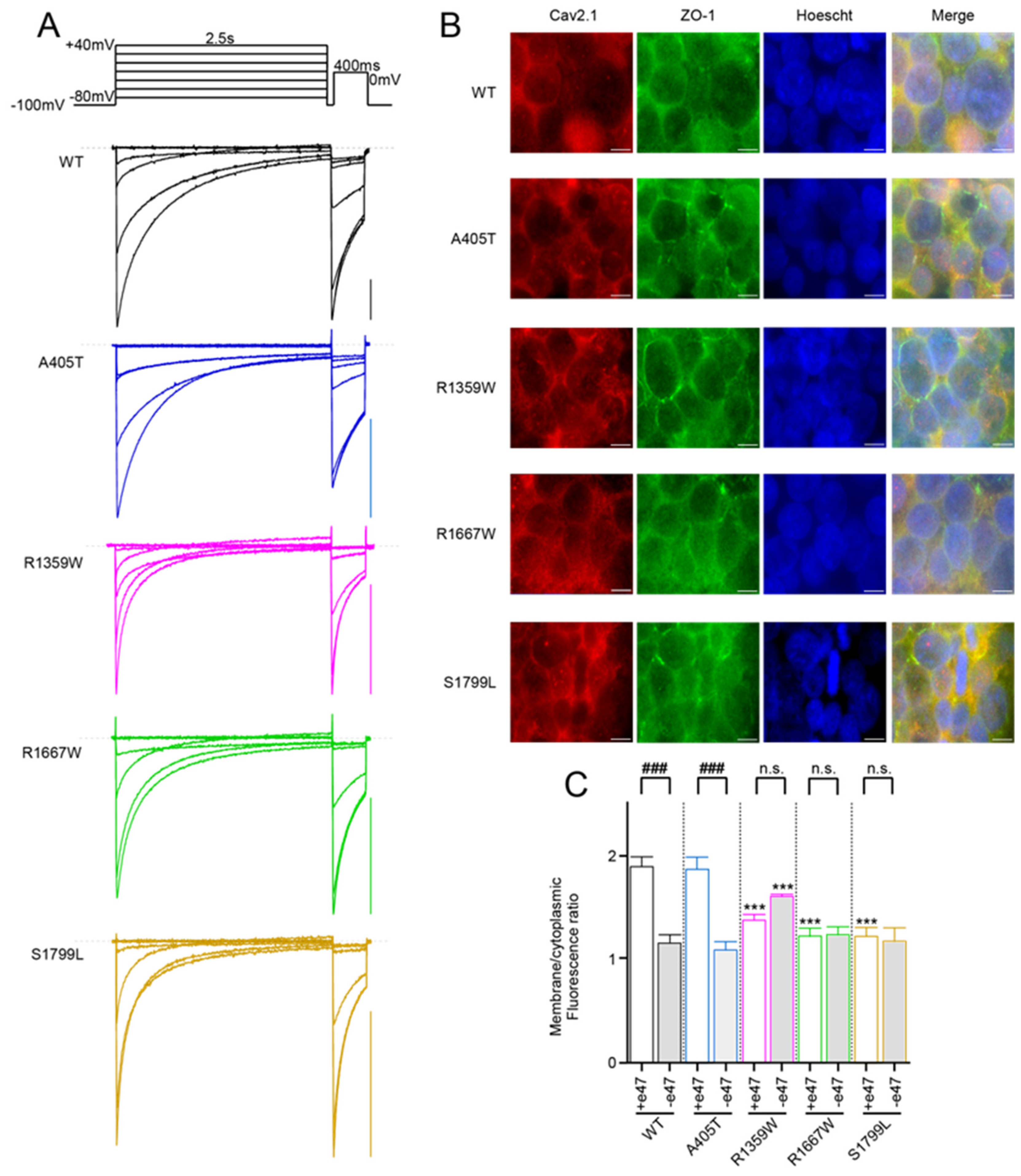
3.2. Functional Expression of Cav2.1 Variants
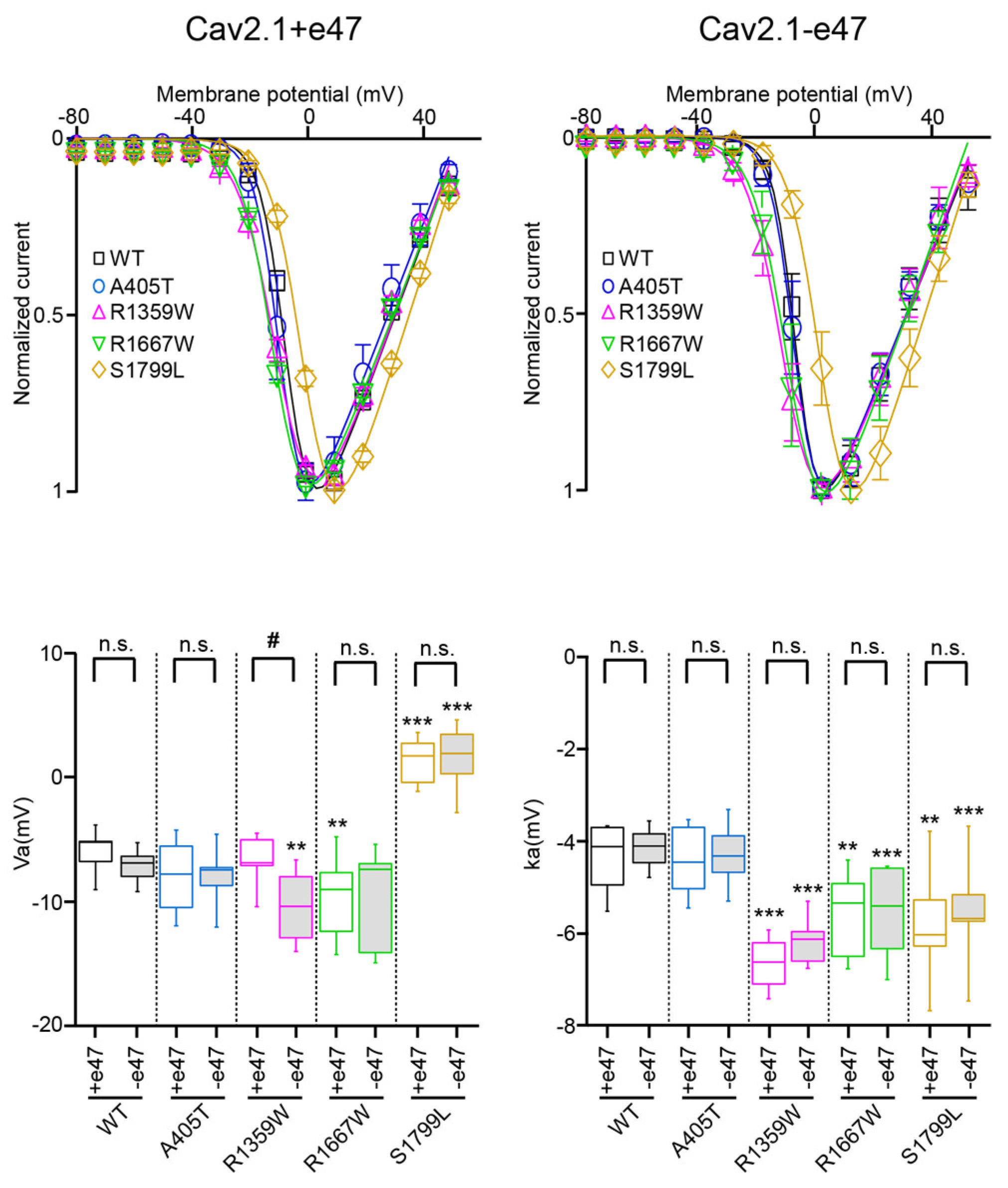
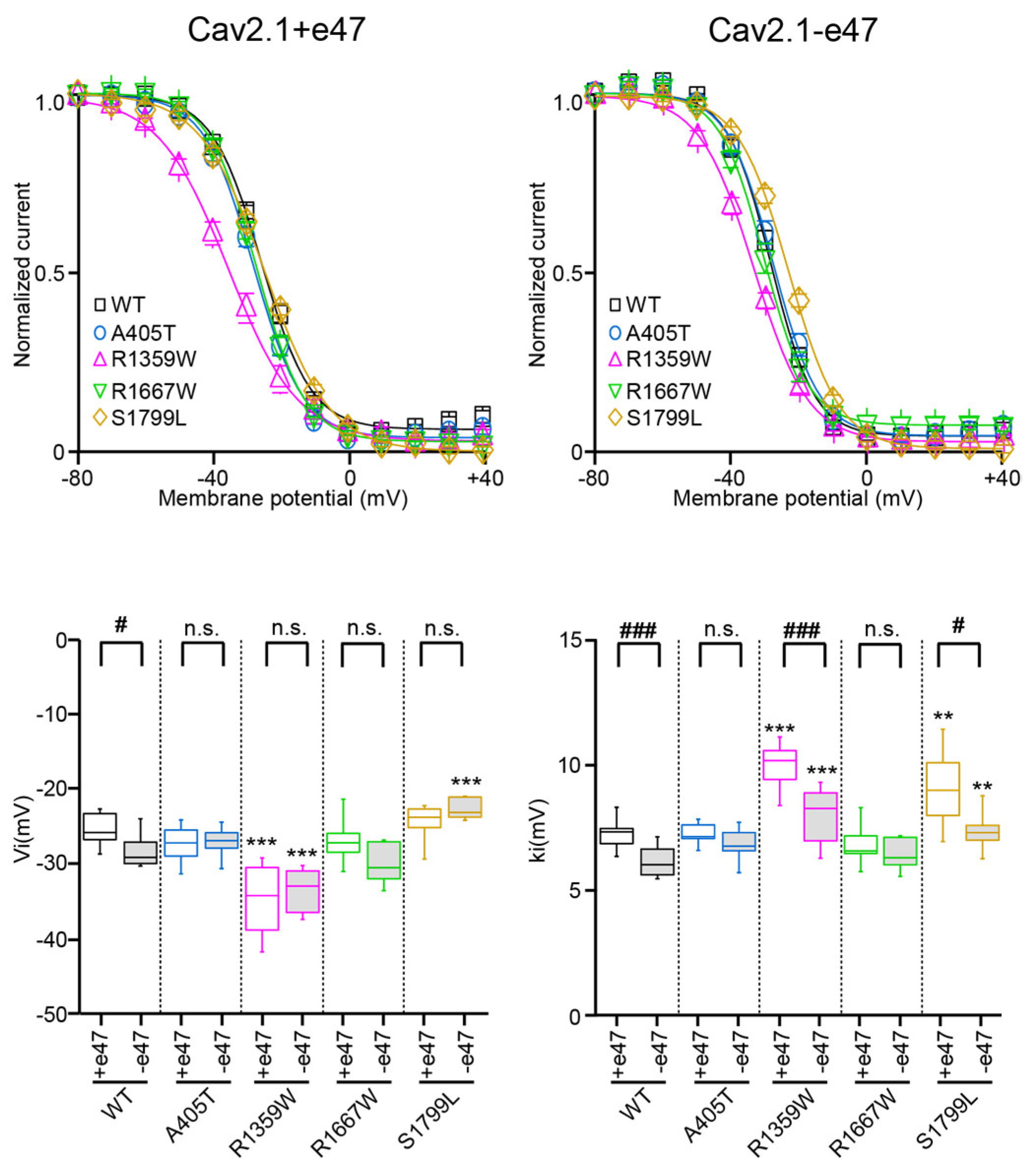
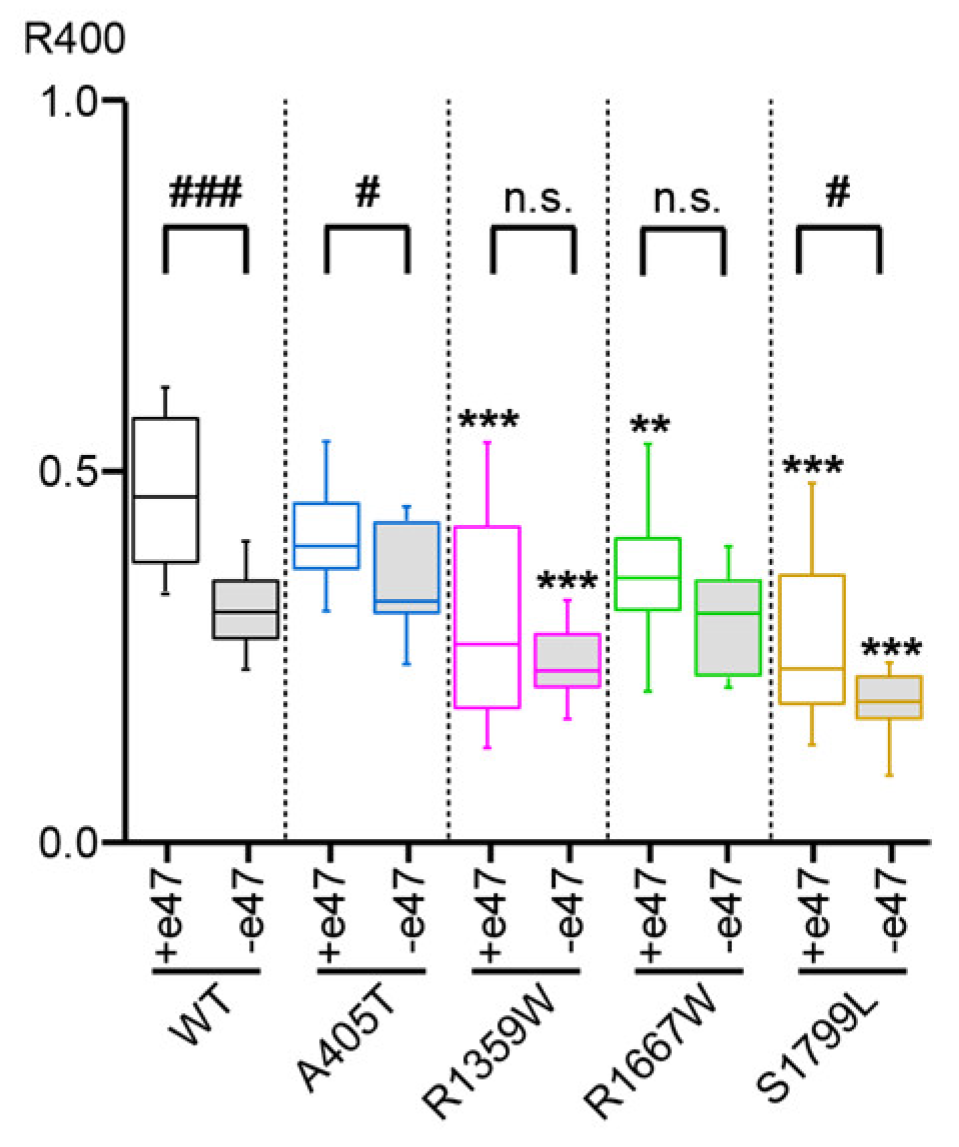
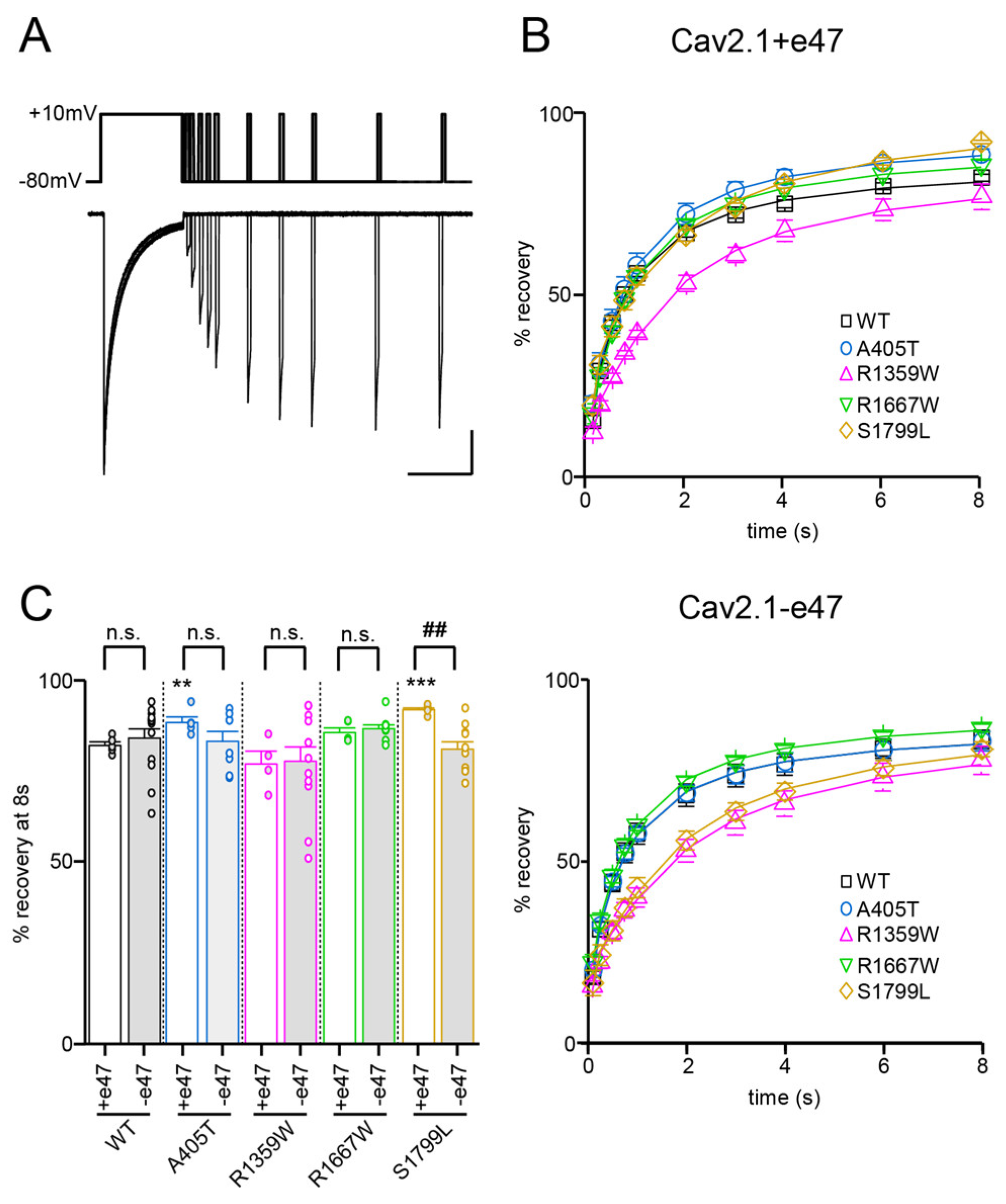
3.3. Functional Characterization of Cav2.1+e47 and Cav2.1−e47
3.4. Functional Characterization of Cav2.1 A405T Variants
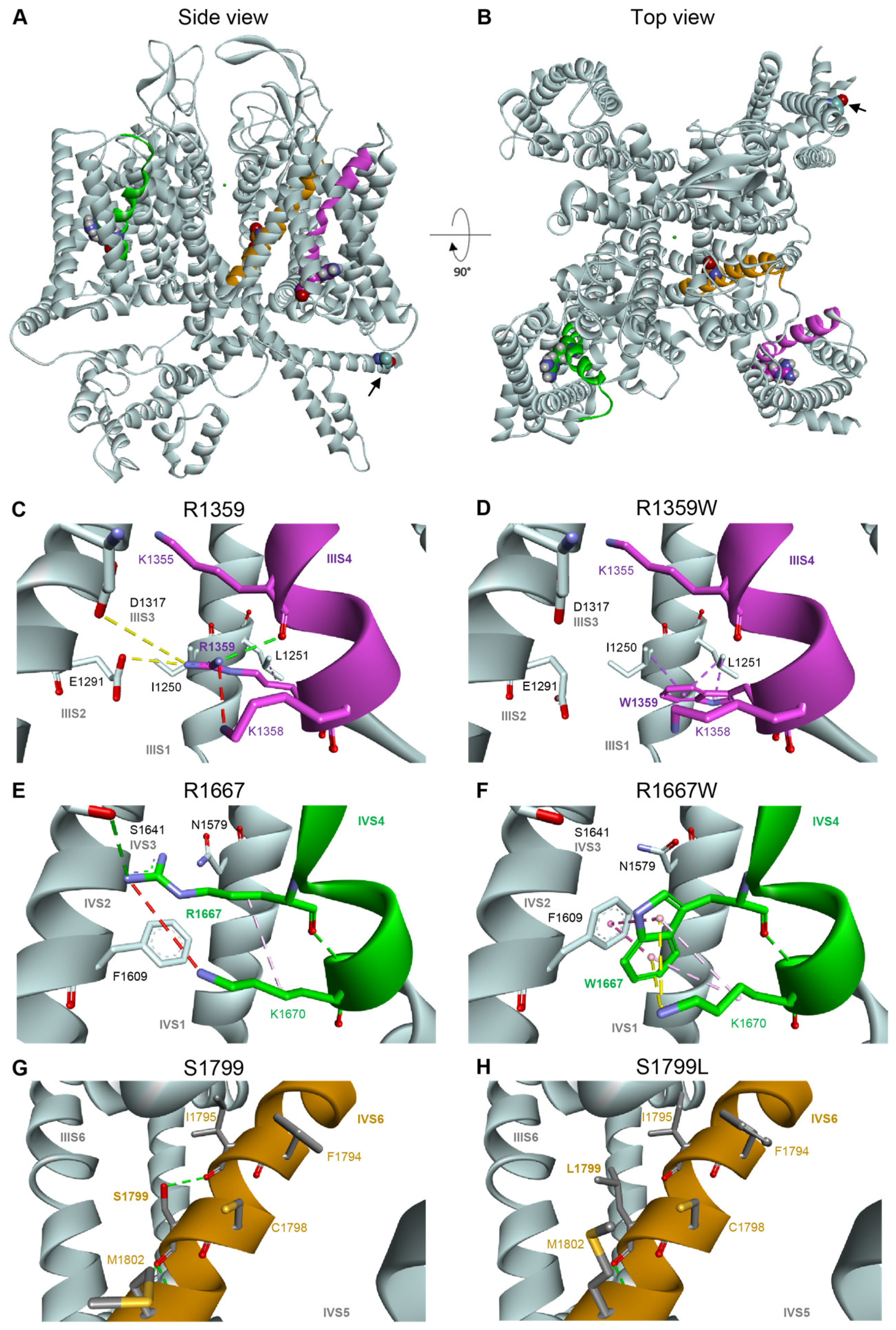
3.5. Functional and Structural Analysis of R1359W and R1667W Variants
3.6. Functional and Structural Analysis of S1799L Variants
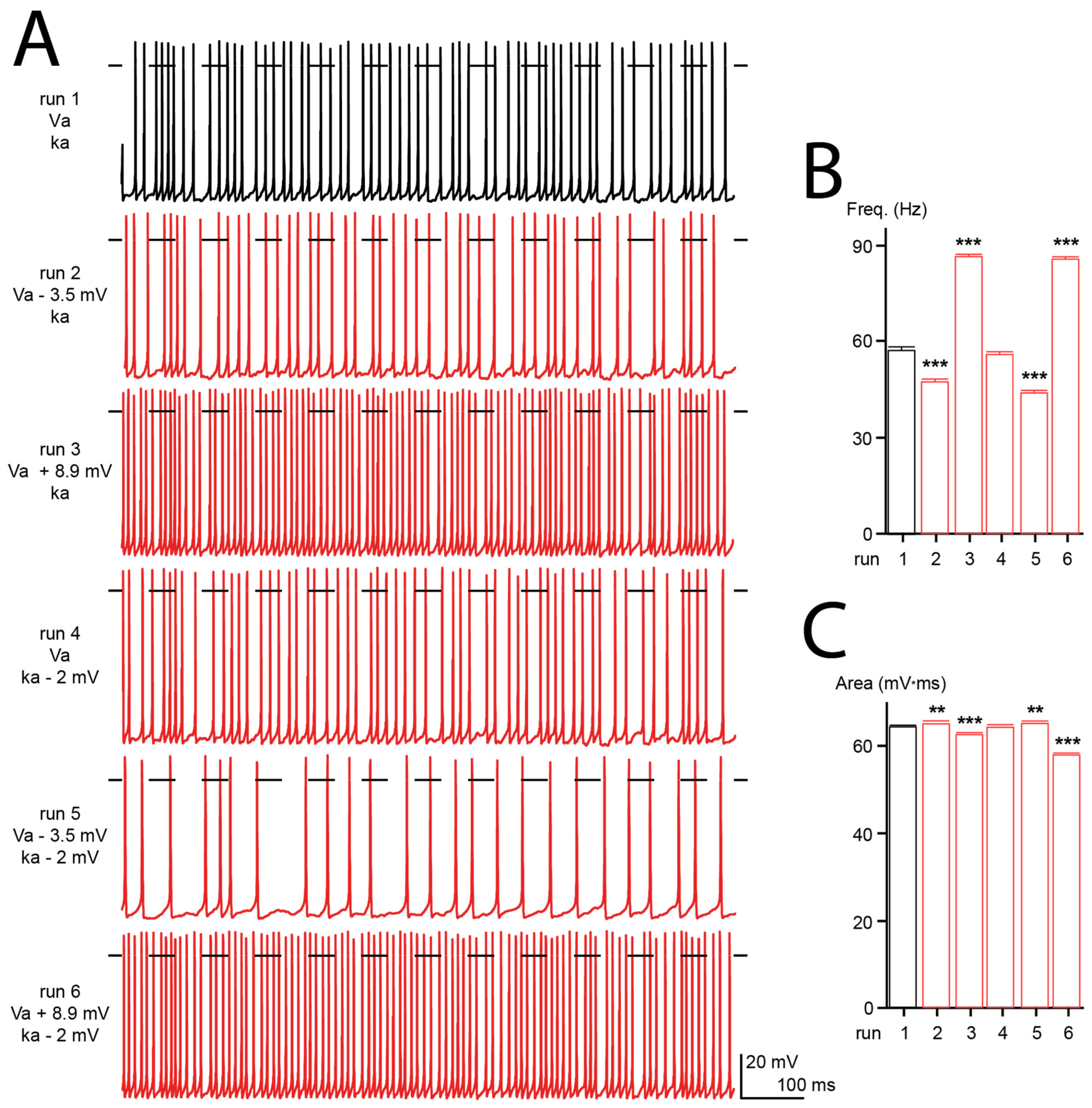
3.7. Computational Simulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolphin, A.C. Voltage-Gated Calcium Channels and Their Auxiliary Subunits: Physiology and Pathophysiology and Pharmacology. J. Physiol. 2016, 594, 5369–5390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soong, T.W.; DeMaria, C.D.; Alvania, R.S.; Zweifel, L.S.; Liang, M.C.; Mittman, S.; Agnew, W.S.; Yue, D.T. Systematic Identification of Splice Variants in Human P/Q-Type Channel Alpha1(2.1) Subunits: Implications for Current Density and Ca2+-Dependent Inactivation. J. Neurosci. 2002, 22, 10142–10152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hering, S.; Zangerl-Plessl, E.-M.; Beyl, S.; Hohaus, A.; Andranovits, S.; Timin, E.N. Calcium Channel Gating. Pflügers Arch.-Eur. J. Physiol. 2018, 470, 1291–1309. [Google Scholar] [CrossRef] [Green Version]
- Cens, T.; Rousset, M.; Kajava, A.; Charnet, P. Molecular Determinant for Specific Ca/Ba Selectivity Profiles of Low and High Threshold Ca2+ Channels. J. Gen. Physiol. 2007, 130, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Yao, X.; Yan, N. Structure of Human Cav2.2 Channel Blocked by the Painkiller Ziconotide. Nature 2021, 596, 143–147. [Google Scholar] [CrossRef]
- Dong, Y.; Gao, Y.; Xu, S.; Wang, Y.; Yu, Z.; Li, Y.; Li, B.; Yuan, T.; Yang, B.; Zhang, X.C.; et al. Closed-State Inactivation and Pore-Blocker Modulation Mechanisms of Human CaV2.2. Cell Rep. 2021, 37, 109931. [Google Scholar] [CrossRef]
- Catterall, W.A.; Wisedchaisri, G.; Zheng, N. The Conformational Cycle of a Prototypical Voltage-Gated Sodium Channel. Nat. Chem. Biol. 2020, 16, 1314–1320. [Google Scholar] [CrossRef]
- Tuluc, P.; Yarov-Yarovoy, V.; Benedetti, B.; Flucher, B.E. Molecular Interactions in the Voltage Sensor Controlling Gating Properties of CaV Calcium Channels. Structure 2016, 24, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Korkosh, V.S.; Kiselev, A.M.; Mikhaylov, E.N.; Kostareva, A.A.; Zhorov, B.S. Atomic Mechanisms of Timothy Syndrome-Associated Mutations in Calcium Channel Cav1.2. Front. Physiol. 2019, 10, 335. [Google Scholar] [CrossRef]
- Dolphin, A.C.; Lee, A. Presynaptic Calcium Channels: Specialized Control of Synaptic Neurotransmitter Release. Nat. Rev. Neurosci. 2020, 21, 213–229. [Google Scholar] [CrossRef]
- Benedetti, B.; Benedetti, A.; Flucher, B.E. Loss of the Calcium Channel Β4 Subunit Impairs Parallel Fibre Volley and Purkinje Cell Firing in Cerebellum of Adult Ataxic Mice. Eur. J. Neurosci. 2016, 43, 1486–1498. [Google Scholar] [CrossRef] [Green Version]
