Antioxidant and Adaptative Response Mediated by Nrf2 during Physical Exercise

 , ,
, , 
Abstract
:1. Introduction
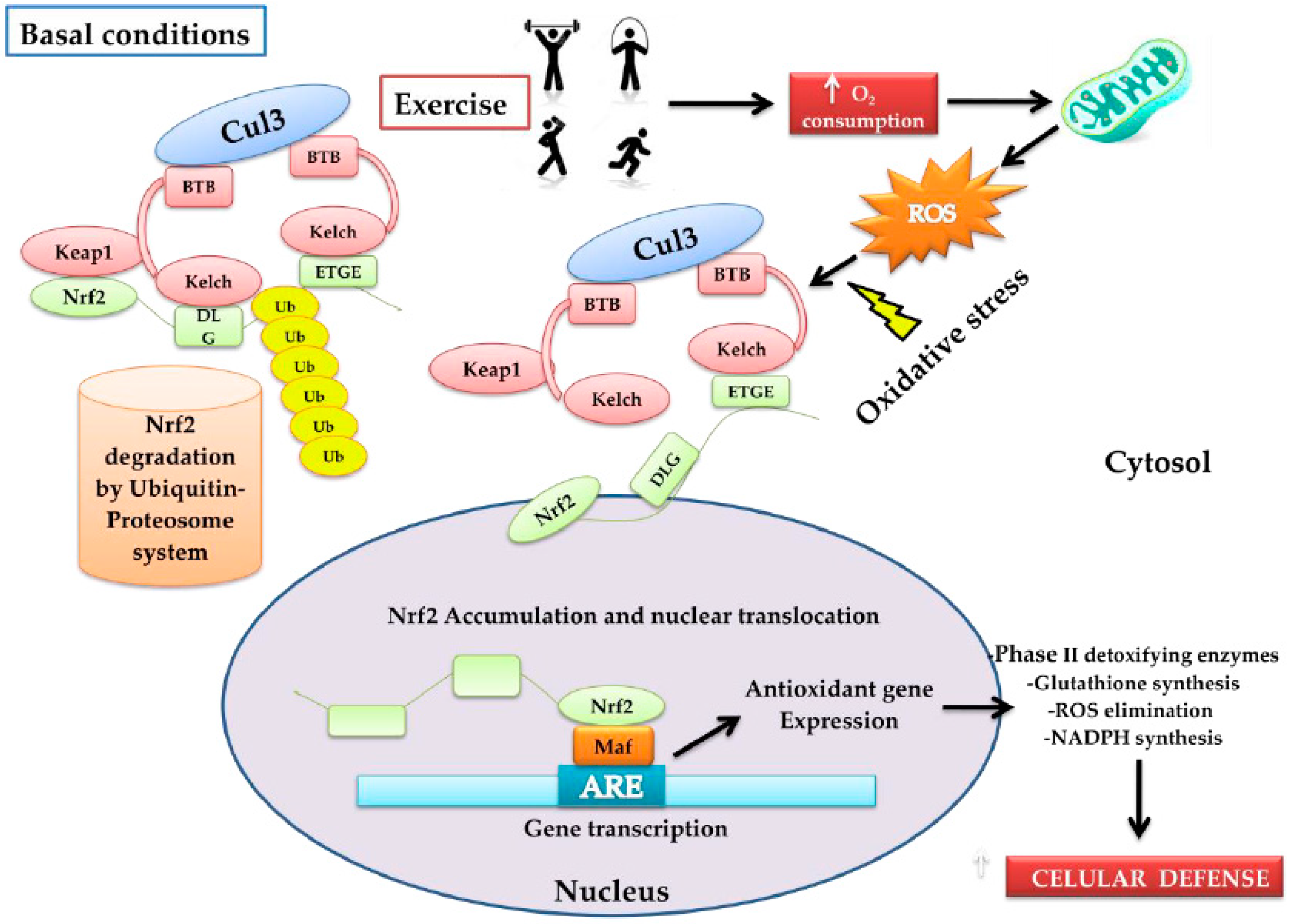
2. Nrf2 Activation and Transcription
3. Nrf2, the Antioxidant Response Master Regulator
4. Physical Exercise and Redox Response
4.1. Oxidative Stress and Exercise
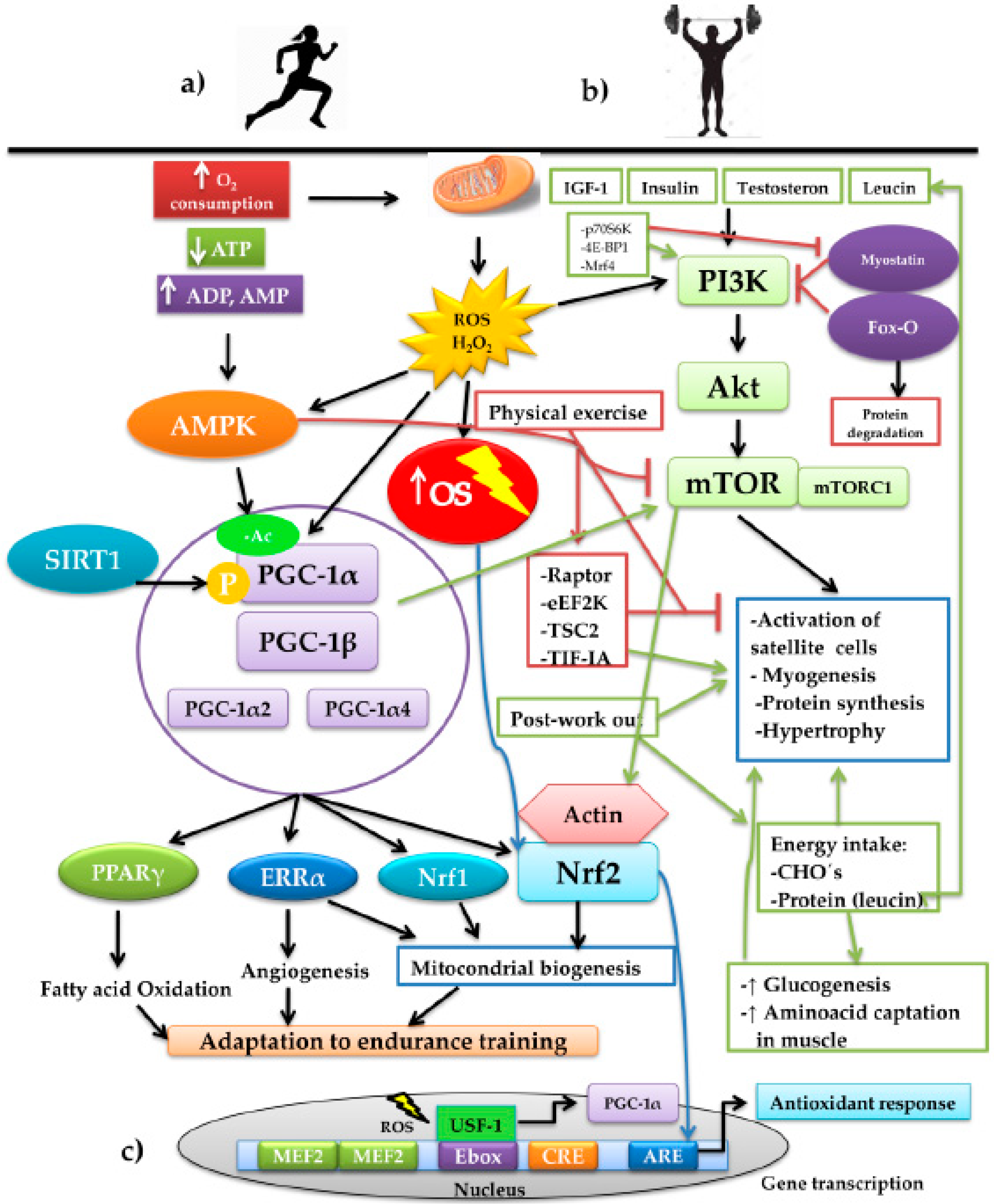
4.2. Signaling Pathways and the Epigenetic Changes Induced by ROS during Physical Exercise
4.3. Adaptative Responses According to the Exercise Training Modality
5. Regulation of Nrf2 by Physical Training
5.1. Nrf2 in Aerobic Exercise Models
5.2. Nfr2 in Resistance Exercise Models
5.3. Physical Exercise and Bioactivador Compounds of Nrf2
6. Nrf2 and Modulation of Energy Metabolism during Exercise
7. Clinical Implications of Therole of Nrf2 in Physical Exercise
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- Yavari, A.; Javadi, M.; Mirmiran, P.; Bahadoran, Z. Exercise-induced oxidative stress and dietary antioxidants. Asian J. Sports Med. 2015, 6, e24898. [Google Scholar] [CrossRef] [PubMed]
- Nosrati, N.; Bakovic, M.; Paliyath, G. Molecular Mechanisms and Pathways as Targets for Cancer Prevention and Progression with Dietary Compounds. Int. J. Mol. Sci. 2017, 18, 2050. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Li, J.; Lin, Q.; Huang, P.; Zhang, L.; Wu, W.; Ma, Y. Research Progress on Signaling Pathway-Associated Oxidative Stress in Endothelial Cells. Oxidative Med. Cell. Longev. 2017, 2017, 7156941. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Jiang, X.; Meng, L.; Dong, X.; Shen, Y.; Xin, Y. Anticancer Activity of Sulforaphane: The Epigenetic Mechanisms and the Nrf2 Signaling Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 5438179. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Johnson, J.A. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim. Et Biophys. Acta 2014, 1842, 1208–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomatto, L.C.D.; Davies, K.J.A. The role of declining adaptive homeostasis in ageing. J. Physiol. 2017, 595, 7275–7309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Done, A.J.; Traustadottir, T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016, 10, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggler, A.L.; Small, E.; Hannink, M.; Mesecar, A.D. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem. J. 2009, 422, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Toyama, T.; Sumi, D.; Shinkai, Y.; Yasutake, A.; Taguchi, K.; Tong, K.I.; Yamamoto, M.; Kumagai, Y. Cytoprotective role of Nrf2/Keap1 system in methylmercury toxicity. Biochem. Biophys. Res. Commun. 2007, 363, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Karapetian, R.N.; Evstafieva, A.G.; Abaeva, I.S.; Chichkova, N.V.; Filonov, G.S.; Rubtsov, Y.P.; Sukhacheva, E.A.; Melnikov, S.V.; Schneider, U.; Wanker, E.E.; et al. Nuclear oncoprotein prothymosin alpha is a partner of Keap1: Implications for expression of oxidative stress-protecting genes. Mol. Cell. Biol. 2005, 25, 1089–1099. [Google Scholar] [CrossRef]
