Inhibition of Heme Oxygenase Antioxidant Activity Exacerbates Hepatic Steatosis and Fibrosis In Vitro

 , ,
, , {kind=link}
{kind=link}
{kind=link}
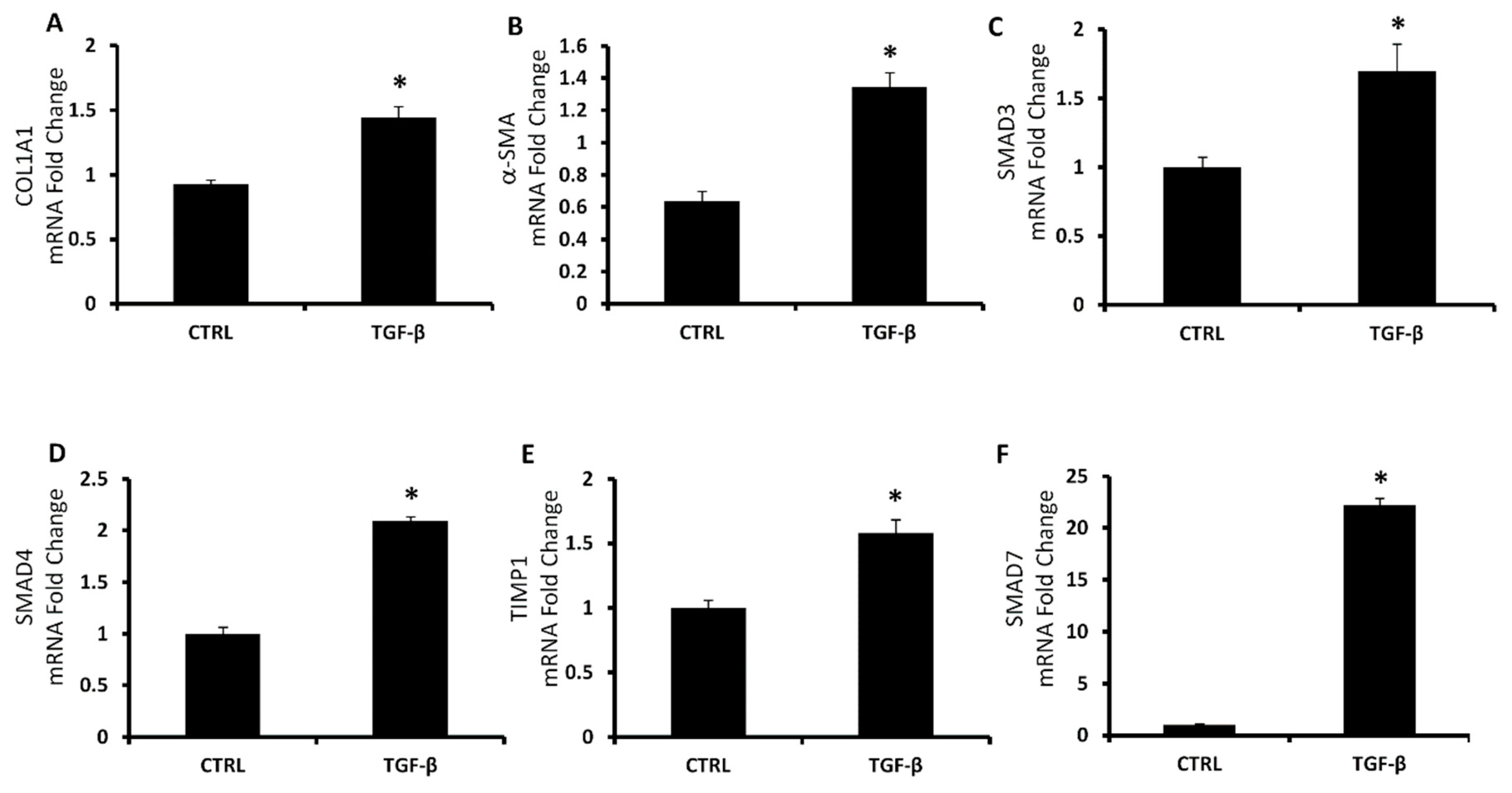{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Oil Red O Staining
2.3. ROS Measurement
2.4. Sircol Collagen Assay
2.5. RNA Extraction and qRT-PCR
2.6. Statistical Analyses
3. Results
3.1. Effect of HO Inhibition on Hepatic Fatty Storage
3.2. Effect of HO Regulation on Lipid Metabolism Pathway
3.3. Hepatic Fibrosis In Vitro Model
3.4. Effect of HO Inhibition on ROS Generation and Soluble Collagen Release
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Clark, J.M.; Brancati, F.L.; Diehl, A.M. Nonalcoholic fatty liver disease. Gastroenterology 2002, 122, 1649–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, I.; Dominguez-Perez, M.; Bucio, L.; Souza, V.; Miranda, R.U.; Clemens, D.L.; Gomez-Quiroz, L.E.; Gutierrez-Ruiz, M.C. Free fatty acids enhance the oxidative damage induced by ethanol metabolism in an in vitro model. Food Chem. 2015, 76, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carulli, L.; Zanca, G.; Schepis, F.; Villa, E. The OMICs Window into Nonalcoholic Fatty Liver Disease (NAFLD). Metabolites 2019, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Gorden, D.L.; Myers, D.S.; Ivanova, P.T.; Fahy, E.; Maurya, M.R.; Gupta, S.; Min, J.; Spann, N.J.; McDonald, J.G.; Kelly, S.L.; et al. Biomarkers of NAFLD progression: A lipidomics approach to an epidemic. J. Lipid Res. 2015, 56, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, C.; Neyrinck, A.M.; Lanthier, N.; Delzenne, N.M. Microbiota and nonalcoholic fatty liver disease: Promising prospects for clinical interventions? Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Licari, M.; Raffaele, M.; Rosman, Z.F.; Schragenheim, J.; Bellner, L.; Vanella, L.; Rezzani, R.; Rodella, L.; Bonomini, F.; Hochhauser, E.; et al. Beneficial Effects of Thymoquinone on Metabolic Function and Fatty Liver in a Murine Model of Obesity. J. Nutr. Food Sci. 2019, 9. [Google Scholar] [CrossRef]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Ma, Y.; Wu, S.; Liu, Y.; Liu, L.; Wu, L. Novel Serum Biomarkers for Noninvasive Diagnosis and Screening of Nonalcoholic Fatty Liver Disease-Related Hepatic Fibrosis. OMICS J. Integr. Biol. 2019, 23, 181–189. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Uhlen, M.; Boren, J. Broad Views of Non-alcoholic Fatty Liver Disease. Cell Syst. 2018, 6, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wruck, W.; Kashofer, K.; Rehman, S.; Daskalaki, A.; Berg, D.; Gralka, E.; Jozefczuk, J.; Drews, K.; Pandey, V.; Regenbrecht, C.; et al. Multi-omic profiles of human non-alcoholic fatty liver disease tissue highlight heterogenic phenotypes. Sci. Data 2015, 2, 150068. [Google Scholar] [CrossRef]
