
In Silico Assessment of the Lipid Fingerprint Signature of ATP2, the Essential P4-ATPase of Malaria Parasites

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brancucci, N.M.B.; Gerdt, J.P.; Wang, C.; De Niz, M.; Philip, N.; Adapa, S.R.; Zhang, M.; Hitz, E.; Niederwieser, I.; Boltryk, S.D.; et al. Lysophosphatidylcholine Regulates Sexual Stage Differentiation in the Human Malaria Parasite Plasmodium falciparum. Cell 2017, 171, 1532–1544.e15. [Google Scholar] [CrossRef] [Green Version]
- Sah, R.K.; Garg, S.; Dangi, P.; Ponnusamy, K.; Singh, S. Phosphatidic acid homeostasis regulated by a type-2 phosphatidic acid phosphatase represents a novel druggable target in malaria intervention. Cell Death Discov. 2019, 5, 107. [Google Scholar] [CrossRef]
- Martin, R.E. The transportome of the malaria parasite. Biol. Rev. Camb. Philos. Soc. 2020, 95, 305–332. [Google Scholar] [CrossRef]
- Andersen, J.P.; Vestergaard, A.L.; Mikkelsen, S.A.; Mogensen, L.S.; Chalat, M.; Molday, R.S. P4-ATPases as Phospholipid Flippases—Structure, Function, and Enigmas. Front. Physiol. 2016, 7, 275. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Nofal, S.D.; Blackman, M.J.; Baker, D.A. CDC50 orthologues in Plasmodium falciparum have distinct roles in merozoite egress and trophozoite maturation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Cowell, A.N.; Istvan, E.S.; Lukens, A.K.; Gomez-Lorenzo, M.G.; Vanaerschot, M.; Sakata-Kato, T.; Flannery, E.L.; Magistrado, P.; Owen, E.; Abraham, M.; et al. Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science 2018, 359, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Lamy, A.; Macarini-Bruzaferro, E.; Dieudonné, T.; Perálvarez-Marín, A.; Lenoir, G.; Montigny, C.; le Maire, M.; Vázquez-Ibar, J.L. ATP2, The essential P4-ATPase of malaria parasites, catalyzes lipid-stimulated ATP hydrolysis in complex with a Cdc50 β-subunit. Emerg. Microbes Infect. 2021, 10, 132–147. [Google Scholar] [CrossRef]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef]
- Fiser, A.; Šali, A. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- de Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef]
