
Transmigration across a Steady-State Blood–Brain Barrier Induces Activation of Circulating Dendritic Cells Partly Mediated by Actin Cytoskeletal Reorganization

 ,
, 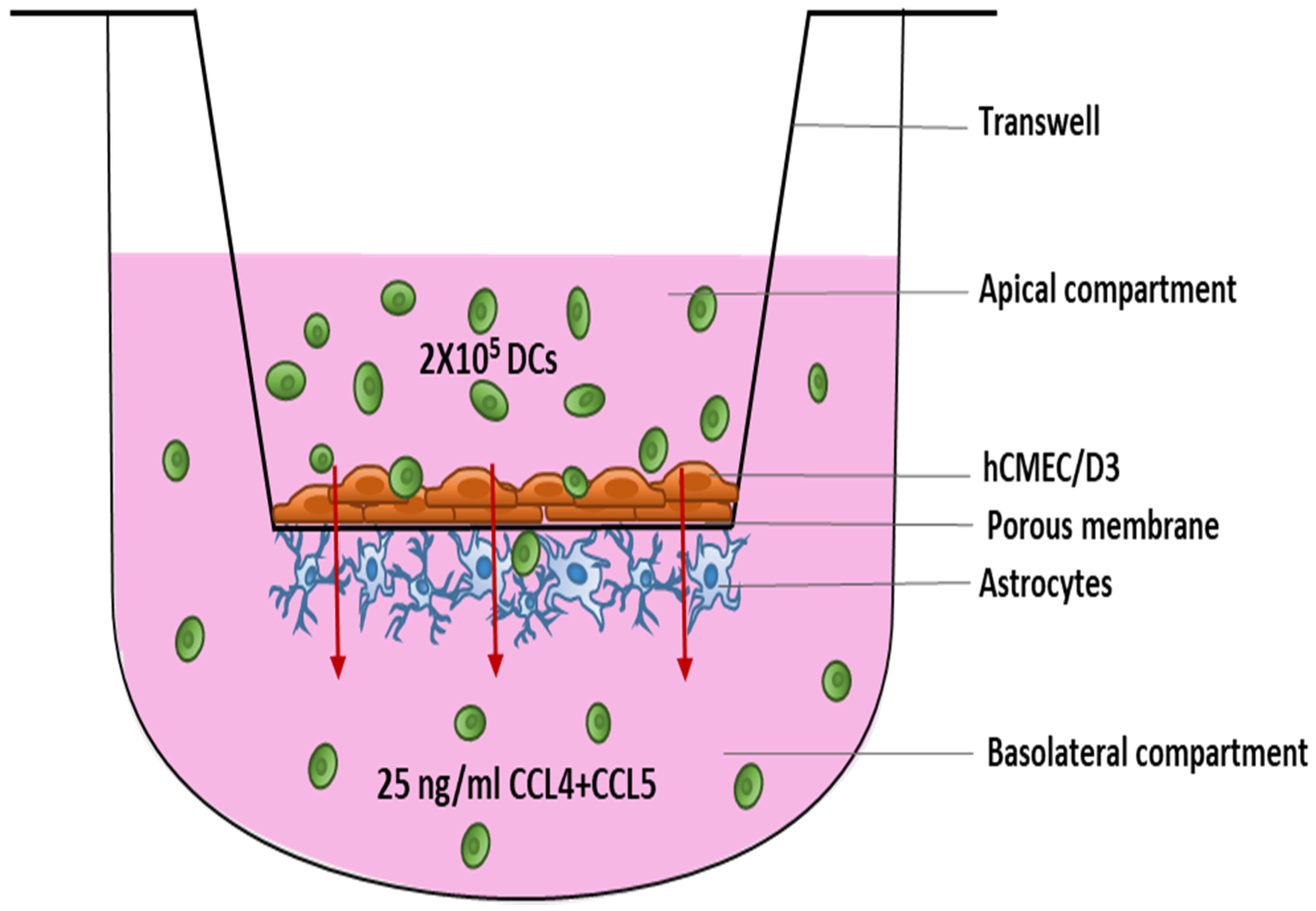{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture Model of Blood-Brain Barrier (BBB)
2.2. Cell Isolation
2.3. Migration Assay
2.4. Fluorescent Labeling of Endothelial Cell Layer
2.5. Flow Cytometry
2.6. Allogeneic Mixed Lymphocyte Reaction
2.7. Statistical Analyses
3. Results
3.1. Dendritic Cells That Migrate across the BBB Are in a More Activated State Than Non-Migrating Dendritic Cells
3.2. Dendritic Cells Do Not Take up Membrane Fragments of Endothelial Cells following Transmigration
3.3. Actin Cytoskeleton Restructuring of DCs Has No Effect on Migration-Induced Phenotypic Activation but Governs DC Migration and T Cell-Stimulatory Capacity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pachter, J.S.; de Vries, H.E.; Fabry, Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J. Neuropathol. Exp. Neurol. 2003, 62, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS immune privilege: Hiding in plain sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Forrester, J.V.; McMenamin, P.G.; Dando, S.J. CNS infection and immune privilege. Nat. Rev. Neurosci. 2018, 19, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A. V Brain endothelial cell-cell junctions: How to “open” the blood brain barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Campos-Bedolla, P.; Walter, F.R.; Veszelka, S.; Deli, M.A. Role of the blood—Brain barrier in the nutrition of the central nervous system. Arch. Med. Res. 2014, 45, 610–638. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood—Brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Török, O.; Schreiner, B.; Schaffenrath, J.; Tsai, H.-C.; Maheshwari, U.; Stifter, S.A.; Welsh, C.; Amorim, A.; Sridhar, S.; Utz, S.G. Pericytes regulate vascular immune homeostasis in the CNS. Proc. Natl. Acad. Sci. USA 2021, 9, 118. [Google Scholar]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef]
- Cabezas, R.; Ávila, M.; Gonzalez, J.; El-Bachá, R.S.; Báez, E.; García-Segura, L.M.; Jurado Coronel, J.C.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic modulation of blood brain barrier: Perspectives on Parkinson’s disease. Front. Cell. Neurosci. 2014, 8, 211. [Google Scholar] [CrossRef]
- Correale, J.; Villa, A. Cellular elements of the blood-brain barrier. Neurochem. Res. 2009, 34, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell 2020, 181, 784–799. [Google Scholar] [CrossRef]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef] [PubMed]
- Wosik, K.; Cayrol, R.; Dodelet-Devillers, A.; Berthelet, F.; Bernard, M.; Moumdjian, R.; Bouthillier, A.; Reudelhuber, T.L.; Prat, A. Angiotensin II controls occludin function and is required for blood–brain barrier maintenance: Relevance to multiple sclerosis. J. Neurosci. 2007, 27, 9032–9042. [Google Scholar] [CrossRef] [PubMed]
- Negi, N.; Das, B.K. CNS: Not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int. Rev. Immunol. 2018, 37, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Mrass, P.; Weninger, W. Immune cell migration as a means to control immune privilege: Lessons from the CNS and tumors. Immunol. Rev. 2006, 213, 195–212. [Google Scholar] [CrossRef]