- Kotronen, A.; Yki-Jarvinen, H. Fatty liver: A novel component of the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Patil, R.; Sood, G.K. Non-alcoholic fatty liver disease and cardiovascular risk. World J. Gastrointest. Pathophysiol. 2017, 8, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [Green Version]
- Idilman, I.S.; Ozdeniz, I.; Karcaaltincaba, M. Hepatic Steatosis: Etiology, Patterns, and Quantification. Semin. Ultrasound CT MR 2016, 37, 501–510. [Google Scholar] [CrossRef]
- Lee, P.C.; Yang, L.Y.; Wang, Y.W.; Huang, S.F.; Lee, K.C.; Hsieh, Y.C.; Yang, Y.Y.; Hsieh, S.L.; Hou, M.C.; Lin, H.C.; et al. Mechanisms of the prevention and inhibition of the progression and development of non-alcoholic steatohepatitis by genetic and pharmacological decoy receptor 3 supplementation. Hepatol. Res. 2017, 47, 1260–1271. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Wallace, K.; Burt, A.D.; Wright, M.C. Liver fibrosis. Biochem. J. 2008, 411, 1–18. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, D.; Wang, G.; Cui, S.; Field, R.A.; Li, J.; Zang, Y. Alogliptin alleviates liver fibrosis via suppression of activated hepatic stellate cell. Biochem. Biophys. Res. Commun. 2019, 511, 387–393. [Google Scholar] [CrossRef]
- Yin, C.; Evason, K.J.; Asahina, K.; Stainier, D.Y. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Investig. 2013, 123, 1902–1910. [Google Scholar] [CrossRef] [Green Version]
- Cordero-Espinoza, L.; Huch, M. The balancing act of the liver: Tissue regeneration versus fibrosis. J. Clin. Investig. 2018, 128, 85–96. [Google Scholar] [CrossRef]
- Fujita, T.; Narumiya, S. Roles of hepatic stellate cells in liver inflammation: A new perspective. Inflamm. Regen. 2016, 36, 1. [Google Scholar] [CrossRef]
- Bauge, C.; Cauvard, O.; Leclercq, S.; Galera, P.; Boumediene, K. Modulation of transforming growth factor beta signalling pathway genes by transforming growth factor beta in human osteoarthritic chondrocytes: Involvement of Sp1 in both early and late response cells to transforming growth factor beta. Arthritis Res. Ther. 2011, 13, R23. [Google Scholar] [CrossRef]
- Qiang, H.; Lin, Y.; Zhang, X.; Zeng, X.; Shi, J.; Chen, Y.X.; Yang, M.F.; Han, Z.G.; Xie, W.F. Differential expression genes analyzed by cDNA array in the regulation of rat hepatic fibrogenesis. Liver Int. 2006, 26, 1126–1137. [Google Scholar] [CrossRef]
- Bi, W.R.; Yang, C.Q.; Shi, Q. Transforming growth factor-beta1 induced epithelial-mesenchymal transition in hepatic fibrosis. Hepatogastroenterology 2012, 59, 1960–1963. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Carcamo, J.; Zentella, A.; Doody, J.; Laiho, M.; Wang, X.F.; Massague, J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell 1992, 71, 1003–1014. [Google Scholar] [CrossRef]
- Lan, H.Y.; Chung, A.C. Transforming growth factor-beta and Smads. Contrib. Nephrol. 2011, 170, 75–82. [Google Scholar] [CrossRef]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef]