- Periole, X.; Cavalli, M.; Marrink, S.-J.; Ceruso, M.A. Combining an Elastic Network With a Coarse-Grained Molecular Force Field: Structure, Dynamics, and Intermolecular Recognition. J. Chem. Theory Comput. 2009, 5, 2531–2543. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Qi, Y.; Ingólfsson, H.I.; Cheng, X.; Lee, J.; Marrink, S.J.; Im, W. CHARMM-GUI Martini Maker for Coarse-Grained Simulations with the Martini Force Field. J. Chem. Theory Comput. 2015, 11, 4486–4494. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Hsu, P.C.; Bruininks, B.; Jefferies, D.; de Souza, P.C.T.; Lee, J.; Patel, D.S.; Marrink, S.J.; Qi, Y.; Khalid, S.; Im, W. CHARMM-GUI Martini Maker for modeling and simulation of complex bacterial membranes with lipopolysaccharides. J. Comput. Chem. 2017, 38, 2354–2363. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar]
- Corradi, V.; Mendez-Villuendas, E.; Ingólfsson, H.I.; Gu, R.-X.; Siuda, I.; Melo, M.N.; Moussatova, A.; DeGagné, L.J.; Sejdiu, B.I.; Singh, G.; et al. Lipid–Protein Interactions Are Unique Fingerprints for Membrane Proteins. ACS Cent. Sci. 2018, 4, 709–717. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, C.A.; Agarwal, A.; Van, Q.N.; Stephen, A.G.; Gnanakaran, S. Unveiling the Dynamics of KRAS4b on Lipid Model Membranes. J. Membr. Biol. 2021, 254, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Prakaash, D.; Cook, G.P.; Acuto, O.; Kalli, A.C. Multi-scale simulations of the T cell receptor reveal its lipid interactions, dynamics and the arrangement of its cytoplasmic region. PLoS Comput. Biol. 2021, 17, e1009232. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, X.; Jafurulla, M.; Sengupta, D.; Chattopadhyay, A. The ganglioside GM1 interacts with the serotonin 1A receptor via the sphingolipid binding domain. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 2818–2826. [Google Scholar] [CrossRef]