- Jun, K.; Piedras-Rentería, E.S.; Smith, S.M.; Wheeler, D.B.; Lee, S.B.; Lee, T.G.; Chin, H.; Adams, M.E.; Scheller, R.H.; Tsien, R.W.; et al. Ablation of P/Q-Type Ca2+ Channel Currents, Altered Synaptic Transmission, and Progressive Ataxia in Mice Lacking the Alpha(1A)-Subunit. Proc. Natl. Acad. Sci. USA 1999, 96, 15245–15250. [Google Scholar] [CrossRef] [Green Version]
- Pietrobon, D. CaV2.1 Channelopathies. Pflugers Arch. 2010, 460, 375–393. [Google Scholar] [CrossRef]
- Hoxha, E.; Balbo, I.; Miniaci, M.C.; Tempia, F. Purkinje Cell Signaling Deficits in Animal Models of Ataxia. Front. Synaptic Neurosci. 2018, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Striessnig, J. Voltage-Gated Ca2+-Channel A1-Subunit de Novo Missense Mutations: Gain or Loss of Function–Implications for Potential Therapies. Front. Synaptic Neurosci. 2021, 13, 634760. [Google Scholar] [CrossRef]
- Hommersom, M.P.; van Prooije, T.H.; Pennings, M.; Schouten, M.I.; van Bokhoven, H.; Kamsteeg, E.-J.; van de Warrenburg, B.P.C. The Complexities of CACNA1A in Clinical Neurogenetics. J. Neurol. 2022, 269, 3094–3108. [Google Scholar] [CrossRef]
- Pradotto, L.; Mencarelli, M.; Bigoni, M.; Milesi, A.; Di Blasio, A.; Mauro, A. Episodic Ataxia and SCA6 within the Same Family Due to the D302N CACNA1A Gene Mutation. J. Neurol. Sci. 2016, 371, 81–84. [Google Scholar] [CrossRef]
- Alonso, I.; Barros, J.; Tuna, A.; Coelho, J.; Sequeiros, J.; Silveira, I.; Coutinho, P. Phenotypes of Spinocerebellar Ataxia Type 6 and Familial Hemiplegic Migraine Caused by a Unique CACNA1A Missense Mutation in Patients from a Large Family. Arch. Neurol. 2003, 60, 610–614. [Google Scholar] [CrossRef] [Green Version]
- Indelicato, E.; Boesch, S. From Genotype to Phenotype: Expanding the Clinical Spectrum of CACNA1A Variants in the Era of Next Generation Sequencing. Front. Neurol. 2021, 12, 639994. [Google Scholar] [CrossRef]
- Epi4K Consortium De Novo Mutations in SLC1A2 and CACNA1A Are Important Causes of Epileptic Encephalopathies. Am. J. Hum. Genet. 2016, 99, 287–298. [CrossRef] [Green Version]
- Izquierdo-Serra, M.; Fernández-Fernández, J.M.; Serrano, M. Rare CACNA1A Mutations Leading to Congenital Ataxia. Pflugers Arch. 2020, 472, 791–809. [Google Scholar] [CrossRef]
- Ngo, K.J.; Rexach, J.E.; Lee, H.; Petty, L.E.; Perlman, S.; Valera, J.M.; Deignan, J.L.; Mao, Y.; Aker, M.; Posey, J.E.; et al. A Diagnostic Ceiling for Exome Sequencing in Cerebellar Ataxia and Related Neurological Disorders. Hum. Mutat. 2020, 41, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Johnson, A.K.; Nelakuditi, V.; Guidugli, L.; Fischer, D.; Arndt, K.; Ma, L.; Sandford, E.; Shakkottai, V.; Boycott, K.; et al. Targeted Exome Analysis Identifies the Genetic Basis of Disease in over 50% of Patients with a Wide Range of Ataxia-Related Phenotypes. Genet. Med. 2019, 21, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Jaudon, F.; Baldassari, S.; Musante, I.; Thalhammer, A.; Zara, F.; Cingolani, L.A. Targeting Alternative Splicing as a Potential Therapy for Episodic Ataxia Type 2. Biomedicines 2020, 8, 332. [Google Scholar] [CrossRef] [PubMed]
- Cens, T.; Mangoni, M.E.; Nargeot, J.; Charnet, P. Modulation of the A1A Ca2+ Channel by β Subunits at Physiological Ca2+ Concentration. FEBS Lett. 1996, 391, 232–237. [Google Scholar] [CrossRef] [Green Version]