- Navarro-Corcuera, A.; Lopez-Zabalza, M.J.; Martinez-Irujo, J.J.; Alvarez-Sola, G.; Avila, M.A.; Iraburu, M.J.; Ansorena, E.; Montiel-Duarte, C. Role of AGAP2 in the profibrogenic effects induced by TGFbeta in LX-2 hepatic stellate cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 673–685. [Google Scholar] [CrossRef]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, M.; Jonscher, K.R.; Friedman, J.E. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging 2011, 3, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Okura, Y.; Namisaki, T.; Moriya, K.; Kitade, M.; Takeda, K.; Kaji, K.; Noguchi, R.; Nishimura, N.; Seki, K.; Kawaratani, H.; et al. Combined treatment with dipeptidyl peptidase-4 inhibitor (sitagliptin) and angiotensin-II type 1 receptor blocker (losartan) suppresses progression in a non-diabetic rat model of steatohepatitis. Hepatol. Res. 2017, 47, 1317–1328. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef]
- Maines, M.D. The heme oxygenase system: Past, present, and future. Antioxid. Redox Signal. 2004, 6, 797–801. [Google Scholar] [CrossRef]
- Signorelli, S.S.; Li Volsi, G.; Fiore, V.; Mangiafico, M.; Barbagallo, I.; Parenti, R.; Rizzo, M.; Li Volti, G. Plasma heme oxygenase-1 is decreased in peripheral artery disease patients. Mol. Med. Rep. 2016, 14, 3459–3463. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Choi, A.M. Heme oxygenase: Colors of defense against cellular stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1029–L1037. [Google Scholar] [CrossRef]
- Zingales, V.; Distefano, A.; Raffaele, M.; Zanghi, A.; Barbagallo, I.; Vanella, L. Metformin: A Bridge between Diabetes and Prostate Cancer. Front. Oncol. 2017, 7, 243. [Google Scholar] [CrossRef]
- Barbagallo, I.; Giallongo, C.; Volti, G.L.; Distefano, A.; Camiolo, G.; Raffaele, M.; Salerno, L.; Pittala, V.; Sorrenti, V.; Avola, R.; et al. Heme Oxygenase Inhibition Sensitizes Neuroblastoma Cells to Carfilzomib. Mol. Neurobiol. 2019, 56, 1451–1460. [Google Scholar] [CrossRef]
- Raffaele, M.; Pittala, V.; Zingales, V.; Barbagallo, I.; Salerno, L.; Li Volti, G.; Romeo, G.; Carota, G.; Sorrenti, V.; Vanella, L. Heme Oxygenase-1 Inhibition Sensitizes Human Prostate Cancer Cells towards Glucose Deprivation and Metformin-Mediated Cell Death. Int. J. Mol. Sci. 2019, 20, 2593. [Google Scholar] [CrossRef]
- Vanella, L.; Di Giacomo, C.; Acquaviva, R.; Barbagallo, I.; Li Volti, G.; Cardile, V.; Abraham, N.G.; Sorrenti, V. Effects of ellagic Acid on angiogenic factors in prostate cancer cells. Cancers 2013, 5, 726–738. [Google Scholar] [CrossRef]
- Carota, G.; Sferrazzo, G.; Spampinato, M.; Sorrenti, V.; Vanella, L. Antiproliferative Effects of Ellagic Acid on DU145 Cells. Open Biochem. J. 2019, 13, 23–31. [Google Scholar] [CrossRef]
- Wilks, A. Heme oxygenase: Evolution, structure, and mechanism. Antioxid. Redox Signal. 2002, 4, 603–614. [Google Scholar] [CrossRef]