- Hayes, J.D.; McLellan, L.I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic. Res. 1999, 31, 273–300. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, S.; Chan, J.Y.; Zhang, D.D. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol. Cell. Biol. 2007, 27, 6334–6349. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yamamoto, M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzym. Regul. 2006, 46, 113–140. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Et Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Vomund, S.; Schafer, A.; Parnham, M.J.; Brune, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef]
- Shen, G.; Kong, A.N. Nrf2 plays an important role in coordinated regulation of Phase II drug metabolism enzymes and Phase III drug transporters. Biopharm. Drug Dispos. 2009, 30, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, H.J.; Karlenius, T.C.; Tonissen, K.F. Regulation of the human thioredoxin gene promoter and its key substrates: A study of functional and putative regulatory elements. Biochim. Et Biophys. Acta 2014, 1840, 303–314. [Google Scholar] [CrossRef]
- Shultz, C.A.; Quinn, A.M.; Park, J.H.; Harvey, R.G.; Bolton, J.L.; Maser, E.; Penning, T.M. Specificity of human aldo-keto reductases, NAD(P)H:quinone oxidoreductase, and carbonyl reductases to redox-cycle polycyclic aromatic hydrocarbon diones and 4-hydroxyequilenin-o-quinone. Chem. Res. Toxicol. 2011, 24, 2153–2166. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
- Cartee, G.D.; Hepple, R.T.; Bamman, M.M.; Zierath, J.R. Exercise Promotes Healthy Aging of Skeletal Muscle. Cell Metab. 2016, 23, 1034–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gounder, S.S.; Kannan, S.; Devadoss, D.; Miller, C.J.; Whitehead, K.J.; Odelberg, S.J.; Firpo, M.A.; Paine, R., 3rd; Hoidal, J.R.; Abel, E.D.; et al. Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PLoS ONE 2012, 7, e45697. [Google Scholar] [CrossRef]
- Boccatonda, A.; Tripaldi, R.; Davi, G.; Santilli, F. Oxidative Stress Modulation Through Habitual Physical Activity. Curr. Pharm. Des. 2016, 22, 3648–3680. [Google Scholar] [CrossRef]
- Ji, L.L.; Gomez-Cabrera, M.C.; Vina, J. Role of nuclear factor kappaB and mitogen-activated protein kinase signaling in exercise-induced antioxidant enzyme adaptation. Appl. Physiol. Nutr. Metab. 2007, 32, 930–935. [Google Scholar] [CrossRef]
- Ji, L.L. Modulation of skeletal muscle antioxidant defense by exercise: Role of redox signaling. Free Radic. Biol. Med. 2008, 44, 142–152. [Google Scholar] [CrossRef]