- Cens, T.; Leyris, J.-P.; Charnet, P. Introduction into Ca(v)2.1 of the Homologous Mutation of Ca(v)1.2 Causing the Timothy Syndrome Questions the Role of V421 in the Phenotypic Definition of P-Type Ca2+ Channel. Pflugers Arch. 2008, 457, 417–430. [Google Scholar] [CrossRef] [Green Version]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a Human Cell Line Transformed by DNA from Human Adenovirus Type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Jiang, X.; Raju, P.K.; D’Avanzo, N.; Lachance, M.; Pepin, J.; Dubeau, F.; Mitchell, W.G.; Bello-Espinosa, L.E.; Pierson, T.M.; Minassian, B.A.; et al. Both Gain-of-Function and Loss-of-Function de Novo CACNA1A Mutations Cause Severe Developmental Epileptic Encephalopathies in the Spectrum of Lennox-Gastaut Syndrome. Epilepsia 2019, 60, 1881–1894. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Studer, G.; Biasini, M.; Schwede, T. Assessing the Local Structural Quality of Transmembrane Protein Models Using Statistical Potentials (QMEANBrane). Bioinformatics 2014, 30, i505–i511. [Google Scholar] [CrossRef] [Green Version]
- Spassov, V.Z.; Yan, L.; Flook, P.K. The Dominant Role of Side-Chain Backbone Interactions in Structural Realization of Amino Acid Code. ChiRotor: A Side-Chain Prediction Algorithm Based on Side-Chain Backbone Interactions. Protein Sci. 2007, 16, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luthman, J.; Hoebeek, F.E.; Maex, R.; Davey, N.; Adams, R.; De Zeeuw, C.I.; Steuber, V. STD-Dependent and Independent Encoding of Input Irregularity as Spike Rate in a Computational Model of a Cerebellar Nucleus Neuron. Cerebellum 2011, 10, 667–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romaniello, R.; Zucca, C.; Tonelli, A.; Bonato, S.; Baschirotto, C.; Zanotta, N.; Epifanio, R.; Righini, A.; Bresolin, N.; Bassi, M.T.; et al. A Wide Spectrum of Clinical, Neurophysiological and Neuroradiological Abnormalities in a Family with a Novel CACNA1A Mutation. J. Neurol. Neurosurg. Psychiatry 2010, 81, 840–843. [Google Scholar] [CrossRef] [PubMed]
- Ohba, C.; Osaka, H.; Iai, M.; Yamashita, S.; Suzuki, Y.; Aida, N.; Shimozawa, N.; Takamura, A.; Doi, H.; Tomita-Katsumoto, A.; et al. Diagnostic Utility of Whole Exome Sequencing in Patients Showing Cerebellar and/or Vermis Atrophy in Childhood. Neurogenetics 2013, 14, 225–232. [Google Scholar] [CrossRef]
- Ziats, M.N.; Ahmad, A.; Bernat, J.A.; Fisher, R.; Glassford, M.; Hannibal, M.C.; Jacher, J.E.; Weiser, N.; Keegan, C.E.; Lee, K.N.; et al. Genotype-Phenotype Analysis of 523 Patients by Genetics Evaluation and Clinical Exome Sequencing. Pediatr. Res. 2020, 87, 735–739. [Google Scholar] [CrossRef]
- Plomp, J.J.; van den Maagdenberg, A.M.J.M.; Kaja, S. The Ataxic Cacna1a-Mutant Mouse Rolling Nagoya: An Overview of Neuromorphological and Electrophysiological Findings. Cerebellum 2009, 8, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Ducros, A.; Denier, C.; Joutel, A.; Cecillon, M.; Lescoat, C.; Vahedi, K.; Darcel, F.; Vicaut, E.; Bousser, M.G.; Tournier-Lasserve, E. The Clinical Spectrum of Familial Hemiplegic Migraine Associated with Mutations in a Neuronal Calcium Channel. N. Engl. J. Med. 2001, 345, 17–24. [Google Scholar] [CrossRef]
- Marti, S.; Baloh, R.W.; Jen, J.C.; Straumann, D.; Jung, H.H. Progressive Cerebellar Ataxia with Variable Episodic Symptoms--Phenotypic Diversity of R1668W CACNA1A Mutation. Eur. Neurol. 2008, 60, 16–20. [Google Scholar] [CrossRef] [Green Version]