- Goda, N.; Suzuki, K.; Naito, M.; Takeoka, S.; Tsuchida, E.; Ishimura, Y.; Tamatani, T.; Suematsu, M. Distribution of heme oxygenase isoforms in rat liver. Topographic basis for carbon monoxide-mediated microvascular relaxation. J. Clin. Investig. 1998, 101, 604–612. [Google Scholar] [CrossRef]
- Abraham, N.G.; Junge, J.M.; Drummond, G.S. Translational Significance of Heme Oxygenase in Obesity and Metabolic Syndrome. Trends Pharmacol. Sci. 2016, 37, 17–36. [Google Scholar] [CrossRef]
- Raffaele, M.; Li Volti, G.; Barbagallo, I.A.; Vanella, L. Therapeutic Efficacy of Stem Cells Transplantation in Diabetes: Role of Heme Oxygenase. Front. Cell Dev. Biol. 2016, 4, 80. [Google Scholar] [CrossRef]
- Liu, L.; Puri, N.; Raffaele, M.; Schragenheim, J.; Singh, S.P.; Bradbury, J.A.; Bellner, L.; Vanella, L.; Zeldin, D.C.; Cao, J.; et al. Ablation of soluble epoxide hydrolase reprogram white fat to beige-like fat through an increase in mitochondrial integrity, HO-1-adiponectin in vitro and in vivo. Prostaglandins Other Lipid Mediat. 2018, 138, 1–8. [Google Scholar] [CrossRef]
- Malaguarnera, L.; Madeddu, R.; Palio, E.; Arena, N.; Malaguarnera, M. Heme oxygenase-1 levels and oxidative stress-related parameters in non-alcoholic fatty liver disease patients. J. Hepatol. 2005, 42, 585–591. [Google Scholar] [CrossRef]
- Sass, G.; Seyfried, S.; Parreira Soares, M.; Yamashita, K.; Kaczmarek, E.; Neuhuber, W.L.; Tiegs, G. Cooperative effect of biliverdin and carbon monoxide on survival of mice in immune-mediated liver injury. Hepatology 2004, 40, 1128–1135. [Google Scholar] [CrossRef]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Barbagallo, I.; Volti, G.L.; Raffaele, M.; Distefano, A.; Palmeri, R.; Parafati, L.; Licari, M.; Zingales, V.; Avola, R.; Vanella, L. The effects of olive leaf extract from a Sicilian cultivar in an experimental model of hepatic steatosis. Rend Lincei 2017, 28, 643–650. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Guicciardi, M.E.; Higuchi, H.; Bronk, S.F.; Gores, G.J. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J. Hepatol. 2003, 39, 978–983. [Google Scholar] [CrossRef]
- Park, M.; Yoo, J.H.; Lee, Y.S.; Lee, H.J. Lonicera caerulea Extract Attenuates Non-Alcoholic Fatty Liver Disease in Free Fatty Acid-Induced HepG2 Hepatocytes and in High Fat Diet-Fed Mice. Nutrients 2019, 11, 494. [Google Scholar] [CrossRef]
- Hempel, S.L.; Buettner, G.R.; O’Malley, Y.Q.; Wessels, D.A.; Flaherty, D.M. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: Comparison with 2′, 7′-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2′, 7′-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic. Biol. Med. 1999, 27, 146–159. [Google Scholar] [CrossRef]
- Abate, A.; Zhao, H.; Wong, R.J.; Stevenson, D.K. The role of Bach1 in the induction of heme oxygenase by tin mesoporphyrin. Biochem. Biophys. Res. Commun. 2007, 354, 757–763. [Google Scholar] [CrossRef] [Green Version]