- Huber, A.K.; Irani, D.N. Is the concept of central nervous system immune privilege irrelevant in the setting of acute infection? Front. Oncol. 2015, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.-J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Guermonprez, P.; Valladeau, J.; Zitvogel, L.; Théry, C.; Amigorena, S. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 2002, 20, 621–667. [Google Scholar] [CrossRef]
- Mohammad, M.G.; Hassanpour, M.; Tsai, V.W.W.; Li, H.; Ruitenberg, M.J.; Booth, D.R.; Serrats, J.; Hart, P.H.; Symonds, G.P.; Sawchenko, P.E.; et al. Dendritic cells and multiple sclerosis: Disease, tolerance and therapy. Int. J. Mol. Sci. 2013, 14, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Ludewig, P.; Gallizioli, M.; Urra, X.; Behr, S.; Brait, V.H.; Gelderblom, M.; Magnus, T.; Planas, A.M. Dendritic cells in brain diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zheng, H. Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–17. [Google Scholar]
- Koutsilieri, E.; Lutz, M.B.; Scheller, C. Autoimmunity, dendritic cells and relevance for Parkinson’s disease. J. Neural Transm. 2013, 120, 75–81. [Google Scholar] [CrossRef]
- Meena, M.; Cools, N. On the road to new treatments for multiple sclerosis: Targeting dendritic cell migration into the central nervous system. Neural Regen. Res. 2019, 14, 2088. [Google Scholar] [CrossRef]
- Anandasabapathy, N.; Victora, G.D.; Meredith, M.; Feder, R.; Dong, B.; Kluger, C.; Yao, K.; Dustin, M.L.; Nussenzweig, M.C.; Steinman, R.M.; et al. Flt3L controls the development of radiosensitive dendritic cells in the meninges and choroid plexus of the steady-state mouse brain. J. Exp. Med. 2011, 208, 1695–1705. [Google Scholar] [CrossRef]
- Sie, C.; Perez, L.G.; Kreutzfeldt, M.; Potthast, M.; Ohnmacht, C.; Merkler, D.; Huber, S.; Krug, A.; Korn, T. Dendritic Cell Accumulation in the Gut and Central Nervous System Is Differentially Dependent on $α$4 Integrins. J. Immunol. 2019, 203, 1417–1427. [Google Scholar] [CrossRef]
- Zozulya, A.L.; Reinke, E.; Baiu, D.C.; Karman, J.; Sandor, M.; Fabry, Z. Dendritic cell transmigration through brain microvessel endothelium is regulated by MIP-1$α$ chemokine and matrix metalloproteinases. J. Immunol. 2007, 178, 520–529. [Google Scholar] [CrossRef]
- Wheway, J.; Latham, S.L.; Combes, V.; Grau, G.E.R. Endothelial microparticles interact with and support the proliferation of T cells. J. Immunol. 2014, 193, 3378–3387. [Google Scholar] [CrossRef] [PubMed]
- Angelot, F.; Seillès, E.; Biichlé, S.; Berda, Y.; Gaugler, B.; Plumas, J.; Chaperot, L.; Dignat-George, F.; Tiberghien, P.; Saas, P.; et al. Endothelial cell-derived microparticles induce plasmacytoid dendritic cell maturation: Potential implications in inflammatory diseases. Haematologica 2009, 94, 1502. [Google Scholar] [CrossRef] [PubMed]