- Ji, L.L. Antioxidant signaling in skeletal muscle: A brief review. Exp. Gerontol. 2007, 42, 582–593. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Choi, K.J.; Kim, J.H.; Han, X.; Piao, Y.; Jeong, J.H.; Choe, W.; Kang, I.; Ha, J.; Forman, H.J.; et al. Endogenous hydrogen peroxide regulates glutathione redox via nuclear factor erythroid 2-related factor 2 downstream of phosphatidylinositol 3-kinase during muscle differentiation. Am. J. Pathol. 2008, 172, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovic, O.; Veenstra, G.J. DNA methylation and methyl-CpG binding proteins: Developmental requirements and function. Chromosoma 2009, 118, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.G.; Lally, J.; Holloway, G.P.; Heigenhauser, G.J.; Bonen, A.; Spriet, L.L. Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J. Physiol. 2010, 588, 4795–4810. [Google Scholar] [CrossRef] [PubMed]
- Barres, R.; Zierath, J.R. The role of diet and exercise in the transgenerational epigenetic landscape of T2DM. Nat. Rev. Endocrinol. 2016, 12, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Nitert, M.D.; Dayeh, T.; Volkov, P.; Elgzyri, T.; Hall, E.; Nilsson, E.; Yang, B.T.; Lang, S.; Parikh, H.; Wessman, Y.; et al. Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 2012, 61, 3322–3332. [Google Scholar] [CrossRef]
- Schenk, A.; Pulverer, W.; Koliamitra, C.; Bauer, C.J.; Ilic, S.; Heer, R.; Schier, R.; Schick, V.; Bottiger, B.W.; Gerhauser, C.; et al. Acute Exercise Increases the Expression of KIR2DS4 by Promoter Demethylation in NK Cells. Int. J. Sports Med. 2019, 40, 62–70. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Kanzleiter, T.; Jahnert, M.; Schulze, G.; Selbig, J.; Hallahan, N.; Schwenk, R.W.; Schurmann, A. Exercise training alters DNA methylation patterns in genes related to muscle growth and differentiation in mice. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E912–E920. [Google Scholar] [CrossRef] [Green Version]
- Barres, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef]
- Sailani, R.M.; Halling, F.J.; Møller, D.H.; Lee, H.; Plomgaard, P.; Pilegaard, H.; Snyder, M.P.; Regenberg, B. Lifelong physical activity is associated with promoter hypomethylation of genes involved in metabolism, myogenesis, contractile properties and oxidative stress resistance in aged human skeletal muscle. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Cosgrove, B.D.; Gilbert, P.M.; Porpiglia, E.; Mourkioti, F.; Lee, S.P.; Corbel, S.Y.; Llewellyn, M.E.; Delp, S.L.; Blau, H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014, 20, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaborne, R.A.; Strauss, J.; Cocks, M.; Shepherd, S.; O’Brien, T.D.; Someren, K.A.V.; Bell, P.G.; Murgatroyd, C.; Morton, J.P.; Stewart, C.E.; et al. Methylome of human skeletal muscle after acute & chronic resistance exercise training, detraining & retraining. Sci. Data 2018, 5, 180213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaborne, R.A.; Strauss, J.; Cocks, M.; Shepherd, S.; O’Brien, T.D.; van Someren, K.A.; Bell, P.G.; Murgatroyd, C.; Morton, J.P.; Stewart, C.E.; et al. Human Skeletal Muscle Possesses an Epigenetic Memory of Hypertrophy. Sci. Rep. 2018, 8, 1898. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.J.; Martinez, P.F.; Pagan, L.U.; Damatto, R.L.; Cezar, M.D.M.; Lima, A.R.R.; Okoshi, K.; Okoshi, M.P. Skeletal muscle aging: Influence of oxidative stress and physical exercise. Oncotarget 2017, 8, 20428–20440. [Google Scholar] [CrossRef]