- Coutelier, M.; Coarelli, G.; Monin, M.-L.; Konop, J.; Davoine, C.-S.; Tesson, C.; Valter, R.; Anheim, M.; Behin, A.; Castelnovo, G.; et al. A Panel Study on Patients with Dominant Cerebellar Ataxia Highlights the Frequency of Channelopathies. Brain 2017, 140, 1579–1594. [Google Scholar] [CrossRef]
- Gauquelin, L.; Hawkins, C.; Tam, E.W.Y.; Miller, S.P.; Yoon, G. Pearls & Oy-Sters: Fatal Brain Edema Is a Rare Complication of Severe CACNA1A-Related Disorder. Neurology 2020, 94, 631–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosso, B.J.; Kramer, A.A.; Tyagi, S.; Bennett, D.F.; Tifft, C.J.; D’Souza, P.; Wangler, M.F.; Macnamara, E.F.; Meza, U.; Bannister, R.A. Complex Effects on CaV2.1 Channel Gating Caused by a CACNA1A Variant Associated with a Severe Neurodevelopmental Disorder. Sci. Rep. 2022, 12, 9186. [Google Scholar] [CrossRef] [PubMed]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal Dominant Cerebellar Ataxia (SCA6) Associated with Small Polyglutamine Expansions in the Alpha 1A-Voltage-Dependent Calcium Channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.J.; Garcia, E.; David, L.S.; Mulatz, K.J.; Spacey, S.D.; Snutch, T.P. Ca(V)2.1 P/Q-Type Calcium Channel Alternative Splicing Affects the Functional Impact of Familial Hemiplegic Migraine Mutations: Implications for Calcium Channelopathies. Channels 2009, 3, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandini, M.A.; Souza, I.A.; Ferron, L.; Innes, A.M.; Zamponi, G.W. The de Novo CACNA1A Pathogenic Variant Y1384C Associated with Hemiplegic Migraine, Early Onset Cerebellar Atrophy and Developmental Delay Leads to a Loss of Cav2.1 Channel Function. Mol. Brain 2021, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Jeng, C.-J.; Chen, Y.-T.; Chen, Y.-W.; Tang, C.-Y. Dominant-Negative Effects of Human P/Q-Type Ca2+ Channel Mutations Associated with Episodic Ataxia Type 2. Am. J. Physiol. Cell Physiol. 2006, 290, C1209–C1220. [Google Scholar] [CrossRef] [Green Version]
- Guida, S.; Trettel, F.; Pagnutti, S.; Mantuano, E.; Tottene, A.; Veneziano, L.; Fellin, T.; Spadaro, M.; Stauderman, K.; Williams, M.; et al. Complete Loss of P/Q Calcium Channel Activity Caused by a CACNA1A Missense Mutation Carried by Patients with Episodic Ataxia Type 2. Am. J. Hum. Genet. 2001, 68, 759–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eltit, J.M.; Bannister, R.A.; Moua, O.; Altamirano, F.; Hopkins, P.M.; Pessah, I.N.; Molinski, T.F.; López, J.R.; Beam, K.G.; Allen, P.D. Malignant Hyperthermia Susceptibility Arising from Altered Resting Coupling between the Skeletal Muscle L-Type Ca2+ Channel and the Type 1 Ryanodine Receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 7923–7928. [Google Scholar] [CrossRef] [Green Version]
- Tottene, A.; Conti, R.; Fabbro, A.; Vecchia, D.; Shapovalova, M.; Santello, M.; van den Maagdenberg, A.M.J.M.; Ferrari, M.D.; Pietrobon, D. Enhanced Excitatory Transmission at Cortical Synapses as the Basis for Facilitated Spreading Depression in Ca(v)2.1 Knockin Migraine Mice. Neuron 2009, 61, 762–773. [Google Scholar] [CrossRef]