- Kedl, R.M.; Lindsay, R.S.; Finlon, J.M.; Lucas, E.D.; Friedman, R.S.; Tamburini, B.A.J. Migratory dendritic cells acquire and present lymphatic endothelial cell-archived antigens during lymph node contraction. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Letort, G.; Ennomani, H.; Gressin, L.; Théry, M.; Blanchoin, L. Dynamic reorganization of the actin cytoskeleton. F1000Research 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Omotade, O.F.; Myers, K.R.; Zheng, J.Q. Actin cytoskeleton in dendritic spine development and plasticity. Curr. Opin. Neurobiol. 2016, 39, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Swaney, K.F.; Li, R. Function and regulation of the Arp2/3 complex during cell migration in diverse environments. Curr. Opin. Cell Biol. 2016, 42, 63–72. [Google Scholar] [CrossRef]
- Schachtner, H.; Weimershaus, M.; Stache, V.; Plewa, N.; Legler, D.F.; Höpken, U.E.; Maritzen, T. Loss of Gadkin affects dendritic cell migration in vitro. PLoS ONE 2015, 10, e0143883. [Google Scholar]
- Ben-Shmuel, A.; Joseph, N.; Sabag, B.; Barda-Saad, M. Lymphocyte mechanotransduction: The regulatory role of cytoskeletal dynamics in signaling cascades and effector functions. J. Leukoc. Biol. 2019, 105, 1261–1273. [Google Scholar] [CrossRef]
- Comrie, W.A.; Li, S.; Boyle, S.; Burkhardt, J.K. The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. J. Cell Biol. 2015, 208, 457–473. [Google Scholar] [CrossRef]
- Pring, M.; Cassimeris, L.; Zigmond, S.H. An unexplained sequestration of latrunculin A is required in neutrophils for inhibition of actin polymerization. Cell Motil. Cytoskeleton 2002, 52, 122–130. [Google Scholar] [CrossRef]
- De Laere, M.; Sousa, C.; Meena, M.; Buckinx, R.; Timmermans, J.-P.; Berneman, Z.; Cools, N. Increased transendothelial transport of CCL3 Is insufficient to drive immune cell transmigration through the blood—Brain barrier under inflammatory conditions in vitro. Mediators Inflamm. 2017, 2017. [Google Scholar] [CrossRef]
- Weksler, B.B.; Subileau, E.A.; Perriere, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef]
- Mahad, D.J.; Howell, S.J.L.; Woodroofe, M.N. Expression of chemokines in the CSF and correlation with clinical disease activity in patients with multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2002, 72, 498–502. [Google Scholar]
- Soleimani, M.; Soleymani, A.; Seyyedirad, N. Elevated CSF concentration of CCL3 and CCL4 in relapsing remitting multiple sclerosis patients. J. Immunoass. Immunochem. 2019, 40, 378–385. [Google Scholar] [CrossRef]
- Matsushita, T.; Tateishi, T.; Isobe, N.; Yonekawa, T.; Yamasaki, R.; Matsuse, D.; Murai, H.; Kira, J. Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS ONE 2013, 8, e61835. [Google Scholar]
- Mitroulis, I.; Alexaki, V.I.; Kourtzelis, I.; Ziogas, A.; Hajishengallis, G.; Chavakis, T. Leukocyte integrins: Role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol. Ther. 2015, 147, 123–135. [Google Scholar] [CrossRef]
- Omari, K.M.; Dorovini-Zis, K. CD40 expressed by human brain endothelial cells regulates CD4+ T cell adhesion to endothelium. J. Neuroimmunol. 2003, 134, 166–178. [Google Scholar] [CrossRef]
- Serafini, B.; Columba-Cabezas, S.; Di Rosa, F.; Aloisi, F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am. J. Pathol. 2000, 157, 1991–2002. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Capello, E.; Mancardi, G.L.; Aloisi, F. Dendritic cells in multiple sclerosis lesions: Maturation stage, myelin uptake, and interaction with proliferating T cells. J. Neuropathol. Exp. Neurol. 2006, 65, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, P.; Corbisiero, V.; Male, D. Transient expression of MIDC-8 in the normal mouse brain. Neurosci. Lett. 2005, 377, 189–194. [Google Scholar] [CrossRef] [PubMed]