- Baumann, C.W.; Kwak, D.; Liu, H.M.; Thompson, L.V. Age-induced oxidative stress: How does it influence skeletal muscle quantity and quality? J. Appl. Physiol. 2016, 121, 1047–1052. [Google Scholar] [CrossRef]
- Baumann, C.W.; Liu, H.M.; Thompson, L.V. Denervation-Induced Activation of the Ubiquitin-Proteasome System Reduces Skeletal Muscle Quantity Not Quality. PLoS ONE 2016, 11, e0160839. [Google Scholar] [CrossRef]
- Spindler, C.; Segabinazi, E.; Meireles, A.L.F.; Piazza, F.V.; Mega, F.; Dos Santos Salvalaggio, G.; Achaval, M.; Elsner, V.R.; Marcuzzo, S. Paternal physical exercise modulates global DNA methylation status in the hippocampus of male rat offspring. Neural Regen. Res. 2019, 14, 491–500. [Google Scholar] [CrossRef]
- Joseph, A.M.; Adhihetty, P.J.; Leeuwenburgh, C. Beneficial effects of exercise on age-related mitochondrial dysfunction and oxidative stress in skeletal muscle. J. Physiol. 2016, 594, 5105–5123. [Google Scholar] [CrossRef]
- Ziaaldini, M.M.; Koltai, E.; Csende, Z.; Goto, S.; Boldogh, I.; Taylor, A.W.; Radak, Z. Exercise training increases anabolic and attenuates catabolic and apoptotic processes in aged skeletal muscle of male rats. Exp. Gerontol. 2015, 67, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Baar, K. Nutrition and the adaptation to endurance training. Sports Med. 2014, 44 (Suppl. 1), S5–S12. [Google Scholar] [CrossRef]
- Leick, L.; Hellsten, Y.; Fentz, J.; Lyngby, S.S.; Wojtaszewski, J.F.; Hidalgo, J.; Pilegaard, H. PGC-1alpha mediates exercise-induced skeletal muscle VEGF expression in mice. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E92–E103. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M. Transcriptional control of energy homeostasis through the PGC1 coactivators. Novartis Found. Symp. 2007, 286, 3–12; 162–163; 196–203. [Google Scholar] [PubMed]
- Spiegelman, B.M. Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found. Symp. 2007, 287, 60–69. [Google Scholar] [PubMed]
- Arany, Z.; Lebrasseur, N.; Morris, C.; Smith, E.; Yang, W.; Ma, Y.; Chin, S.; Spiegelman, B.M. The transcriptional coactivator PGC-1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab. 2007, 5, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Gilde, A.J.; Van Bilsen, M. Peroxisome proliferator-activated receptors (PPARS): Regulators of gene expression in heart and skeletal muscle. Acta Physiol. Scand. 2003, 178, 425–434. [Google Scholar] [CrossRef]