- De Vecchis, D.; Reithmeier, R.A.F.; Kalli, A.C. Molecular Simulations of Intact Anion Exchanger 1 Reveal Specific Domain and Lipid Interactions. Biophys. J. 2019, 117, 1364–1379. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Wagh, K.; Gnanakaran, S.; López, C.A. Development of Martini 2.2 parameters for N-glycans: A case study of the HIV-1 Env glycoprotein dynamics. Glycobiology 2021, 31, 787–799. [Google Scholar] [CrossRef]
- Song, W.; Corey, R.A.; Ansell, T.B.; Cassidy, C.K.; Horrell, M.R.; Duncan, A.L.; Stansfeld, P.J.; Sansom, M.S.P. PyLipID: A Python Package for Analysis of Protein–Lipid Interactions from Molecular Dynamics Simulations. J. Chem. Theory Comput. 2022, 18, 1188–1201. [Google Scholar] [CrossRef]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2020, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Kenthirapalan, S.; Waters, A.P.; Matuschewski, K.; Kooij, T.W.A. Functional profiles of orphan membrane transporters in the life cycle of the malaria parasite. Nat. Commun. 2016, 7, 10519. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Ekland, E.H.; Ruggles, K.V.; Chan, R.B.; Jayabalasingham, B.; Zhou, B.; Mantel, P.-Y.; Lee, M.C.; Spottiswoode, N.; Coburn-Flynn, O.; et al. Profiling the Essential Nature of Lipid Metabolism in Asexual Blood and Gametocyte Stages of Plasmodium falciparum. Cell Host Microbe 2015, 18, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimzadeh, Z.; Mukherjee, A.; Richard, D. A map of the subcellular distribution of phosphoinositides in the erythrocytic cycle of the malaria parasite Plasmodium falciparum. Int. J. Parasitol. 2018, 48, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Elmendorf, H.G.; Haldar, K. Plasmodium falciparum exports the Golgi marker sphingomyelin synthase into a tubovesicular network in the cytoplasm of mature erythrocytes. J. Cell Biol. 1994, 124, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Buyan, A.; Cox, C.D.; Barnoud, J.; Li, J.; Chan, H.S.; Martinac, B.; Marrink, S.J.; Corry, B. Piezo1 Forms Specific, Functionally Important Interactions with Phosphoinositides and Cholesterol. Biophys. J. 2020, 119, 1683–1697. [Google Scholar] [CrossRef] [PubMed]