- McMahon, E.J.; Bailey, S.L.; Miller, S.D. CNS dendritic cells: Critical participants in CNS inflammation? Neurochem. Int. 2006, 49, 195–203. [Google Scholar] [CrossRef] [PubMed]
- GanjiBakhsh, M.; Nejati, V.; Delirezh, N.; Asadi, M.; Gholami, K. Mixture of fibroblast, epithelial and endothelial cells conditioned media induce monocyte-derived dendritic cell maturation. Cell. Immunol. 2011, 272, 18–24. [Google Scholar] [CrossRef]
- Tian, F.; Grimaldo, S.; Fugita, M.; Cutts, J.; Vujanovic, N.L.; Li, L.-Y. The endothelial cell-produced antiangiogenic cytokine vascular endothelial growth inhibitor induces dendritic cell maturation. J. Immunol. 2007, 179, 3742–3751. [Google Scholar] [CrossRef]
- Weis, M.; Schlichting, C.L.; Engleman, E.G.; Cooke, J.P. Endothelial determinants of dendritic cell adhesion and migration: New implications for vascular diseases. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1817–1823. [Google Scholar] [CrossRef]
- Moldenhauer, A.; Nociari, M.; Lam, G.; Salama, A.; Rafii, S.; Moore, M.A.S. Tumor necrosis factor alpha-stimulated endothelium: An inducer of dendritic cell development from hematopoietic progenitors and myeloid leukemic cells. Stem Cells 2004, 22, 144–157. [Google Scholar] [CrossRef]
- Randolph, G.J.; Beaulieu, S.; Lebecque, S.; Steinman, R.M.; Muller, W.A. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science 1998, 282, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Methe, H.; Hess, S.; Edelman, E.R. Endothelial cell-matrix interactions determine maturation of dendritic cells. Eur. J. Immunol. 2007, 37, 1773–1784. [Google Scholar] [CrossRef] [PubMed]
- Touil, H.; Kobert, A.; Lebeurrier, N.; Rieger, A.; Saikali, P.; Lambert, C.; Fawaz, L.; Moore, C.S.; Prat, A.; Gommerman, J.; et al. Human central nervous system astrocytes support survival and activation of B cells: Implications for MS pathogenesis. J. Neuroinflamm. 2018, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cornet, A.; Bettelli, E.; Oukka, M.; Cambouris, C.; Avellana-Adalid, V.; Kosmatopoulos, K.; Liblau, R.S. Role of astrocytes in antigen presentation and naive T-cell activation. J. Neuroimmunol. 2000, 106, 69–77. [Google Scholar] [CrossRef]
- Burns, S.; Hardy, S.J.; Buddle, J.; Yong, K.L.; Jones, G.E.; Thrasher, A.J. Maturation of DC is associated with changes in motile characteristics and adherence. Cell Motil. Cytoskelet. 2004, 57, 118–132. [Google Scholar] [CrossRef]
- Burns, S.; Thrasher, A.J.; Blundell, M.P.; Machesky, L.; Jones, G.E. Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood 2001, 98, 1142–1149. [Google Scholar] [CrossRef]
- Garrett, W.S.; Chen, L.-M.; Kroschewski, R.; Ebersold, M.; Turley, S.; Trombetta, S.; Galán, J.E.; Mellman, I. Developmental control of endocytosis in dendritic cells by Cdc42. Cell 2000, 102, 325–334. [Google Scholar] [CrossRef]
- West, M.A.; Prescott, A.R.; Eskelinen, E.-L.; Ridley, A.J.; Watts, C. Rac is required for constitutive macropinocytosis by dendritic cells but does not control its downregulation. Curr. Biol. 2000, 10, 839–848. [Google Scholar] [CrossRef]