- Fioretti, B.; Catacuzzeno, L.; Sforna, L.; Gerke-Duncan, M.B.; van den Maagdenberg, A.M.J.M.; Franciolini, F.; Connor, M.; Pietrobon, D. Trigeminal Ganglion Neuron Subtype-Specific Alterations of Ca(V)2.1 Calcium Current and Excitability in a Cacna1a Mouse Model of Migraine. J. Physiol. 2011, 589, 5879–5895. [Google Scholar] [CrossRef]
- Ménard, C.; Charnet, P.; Rousset, M.; Vignes, M.; Cens, T. Cav2.1 C-Terminal Fragments Produced in Xenopus Laevis Oocytes Do Not Modify the Channel Expression and Functional Properties. Eur. J. Neurosci. 2020, 51, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Chang, S.-Y.; DeMaria, C.D.; Alvania, R.S.; Soong, T.W.; Yue, D.T. Alternative Splicing as a Molecular Switch for Ca2+/Calmodulin-Dependent Facilitation of P/Q-Type Ca2+ Channels. J. Neurosci. 2004, 24, 6334–6342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pragnell, M.; De Waard, M.; Mori, Y.; Tanabe, T.; Snutch, T.P.; Campbell, K.P. Calcium Channel Beta-Subunit Binds to a Conserved Motif in the I-II Cytoplasmic Linker of the Alpha 1-Subunit. Nature 1994, 368, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Bichet, D.; Cornet, V.; Geib, S.; Carlier, E.; Volsen, S.; Hoshi, T.; Mori, Y.; De Waard, M. The I-II Loop of the Ca2+ Channel Alpha1 Subunit Contains an Endoplasmic Reticulum Retention Signal Antagonized by the Beta Subunit. Neuron 2000, 25, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Cricchi, F.; Di Lorenzo, C.; Grieco, G.S.; Rengo, C.; Cardinale, A.; Racaniello, M.; Santorelli, F.M.; Nappi, G.; Pierelli, F.; Casali, C. Early-Onset Progressive Ataxia Associated with the First CACNA1A Mutation Identified within the I-II Loop. J. Neurol. Sci. 2007, 254, 69–71. [Google Scholar] [CrossRef]
- Serra, S.A.; Cuenca-León, E.; Llobet, A.; Rubio-Moscardo, F.; Plata, C.; Carreño, O.; Fernàndez-Castillo, N.; Corominas, R.; Valverde, M.A.; Macaya, A.; et al. A Mutation in the First Intracellular Loop of CACNA1A Prevents P/Q Channel Modulation by SNARE Proteins and Lowers Exocytosis. Proc. Natl. Acad. Sci. USA 2010, 107, 1672–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rettig, J.; Sheng, Z.H.; Kim, D.K.; Hodson, C.D.; Snutch, T.P.; Catterall, W.A. Isoform-Specific Interaction of the Alpha1A Subunits of Brain Ca2+ Channels with the Presynaptic Proteins Syntaxin and SNAP-25. Proc. Natl. Acad. Sci. USA 1996, 93, 7363–7368. [Google Scholar] [CrossRef] [Green Version]
- Moreau, A.; Gosselin-Badaroudine, P.; Chahine, M. Biophysics, Pathophysiology, and Pharmacology of Ion Channel Gating Pores. Front. Pharmacol. 2014, 5, 53. [Google Scholar] [CrossRef] [Green Version]
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.E.; Mohrenweiser, H.W.; Bulman, D.E.; Ferrari, M.; et al. Familial Hemiplegic Migraine and Episodic Ataxia Type-2 Are Caused by Mutations in the Ca2+ Channel Gene CACNL1A4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Kraus, R.L.; Sinnegger, M.J.; Glossmann, H.; Hering, S.; Striessnig, J. Familial Hemiplegic Migraine Mutations Change Alpha1A Ca2+ Channel Kinetics. J. Biol. Chem. 1998, 273, 5586–5590. [Google Scholar] [CrossRef]
- Hans, M.; Luvisetto, S.; Williams, M.E.; Spagnolo, M.; Urrutia, A.; Tottene, A.; Brust, P.F.; Johnson, E.C.; Harpold, M.M.; Stauderman, K.A.; et al. Functional Consequences of Mutations in the Human Alpha1A Calcium Channel Subunit Linked to Familial Hemiplegic Migraine. J. Neurosci. 1999, 19, 1610–1619. [Google Scholar] [CrossRef] [Green Version]