- Sardana, M.K.; Kappas, A. Dual control mechanism for heme oxygenase: Tin(IV)-protoporphyrin potently inhibits enzyme activity while markedly increasing content of enzyme protein in liver. Proc. Natl. Acad. Sci. USA 1987, 84, 2464–2468. [Google Scholar] [CrossRef]
- Liu, X.; Gao, Y.; Li, M.; Geng, C.; Xu, H.; Yang, Y.; Guo, Y.; Jiao, T.; Fang, F.; Chang, Y. Sirt1 mediates the effect of the heme oxygenase inducer, cobalt protoporphyrin, on ameliorating liver metabolic damage caused by a high-fat diet. J. Hepatol. 2015, 63, 713–721. [Google Scholar] [CrossRef]
- Presser, L.D.; McRae, S.; Waris, G. Activation of TGF-beta1 promoter by hepatitis C virus-induced AP-1 and Sp1: Role of TGF-beta1 in hepatic stellate cell activation and invasion. PLoS ONE 2013, 8, e56367. [Google Scholar] [CrossRef]
- Koutsari, C.; Mundi, M.S.; Ali, A.H.; Patterson, B.W.; Jensen, M.D. Systemic free fatty acid disposal into very low-density lipoprotein triglycerides. Diabetes 2013, 62, 2386–2395. [Google Scholar] [CrossRef]
- Dhami-Shah, H.; Vaidya, R.; Udipi, S.; Raghavan, S.; Abhijit, S.; Mohan, V.; Balasubramanyam, M.; Vaidya, A. Picroside II attenuates fatty acid accumulation in HepG2 cells via modulation of fatty acid uptake and synthesis. Clin. Mol. Hepatol. 2018, 24, 77–87. [Google Scholar] [CrossRef]
- Gomez-Lechon, M.J.; Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Yahagi, N.; Shimano, H.; Hasty, A.H.; Matsuzaka, T.; Ide, T.; Yoshikawa, T.; Amemiya-Kudo, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J. Biol. Chem. 2002, 277, 19353–19357. [Google Scholar] [CrossRef]
- Pettinelli, P.; Obregon, A.M.; Videla, L.A. Molecular mechanisms of steatosis in nonalcoholic fatty liver disease. Nutr. Hosp. 2011, 26, 441–450. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Liang, F.; Kume, S.; Koya, D. SIRT1 and insulin resistance. Nat. Rev. Endocrinol. 2009, 5, 367–373. [Google Scholar] [CrossRef]
- Ponugoti, B.; Kim, D.H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.Y.; Chiang, C.M.; Veenstra, T.D.; Kemper, J.K. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 2010, 285, 33959–33970. [Google Scholar] [CrossRef]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschop, M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef] [Green Version]
- Sacerdoti, D.; Singh, S.P.; Schragenheim, J.; Bellner, L.; Vanella, L.; Raffaele, M.; Meissner, A.; Grant, I.; Favero, G.; Rezzani, R.; et al. Development of NASH in Obese Mice is Confounded by Adipose Tissue Increase in Inflammatory NOV and Oxidative Stress. Int. J. Hepatol. 2018, 2018, 3484107. [Google Scholar] [CrossRef]
- Raffaele, M.; Bellner, L.; Singh, S.P.; Favero, G.; Rezzani, R.; Rodella, L.F.; Falck, J.R.; Abraham, N.G.; Vanella, L. Epoxyeicosatrienoic intervention improves NAFLD in leptin receptor deficient mice by an increase in PGC1alpha-HO-1-PGC1alpha-mitochondrial signaling. Exp. Cell Res. 2019, 380, 180–187. [Google Scholar] [CrossRef]