- Penninger, J.M.; Crabtree, G.R. The actin cytoskeleton and lymphocyte activation. Cell 1999, 96, 9–12. [Google Scholar] [CrossRef]
- Billadeau, D.D.; Nolz, J.C.; Gomez, T.S. Regulation of T-cell activation by the cytoskeleton. Nat. Rev. Immunol. 2007, 7, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Beemiller, P.; Krummel, M.F. Mediation of T-cell activation by actin meshworks. Cold Spring Harb. Perspect. Biol. 2010, 2, a002444. [Google Scholar] [CrossRef]
- Batista, F.D.; Treanor, B.; Harwood, N.E. Visualizing a role for the actin cytoskeleton in the regulation of B-cell activation. Immunol. Rev. 2010, 237, 191–204. [Google Scholar] [CrossRef]
- Blumenthal, D.; Chandra, V.; Avery, L.; Burkhardt, J.K. Mouse T cell priming is enhanced by maturation-dependent stiffening of the dendritic cell cortex. Elife 2020, 9, e55995. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; De Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T. Design and validation of a human brain endothelial microvessel-on-a-chip open microfluidic model enabling advanced optical imaging. Front. Bioeng. Biotechnol. 2020, 8, 1077. [Google Scholar] [CrossRef]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; Van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G. A perfused human blood–brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 1–12. [Google Scholar] [CrossRef]
- Wang, H. Modeling neurological diseases with human brain organoids. Front. Synaptic Neurosci. 2018, 10, 15. [Google Scholar] [CrossRef]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wan, Z.; Kamm, R.D. Vascularized organoids on a chip: Strategies for engineering organoids with functional vasculature. Lab Chip 2021, 21, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Al-Obaidi, Z.; Kitchen, P.; Loreto, A.; Bill, R.M.; Wade-Martins, R. Advances in applying computer-aided drug design for neurodegenerative diseases. Int. J. Mol. Sci. 2021, 22, 4688. [Google Scholar] [CrossRef] [PubMed]
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-throughput screening platforms in the discovery of novel drugs for neurodegenerative diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meena, M.; Van Delen, M.; De Laere, M.; Sterkens, A.; Costas Romero, C.; Berneman, Z.; Cools, N. Transmigration across a Steady-State Blood–Brain Barrier Induces Activation of Circulating Dendritic Cells Partly Mediated by Actin Cytoskeletal Reorganization. Membranes 2021, 11, 700. https://doi.org/10.3390/membranes11090700
Meena M, Van Delen M, De Laere M, Sterkens A, Costas Romero C, Berneman Z, Cools N. Transmigration across a Steady-State Blood–Brain Barrier Induces Activation of Circulating Dendritic Cells Partly Mediated by Actin Cytoskeletal Reorganization. Membranes. 2021; 11(9):700. https://doi.org/10.3390/membranes11090700
Chicago/Turabian StyleMeena, Megha, Mats Van Delen, Maxime De Laere, Ann Sterkens, Coloma Costas Romero, Zwi Berneman, and Nathalie Cools. 2021. "Transmigration across a Steady-State Blood–Brain Barrier Induces Activation of Circulating Dendritic Cells Partly Mediated by Actin Cytoskeletal Reorganization" Membranes 11, no. 9: 700. https://doi.org/10.3390/membranes11090700