- Melliti, K.; Grabner, M.; Seabrook, G.R. The Familial Hemiplegic Migraine Mutation R192Q Reduces G-Protein-Mediated Inhibition of P/Q-Type (Ca(V)2.1) Calcium Channels Expressed in Human Embryonic Kidney Cells. J. Physiol. 2003, 546, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.-Q.; Tsien, R.W. Effects of Familial Hemiplegic Migraine Type 1 Mutations on Neuronal P/Q-Type Ca2+ Channel Activity and Inhibitory Synaptic Transmission. Proc. Natl. Acad. Sci. USA 2005, 102, 2590–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Liu, P.; Xiao, Z.; Zhao, H.; Gerber, B.R.; Cao, Y.-Q. Effects of Familial Hemiplegic Migraine Type 1 Mutation T666M on Voltage-Gated Calcium Channel Activities in Trigeminal Ganglion Neurons. J. Neurophysiol. 2012, 107, 1666–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, R.L.; Sinnegger, M.J.; Koschak, A.; Glossmann, H.; Stenirri, S.; Carrera, P.; Striessnig, J. Three New Familial Hemiplegic Migraine Mutants Affect P/Q-Type Ca2+ Channel Kinetics. J. Biol. Chem. 2000, 275, 9239–9243. [Google Scholar] [CrossRef] [Green Version]
- Miki, T.; Zwingman, T.A.; Wakamori, M.; Lutz, C.M.; Cook, S.A.; Hosford, D.A.; Herrup, K.; Fletcher, C.F.; Mori, Y.; Frankel, W.N.; et al. Two Novel Alleles of Tottering with Distinct Ca(v)2.1 Calcium Channel Neuropathologies. Neuroscience 2008, 155, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Bendrick, T.R.; Filipova, D.; Papadopoulos, S.; Bannister, R.A. A Mutation in CaV2.1 Linked to a Severe Neurodevelopmental Disorder Impairs Channel Gating. J. Gen. Physiol. 2019, 151, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Quintero, M.L.; El Ghaleb, Y.; Tuluc, P.; Campiglio, M.; Liedl, K.R.; Flucher, B.E. Structural Determinants of Voltage-Gating Properties in Calcium Channels. eLife 2021, 10, e64087. [Google Scholar] [CrossRef]
- Beyl, S.; Hohaus, A.; Andranovits, S.; Timin, E.; Hering, S. Upward Movement of IS4 and IIIS4 Is a Rate-Limiting Stage in Cav1.2 Activation. Pflugers Arch. 2016, 468, 1895–1907. [Google Scholar] [CrossRef] [Green Version]
- Andranovits, S.; Beyl, S.; Hohaus, A.; Zangerl-Plessl, E.M.; Timin, E.; Hering, S. Key Role of Segment IS4 in Cav1.2 Inactivation: Link between Activation and Inactivation. Pflügers Arch.-Eur. J. Physiol. 2017, 469, 1485–1493. [Google Scholar] [CrossRef]
- Pantazis, A.; Savalli, N.; Sigg, D.; Neely, A.; Olcese, R. Functional Heterogeneity of the Four Voltage Sensors of a Human L-Type Calcium Channel. Proc. Natl. Acad. Sci. USA 2014, 111, 18381–18386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savalli, N.; Angelini, M.; Steccanella, F.; Wier, J.; Wu, F.; Quinonez, M.; DiFranco, M.; Neely, A.; Cannon, S.C.; Olcese, R. The Distinct Role of the Four Voltage Sensors of the Skeletal CaV1.1 Channel in Voltage-Dependent Activation. J. Gen. Physiol. 2021, 153, e202112915. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yan, Z.; Li, Z.; Yan, C.; Lu, S.; Dong, M.; Yan, N. Structure of the Voltage-Gated Calcium Channel Cav1.1 Complex. Science 2015, 350, aad2395. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yan, Z.; Li, Z.; Qian, X.; Lu, S.; Dong, M.; Zhou, Q.; Yan, N. Structure of the Voltage-Gated Calcium Channel Ca(v)1.1 at 3.6 Å Resolution. Nature 2016, 537, 191–196. [Google Scholar] [CrossRef]