- Sejdiu, B.I.; Tieleman, D.P. Lipid-Protein Interactions Are a Unique Property and Defining Feature of G Protein-Coupled Receptors. Biophys. J. 2020, 118, 1887–1900. [Google Scholar] [CrossRef]
- Cabezudo, A.C.; Khoualdi, A.F.; D’Avanzo, N. Computational Prediction of Phosphoinositide Binding to Hyperpolarization-Activated Cyclic-Nucleotide Gated Channels. Front. Physiol. 2022, 13, 485. [Google Scholar] [CrossRef]
- Timcenko, M.; Lyons, J.A.; Januliene, D.; Ulstrup, J.J.; Dieudonné, T.; Montigny, C.; Ash, M.-R.; Karlsen, J.L.; Boesen, T.; Kühlbrandt, W.; et al. Structure and autoregulation of a P4-ATPase lipid flippase. Nature 2019, 571, 366–370. [Google Scholar] [CrossRef]
- Suh, B.-C.; Hille, B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Neurobiol. 2005, 15, 370–378. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, M.A.; Wilson, C.; D’Angelo, G. Phosphatidylinositol-4-phosphate: The Golgi and beyond. BioEssays 2013, 35, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Dickson, E.J.; Hille, B. Understanding phosphoinositides: Rare, dynamic, and essential membrane phospholipids. Biochem. J. 2019, 476, 1–23. [Google Scholar] [CrossRef]
- Natarajan, P.; Liu, K.; Patil, D.V.; Sciorra, V.A.; Jackson, C.; Graham, T.R. Regulation of a Golgi flippase by phosphoinositides and an ArfGEF. Nat. Cell Biol. 2009, 11, 1421–1426. [Google Scholar] [CrossRef]
- Jacquot, A.; Montigny, C.; Hennrich, H.; Barry, R.; le Maire, M.; Jaxel, C.; Holthuis, J.; Champeil, P.; Lenoir, G. Phosphatidylserine Stimulation of Drs2p·Cdc50p Lipid Translocase Dephosphorylation Is Controlled by Phosphatidylinositol-4-phosphate. J. Biol. Chem. 2012, 287, 13249–13261. [Google Scholar] [CrossRef] [Green Version]
- McNamara, C.W.; Lee, M.C.S.; Lim, C.S.; Lim, S.H.; Roland, J.; Nagle, A.; Simon, O.; Yeung, B.K.S.; Chatterjee, A.K.; McCormack, S.L.; et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature 2013, 504, 248–253. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Kovach, A.; You, Q.; Hsu, H.-C.; Zhao, G.; Li, H. Autoinhibition and activation mechanisms of the eukaryotic lipid flippase Drs2p-Cdc50p. Nat. Commun. 2019, 10, 4142. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- Machado, M.R.; Barrera, E.E.; Klein, F.; Sóñora, M.; Silva, S.; Pantano, S. The SIRAH 2.0 Force Field: Altius, Fortius, Citius. J. Chem. Theory Comput. 2019, 15, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Gahbauer, S.; Böckmann, R.A. Comprehensive Characterization of Lipid-Guided G Protein-Coupled Receptor Dimerization. J. Phys. Chem. B 2020, 124, 2823–2834. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.; Patrice, G. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]

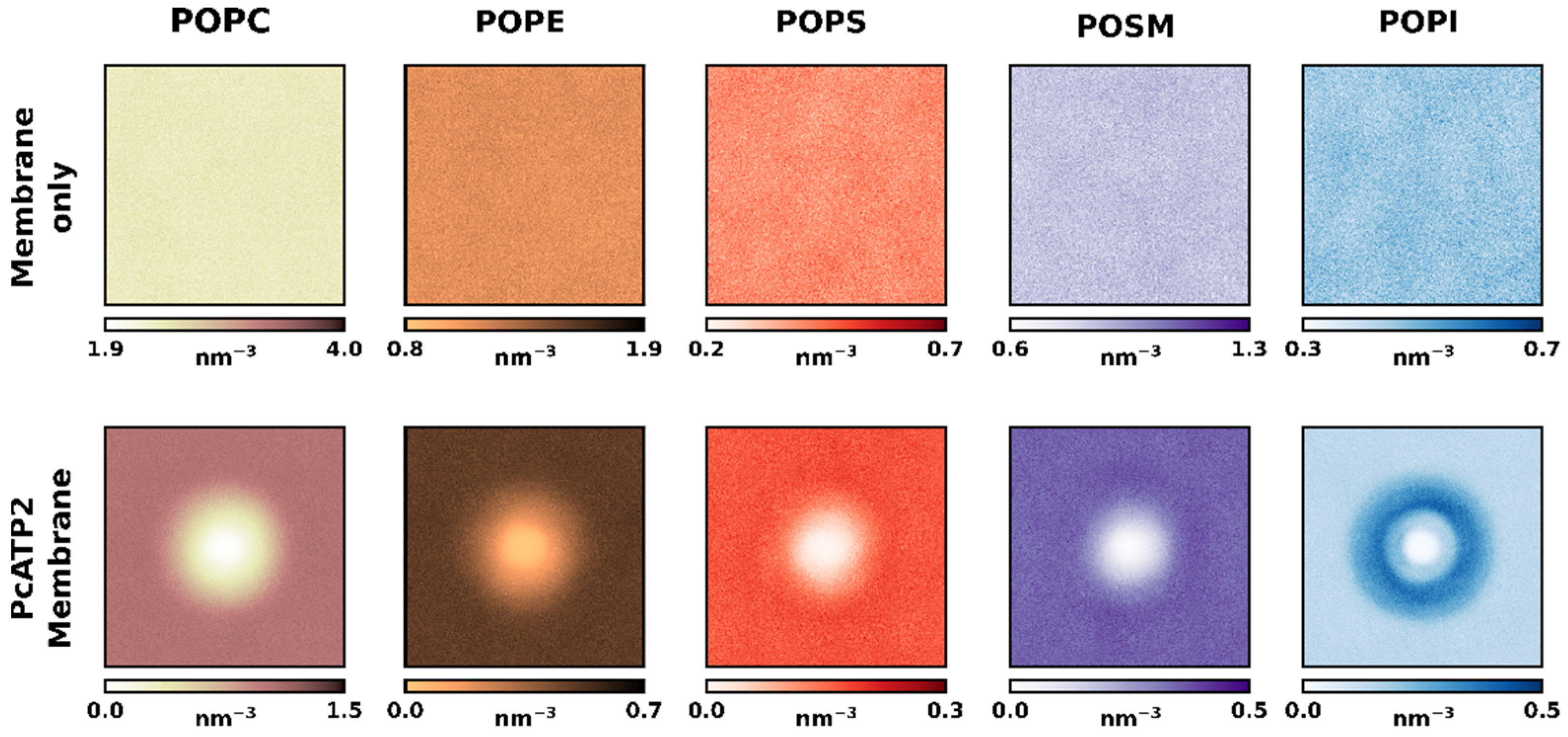

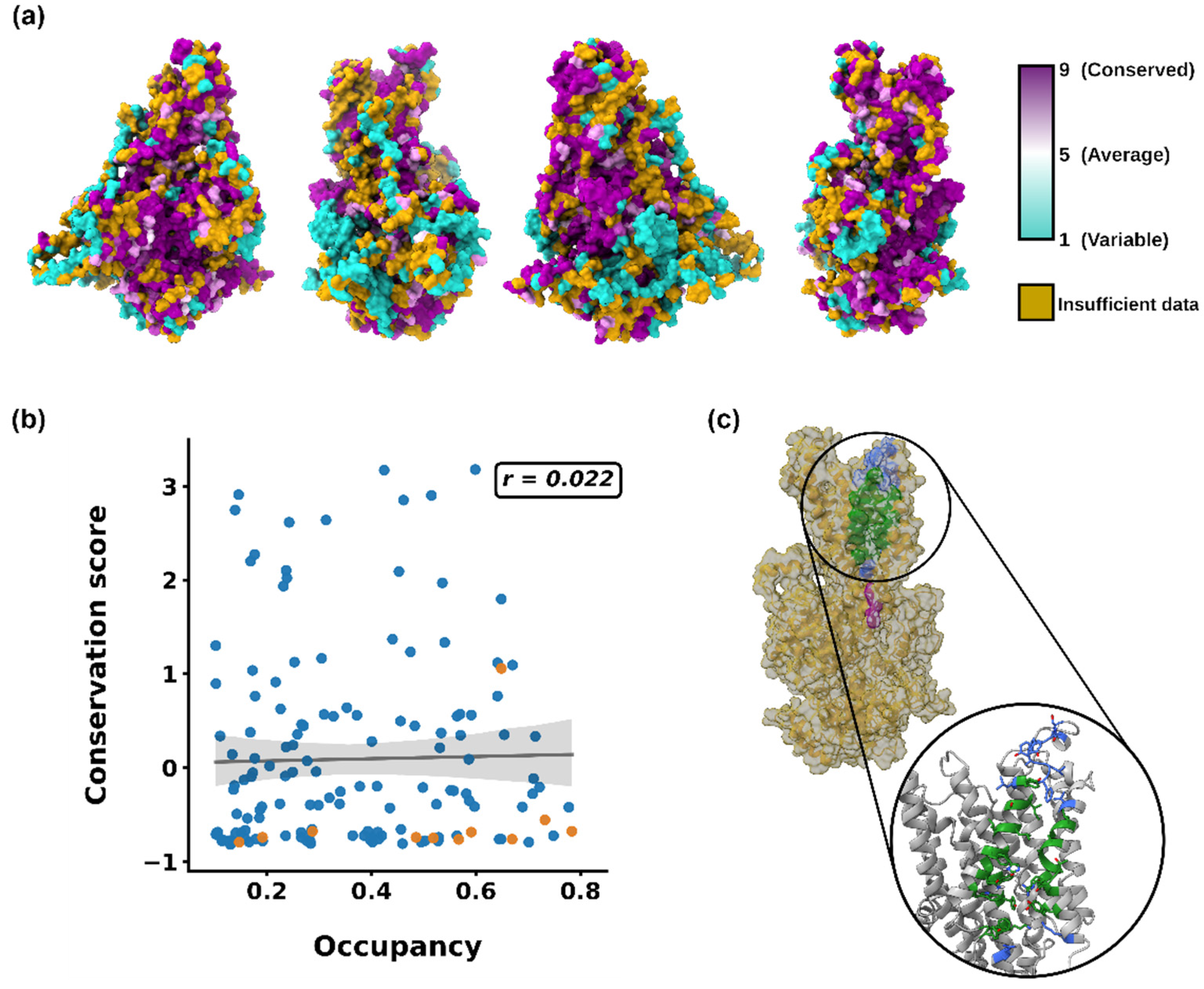

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PcATP2–Membrane | Membrane Only | |||
|---|---|---|---|---|
| Number of replicas | 4 | 4 | ||
| Simulation time per replica | 25 μs | 25 μs | ||
| Time step | 20 fs | 20 fs | ||
| Number of atoms | 32,181 | 12,591 | ||
| Initial box size (xyz) | 14.68 nm × 14.68 nm × 17.99 nm | 14.05 nm × 14.05 nm × 8.57 nm | ||
| Outer leaflet | Inner leaflet | Outer leaflet | Inner leaflet | |
| POPC | 175 | 100 | 175 | 100 |
| POPS | 10 | 36 | 10 | 36 |
| POPE | 26 | 103 | 26 | 103 |
| POSM | 86 | 21 | 86 | 21 |
| POPI | 0 | 45 | 0 | 45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Martín, M.; Renault, P.; Giraldo, J.; Vázquez-Ibar, J.L.; Perálvarez-Marín, A. In Silico Assessment of the Lipid Fingerprint Signature of ATP2, the Essential P4-ATPase of Malaria Parasites. Membranes 2022, 12, 702. https://doi.org/10.3390/membranes12070702
López-Martín M, Renault P, Giraldo J, Vázquez-Ibar JL, Perálvarez-Marín A. In Silico Assessment of the Lipid Fingerprint Signature of ATP2, the Essential P4-ATPase of Malaria Parasites. Membranes. 2022; 12(7):702. https://doi.org/10.3390/membranes12070702
Chicago/Turabian StyleLópez-Martín, Mario, Pedro Renault, Jesus Giraldo, José Luis Vázquez-Ibar, and Alex Perálvarez-Marín. 2022. "In Silico Assessment of the Lipid Fingerprint Signature of ATP2, the Essential P4-ATPase of Malaria Parasites" Membranes 12, no. 7: 702. https://doi.org/10.3390/membranes12070702