- Chinsomboon, J.; Ruas, J.; Gupta, R.K.; Thom, R.; Shoag, J.; Rowe, G.C.; Sawada, N.; Raghuram, S.; Arany, Z. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2009, 106, 21401–21406. [Google Scholar] [CrossRef]
- Akimoto, T.; Sorg, B.S.; Yan, Z. Real-time imaging of peroxisome proliferator-activated receptor-gamma coactivator-1alpha promoter activity in skeletal muscles of living mice. Am. J. Physiol. Cell Physiol. 2004, 287, C790–C796. [Google Scholar] [CrossRef]
- Irrcher, I.; Ljubicic, V.; Hood, D.A. Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2009, 296, C116–C123. [Google Scholar] [CrossRef]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.L.; Hargreaves, M. AMPK and transcriptional regulation. Front. Biosci. A J. Virtual Libr. 2008, 13, 3022–3033. [Google Scholar] [CrossRef]
- Schenk, S.; McCurdy, C.E.; Philp, A.; Chen, M.Z.; Holliday, M.J.; Bandyopadhyay, G.K.; Osborn, O.; Baar, K.; Olefsky, J.M. Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. J. Clin. Investig. 2011, 121, 4281–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann. N. Y. Acad. Sci. 2008, 1147, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.E.; Mackenzie, R.E. Mitochondrial methylenetetrahydrofolate dehydrogenase, methenyltetrahydrofolate cyclohydrolase, and formyltetrahydrofolate synthetases. Vitam. Horm. 2008, 79, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Wenz, T.; Rossi, S.G.; Rotundo, R.L.; Spiegelman, B.M.; Moraes, C.T. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA 2009, 106, 20405–20410. [Google Scholar] [CrossRef]
- Wenz, T. PGC-1alpha activation as a therapeutic approach in mitochondrial disease. IUBMB Life 2009, 61, 1051–1062. [Google Scholar] [CrossRef]
- Sun, Y.; Cui, D.; Zhang, Z.; Zhang, T.; Shi, J.; Jin, H.; Ge, Z.; Ji, L.; Ding, S. Attenuated Oxidative Stress following Acute Exhaustive Swimming Exercise Was Accompanied with Modified Gene Expression Profiles of Apoptosis in the Skeletal Muscle of Mice. Oxidative Med. Cell. Longev. 2016, 2016, 8381242. [Google Scholar] [CrossRef]
- Schoenfeld, B.J.; Contreras, B.; Krieger, J.; Grgic, J.; Delcastillo, K.; Belliard, R.; Alto, A. Resistance Training Volume Enhances Muscle Hypertrophy but Not Strength in Trained Men. Med. Sci. Sports Exerc. 2019, 51, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Morton, R.W.; McGlory, C.; Phillips, S.M. Nutritional interventions to augment resistance training-induced skeletal muscle hypertrophy. Front. Physiol. 2015, 6, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tipton, K.D.; Hamilton, D.L.; Gallagher, I.J. Assessing the Role of Muscle Protein Breakdown in Response to Nutrition and Exercise in Humans. Sports Med. 2018, 48, 53–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egerman, M.A.; Glass, D.J. Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Mammucari, C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: Insights from genetic models. Skelet. Muscle 2011, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.P.; Gao, S.; Puppa, M.J.; Sato, S.; Welle, S.L.; Carson, J.A. Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol. Cell. Endocrinol. 2013, 365, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, M.T.; Herda, T.J.; Fry, A.C.; Cooper, M.A.; Andre, M.J.; Gallagher, P.M. Endocrine responses and acute mTOR pathway phosphorylation to resistance exercise with leucine and whey. Biol. Sport 2017, 34, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Glass, D.J. PI3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr. Top. Microbiol. Immunol. 2010, 346, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Fry, C.S.; Lee, J.D.; Mula, J.; Kirby, T.J.; Jackson, J.R.; Liu, F.; Yang, L.; Mendias, C.L.; Dupont-Versteegden, E.E.; McCarthy, J.J.; et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med. 2015, 21, 76–80. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Shioi, T.; Zhang, L.; Tarnavski, O.; Sherwood, M.C.; Kang, P.M.; Izumo, S. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2003, 100, 12355–12360. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Konhilas, J.P.; Kelly, D.P.; Leinwand, L.A. Molecular Mechanisms Underlying Cardiac Adaptation to Exercise. Cell Metab. 2017, 25, 1012–1026. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Bugger, H.; Sena, S.; O’Neill, B.T.; Zaha, V.G.; Ilkun, O.; Wright, J.J.; Mazumder, P.K.; Palfreyman, E.; Tidwell, T.J.; et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 2009, 119, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.W.; Lee, S.J.; Kim, S.G. Molecular mechanism of nrf2 activation by oxidative stress. Antioxid. Redox Signal. 2005, 7, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A.R.; Sreekumaran Nair, K. Mitochondrial and skeletal muscle health with advancing age. Mol. Cell. Endocrinol. 2013, 379, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopka, A.R.; Suer, M.K.; Wolff, C.A.; Harber, M.P. Markers of human skeletal muscle mitochondrial biogenesis and quality control: Effects of age and aerobic exercise training. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 371–378. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Horman, S.; Browne, G.; Krause, U.; Patel, J.; Vertommen, D.; Bertrand, L.; Lavoinne, A.; Hue, L.; Proud, C.; Rider, M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr. Biol. 2002, 12, 1419–1423. [Google Scholar] [CrossRef]
- Hoppe, S.; Bierhoff, H.; Cado, I.; Weber, A.; Tiebe, M.; Grummt, I.; Voit, R. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc. Natl. Acad. Sci. USA 2009, 106, 17781–17786. [Google Scholar] [CrossRef] [Green Version]