- Hemara-Wahanui, A.; Berjukow, S.; Hope, C.I.; Dearden, P.K.; Wu, S.-B.; Wilson-Wheeler, J.; Sharp, D.M.; Lundon-Treweek, P.; Clover, G.M.; Hoda, J.-C.; et al. A CACNA1F Mutation Identified in an X-Linked Retinal Disorder Shifts the Voltage Dependence of Cav1.4 Channel Activation. Proc. Natl. Acad. Sci. USA 2005, 102, 7553–7558. [Google Scholar] [CrossRef] [Green Version]
- Hohaus, A.; Beyl, S.; Kudrnac, M.; Berjukow, S.; Timin, E.N.; Marksteiner, R.; Maw, M.A.; Hering, S. Structural Determinants of L-Type Channel Activation in Segment IIS6 Revealed by a Retinal Disorder. J. Biol. Chem. 2005, 280, 38471–38477. [Google Scholar] [CrossRef] [Green Version]
- Marcantoni, A.; Calorio, C.; Hidisoglu, E.; Chiantia, G.; Carbone, E. Cav1.2 Channelopathies Causing Autism: New Hallmarks on Timothy Syndrome. Pflugers Arch. 2020, 472, 775–789. [Google Scholar] [CrossRef]
- Raybaud, A.; Dodier, Y.; Bissonnette, P.; Simoes, M.; Bichet, D.G.; Sauvé, R.; Parent, L. The Role of the GX9GX3G Motif in the Gating of High Voltage-Activated Ca2+ Channels. J. Biol. Chem. 2006, 281, 39424–39436. [Google Scholar] [CrossRef] [Green Version]
- Hans, M.; Urrutia, A.; Deal, C.; Brust, P.F.; Stauderman, K.; Ellis, S.B.; Harpold, M.M.; Johnson, E.C.; Williams, M.E. Structural Elements in Domain IV That Influence Biophysical and Pharmacological Properties of Human Alpha1A-Containing High-Voltage-Activated Calcium Channels. Biophys. J. 1999, 76, 1384–1400. [Google Scholar] [CrossRef] [Green Version]
- Müllner, C.; Broos, L.A.M.; van den Maagdenberg, A.M.J.M.; Striessnig, J. Familial Hemiplegic Migraine Type 1 Mutations K1336E, W1684R, and V1696I Alter Cav2.1 Ca2+ Channel Gating: Evidence for Beta-Subunit Isoform-Specific Effects. J. Biol. Chem. 2004, 279, 51844–51850. [Google Scholar] [CrossRef]
- Barrett, C.F.; Cao, Y.-Q.; Tsien, R.W. Gating Deficiency in a Familial Hemiplegic Migraine Type 1 Mutant P/Q-Type Calcium Channel. J. Biol. Chem. 2005, 280, 24064–24071. [Google Scholar] [CrossRef] [Green Version]
- Rajakulendran, S.; Graves, T.D.; Labrum, R.W.; Kotzadimitriou, D.; Eunson, L.; Davis, M.B.; Davies, R.; Wood, N.W.; Kullmann, D.M.; Hanna, M.G.; et al. Genetic and Functional Characterisation of the P/Q Calcium Channel in Episodic Ataxia with Epilepsy. J. Physiol. 2010, 588, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.T.; Alviña, K.; Womack, M.D.; Chevez, C.; Khodakhah, K. Decreases in the Precision of Purkinje Cell Pacemaking Cause Cerebellar Dysfunction and Ataxia. Nat. Neurosci. 2006, 9, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Dorgans, K.; Salvi, J.; Bertaso, F.; Bernard, L.; Lory, P.; Doussau, F.; Mezghrani, A. Characterization of the Dominant Inheritance Mechanism of Episodic Ataxia Type 2. Neurobiol. Dis. 2017, 106, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Tara, E.; Vitenzon, A.; Hess, E.; Khodakhah, K. Aberrant Cerebellar Purkinje Cell Activity as the Cause of Motor Attacks in a Mouse Model of Episodic Ataxia Type 2. Dis. Model. Mech. 2018, 11, dmm034181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Womack, M.D.; Chevez, C.; Khodakhah, K. Calcium-Activated Potassium Channels Are Selectively Coupled to P/Q-Type Calcium Channels in Cerebellar Purkinje Neurons. J. Neurosci. 2004, 24, 8818–8822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alviña, K.; Khodakhah, K. The Therapeutic Mode of Action of 4-Aminopyridine in Cerebellar Ataxia. J. Neurosci. 2010, 30, 7258–7268. [Google Scholar] [CrossRef] [Green Version]