- Sodhi, K.; Puri, N.; Favero, G.; Stevens, S.; Meadows, C.; Abraham, N.G.; Rezzani, R.; Ansinelli, H.; Lebovics, E.; Shapiro, J.I. Fructose Mediated Non-Alcoholic Fatty Liver Is Attenuated by HO-1-SIRT1 Module in Murine Hepatocytes and Mice Fed a High Fructose Diet. PLoS ONE 2015, 10, e0128648. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Sodhi, K.; Meadows, C.; Fedorova, L.; Puri, N.; Kim, D.H.; Peterson, S.J.; Shapiro, J.; Abraham, N.G.; Kappas, A. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity 2014, 22, 705–712. [Google Scholar] [CrossRef]
- Mostafa, M.E.; Shaaban, A.A.; Salem, H.A. Dimethylfumarate ameliorates hepatic injury and fibrosis induced by carbon tetrachloride. Chem. Biol. Interact. 2019, 302, 53–60. [Google Scholar] [CrossRef]
- Yang, K.L.; Chang, W.T.; Hong, M.Y.; Hung, K.C.; Chuang, C.C. Prevention of TGF-beta-induced early liver fibrosis by a maleic acid derivative anti-oxidant through suppression of ROS, inflammation and hepatic stellate cells activation. PLoS ONE 2017, 12, e0174008. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Redox Functions of Heme Oxygenase-1 and Biliverdin Reductase in Diabetes. Trends Endocrinol. Metab. TEM 2018, 29, 74–85. [Google Scholar] [CrossRef]
- Liu, X.; Cui, Y.; Li, M.; Xu, H.; Zuo, J.; Fang, F.; Chang, Y. Cobalt protoporphyrin induces HO-1 expression mediated partially by FOXO1 and reduces mitochondria-derived reactive oxygen species production. PLoS ONE 2013, 8, e80521. [Google Scholar] [CrossRef]
- Waldman, M.; Nudelman, V.; Shainberg, A.; Zemel, R.; Kornwoski, R.; Aravot, D.; Peterson, S.J.; Arad, M.; Hochhauser, E. The Role of Heme Oxygenase 1 in the Protective Effect of Caloric Restriction against Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 2427. [Google Scholar] [CrossRef]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappala, A.; Li Volti, G. The non-canonical functions of the heme oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raffaele, M.; Carota, G.; Sferrazzo, G.; Licari, M.; Barbagallo, I.; Sorrenti, V.; Signorelli, S.S.; Vanella, L. Inhibition of Heme Oxygenase Antioxidant Activity Exacerbates Hepatic Steatosis and Fibrosis In Vitro. Antioxidants 2019, 8, 277. https://doi.org/10.3390/antiox8080277
Raffaele M, Carota G, Sferrazzo G, Licari M, Barbagallo I, Sorrenti V, Signorelli SS, Vanella L. Inhibition of Heme Oxygenase Antioxidant Activity Exacerbates Hepatic Steatosis and Fibrosis In Vitro. Antioxidants. 2019; 8(8):277. https://doi.org/10.3390/antiox8080277
Chicago/Turabian StyleRaffaele, Marco, Giuseppe Carota, Giuseppe Sferrazzo, Maria Licari, Ignazio Barbagallo, Valeria Sorrenti, Salvatore S. Signorelli, and Luca Vanella. 2019. "Inhibition of Heme Oxygenase Antioxidant Activity Exacerbates Hepatic Steatosis and Fibrosis In Vitro" Antioxidants 8, no. 8: 277. https://doi.org/10.3390/antiox8080277