- Richter, E.A.; Ruderman, N.B. AMPK and the biochemistry of exercise: Implications for human health and disease. Biochem. J. 2009, 418, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Kjobsted, R.; Hingst, J.R.; Fentz, J.; Foretz, M.; Sanz, M.N.; Pehmoller, C.; Shum, M.; Marette, A.; Mounier, R.; Treebak, J.T.; et al. AMPK in skeletal muscle function and metabolism. FASEB J. 2018, 32, 1741–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.S.; Jeon, J.; An, J.J.; Yi, H.K. Interval running training improves age-related skeletal muscle wasting and bone loss: Experiments with ovariectomized rats. Exp. Physiol. 2019, 104, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.K.; Matthews, K.G.; Oldham, J.M.; Jeanplong, F.; Falconer, S.J.; Bass, J.J.; Senna-Salerno, M.; Bracegirdle, J.W.; McMahon, C.D. Translational signalling, atrogenic and myogenic gene expression during unloading and reloading of skeletal muscle in myostatin-deficient mice. PLoS ONE 2014, 9, e94356. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.S.; Schuler, G.; Adams, V. Skeletal muscle wasting in cachexia and sarcopenia: Molecular pathophysiology and impact of exercise training. J. Cachexia Sarcopenia Muscle 2015, 6, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.J. Signaling pathways perturbing muscle mass. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 225–229. [Google Scholar] [CrossRef]
- Choi, D.H.; Yang, J.; Kim, Y.S. Rapamycin suppresses postnatal muscle hypertrophy induced by myostatin-inhibition accompanied by transcriptional suppression of the Akt/mTOR pathway. Biochem. Biophys. Rep. 2019, 17, 182–190. [Google Scholar] [CrossRef]
- Degens, H. The role of systemic inflammation in age-related muscle weakness and wasting. Scand. J. Med. Sci. Sports 2010, 20, 28–38. [Google Scholar] [CrossRef]
- Brown, E.L.; Foletta, V.C.; Wright, C.R.; Sepulveda, P.V.; Konstantopoulos, N.; Sanigorski, A.; Della Gatta, P.; Cameron-Smith, D.; Kralli, A.; Russell, A.P. PGC-1alpha and PGC-1beta Increase Protein Synthesis via ERRalpha in C2C12 Myotubes. Front. Physiol. 2018, 9, 1336. [Google Scholar] [CrossRef] [PubMed]
- Merry, T.L.; Ristow, M. Mitohormesis in exercise training. Free Radic. Biol. Med. 2016, 98, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Maughan, R.J.; Burke, L.M.; Dvorak, J.; Larson-Meyer, D.E.; Peeling, P.; Phillips, S.M.; Rawson, E.S.; Walsh, N.P.; Garthe, I.; Geyer, H.; et al. IOC Consensus Statement: Dietary Supplements and the High-Performance Athlete. Int. J. Sport Nutr. Exerc. Metab. 2018, 28, 104–125. [Google Scholar] [CrossRef] [PubMed]
- Maughan, R.J.; Shirreffs, S.M.; Vernec, A. Making Decisions About Supplement Use. Int. J. Sport Nutr. Exerc. Metab. 2018, 28, 212–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S.; Spoletini, I.; Rosano, G. Exercise-induced skeletal muscle remodeling and metabolic adaptation: Redox signaling and role of autophagy. Antioxid. Redox Signal. 2014, 21, 154–176. [Google Scholar] [CrossRef] [PubMed]