- Jayabal, S.; Chang, H.H.V.; Cullen, K.E.; Watt, A.J. 4-Aminopyridine Reverses Ataxia and Cerebellar Firing Deficiency in a Mouse Model of Spinocerebellar Ataxia Type 6. Sci. Rep. 2016, 6, 29489. [Google Scholar] [CrossRef] [Green Version]
- Cook, A.A.; Fields, E.; Watt, A.J. Losing the Beat: Contribution of Purkinje Cell Firing Dysfunction to Disease, and Its Reversal. Neuroscience 2021, 462, 247–261. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Va (mV) | ka (mV) | n | Vi (mV) | ki (mV) | n | ||
|---|---|---|---|---|---|---|---|
| WT | +e47 | −6.0 ± 0.6 | −4.5 ± 0.2 | 12 | −25.8 ± 0.6 | 7.3 ± 0.2 | 9 |
| −e47 | −7.2 ± 1.6 | −4.2 ± 0.5 | 12 | −28.2 ± 0.7 # | 6.2 ± 0.2 ### | 13 | |
| A405T | +e47 | −7.7 ± 0.8 | −4.4 ± 0.2 | 14 | −27.8 ± 0.9 | 7.1 ± 0.2 | 11 |
| −e47 | −7.9 ± 2.3 | −4.3 ± 0.7 | 8 | −27.5 ± 0.9 | 6.7 ± 0.3 | 7 | |
| R1359W | +e47 | −7.9 ± 0.9 | −6.6 ± 0.2 *** | 12 | −34.8 ± 1.4 *** | 9.9 ± 0.3 *** | 12 |
| −e47 | −10.7 ± 2.8 #, ** | −6.2 ± 0.5 *** | 11 | −33.6 ± 0.9 *** | 8.0 ± 0.4 ###, *** | 12 | |
| R1667W | +e47 | −9.5 ± 0.7 ** | −5.6 ± 0.2 ** | 21 | −26.9 ± 0.8 | 6.8 ± 0.2 | 15 |
| −e47 | −9.7 ± 3.9 | −5.6 ± 1.0 *** | 6 | −30.0 ± 1.1 | 6.4 ± 0.2 | 7 | |
| S1799L | +e47 | 1.2 ± 0.5 *** | −5.8 ± 0.3 ** | 15 | −24.5 ± 0.7 | 9.1 ± 0.5 ** | 13 |
| −e47 | 1.7 ± 2.5 *** | −5.5 ± 1.0 *** | 9 | −22.7 ± 0.6 *** | 7.4 ± 0.3 #, ** | 6 |
| R400 | n | % Recovery at 8 s | n | ||
|---|---|---|---|---|---|
| WT | +e47 | 0.47 ± 0.02 | 22 | 82 ± 1 | 5 |
| −e47 | 0.32 ± 0.02 ### | 17 | 85 ± 2 | 11 | |
| A405T | +e47 | 0.41 ± 0.01 | 28 | 88 ± 2 ** | 5 |
| −e47 | 0.35 ± 0.02 # | 11 | 83 ± 3 | 8 | |
| R1359W | +e47 | 0.31 ± 0.03 *** | 29 | 77 ± 4 | 4 |
| −e47 | 0.24 ± 0.01 *** | 18 | 78 ± 4 | 12 | |
| R1667W | +e47 | 0.36 ± 0.02 ** | 44 | 85 ± 1 | 5 |
| −e47 | 0.30 ± 0.02 | 13 | 86 ± 1 | 9 | |
| S1799L | +e47 | 0.27 ± 0.02 *** | 38 | 92 ± 1 *** | 6 |
| −e47 | 0.19 ± 0.02 #, *** | 13 | 81 ± 2 ## | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folacci, M.; Estaran, S.; Ménard, C.; Bertaud, A.; Rousset, M.; Roussel, J.; Thibaud, J.-B.; Vignes, M.; Chavanieu, A.; Charnet, P.; et al. Functional Characterization of Four Known Cav2.1 Variants Associated with Neurodevelopmental Disorders. Membranes 2023, 13, 96. https://doi.org/10.3390/membranes13010096
Folacci M, Estaran S, Ménard C, Bertaud A, Rousset M, Roussel J, Thibaud J-B, Vignes M, Chavanieu A, Charnet P, et al. Functional Characterization of Four Known Cav2.1 Variants Associated with Neurodevelopmental Disorders. Membranes. 2023; 13(1):96. https://doi.org/10.3390/membranes13010096
Chicago/Turabian StyleFolacci, Mathilde, Sébastien Estaran, Claudine Ménard, Anaïs Bertaud, Matthieu Rousset, Julien Roussel, Jean-Baptiste Thibaud, Michel Vignes, Alain Chavanieu, Pierre Charnet, and et al. 2023. "Functional Characterization of Four Known Cav2.1 Variants Associated with Neurodevelopmental Disorders" Membranes 13, no. 1: 96. https://doi.org/10.3390/membranes13010096