- Done, A.J.; Gage, M.J.; Nieto, N.C.; Traustadottir, T. Exercise-induced Nrf2-signaling is impaired in aging. Free Radic. Biol. Med. 2016, 96, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, C.G.; Qi, Z.; Cui, D.; Ding, S. Acute exercise stress promotes Ref1/Nrf2 signalling and increases mitochondrial antioxidant activity in skeletal muscle. Exp. Physiol. 2016, 101, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Elokda, A.S.; Nielsen, D.H. Effects of exercise training on the glutathione antioxidant system. Eur. J. Cardiovasc. Prev. Rehabil. 2007, 14, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K.; Gounder, S.S.; Davidson, C.J.; Boeheme, C.; Hoidal, J.R.; Wang, L.; Rajasekaran, N.S. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 2012, 52, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holley, A.K.; Dhar, S.K.; St Clair, D.K. Manganese superoxide dismutase vs. p53: Regulation of mitochondrial ROS. Mitochondrion 2010, 10, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, C.G.; Qi, Z.; Cui, D.; Ding, S. Acute exercise induced mitochondrial H(2)O(2) production in mouse skeletal muscle: Association with p(66Shc) and FOXO3a signaling and antioxidant enzymes. Oxidative Med. Cell. Longev. 2015, 2015, 536456. [Google Scholar] [CrossRef] [PubMed]
- Crilly, M.J.; Tryon, L.D.; Erlich, A.T.; Hood, D.A. The role of Nrf2 in skeletal muscle contractile and mitochondrial function. J. Appl. Physiol. 2016, 121, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Komine, S.; Warabi, E.; Akiyama, K.; Ishii, A.; Ishige, K.; Mizokami, Y.; Kuga, K.; Horie, M.; Miwa, Y.; et al. Nuclear factor (erythroid derived 2)-like 2 activation increases exercise endurance capacity via redox modulation in skeletal muscles. Sci. Rep. 2017, 7, 12902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Done, A.J.; Newell, M.J.; Traustadottir, T. Effect of exercise intensity on Nrf2 signalling in young men. Free Radic. Res. 2017, 51, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.F.; Guo, Y.; Li, J.W.; Yuan, K. Antifatigue Effect of Luteolin-6-C-Neohesperidoside on Oxidative Stress Injury Induced by Forced Swimming of Rats through Modulation of Nrf2/ARE Signaling Pathways. Oxidative Med. Cell. Longev. 2017, 2017, 3159358. [Google Scholar] [CrossRef] [PubMed]
- Merry, T.L.; Ristow, M. Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and the anti-oxidant response in mice. J. Physiol. 2016, 594, 5195–5207. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.R.; Narasimhan, M.; Shanmugam, G.; Hong, J.; Devarajan, A.; Palaniappan, S.; Zhang, J.; Halade, G.V.; Darley-Usmar, V.M.; Hoidal, J.R.; et al. Abrogation of Nrf2 impairs antioxidant signaling and promotes atrial hypertrophy in response to high-intensity exercise stress. J. Transl. Med. 2016, 14, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pala, R.; Orhan, C.; Tuzcu, M.; Sahin, N.; Ali, S.; Cinar, V.; Atalay, M.; Sahin, K. Coenzyme Q10 Supplementation Modulates NFkappaB and Nrf2 Pathways in Exercise Training. J. Sports Sci. Med. 2016, 15, 196–203. [Google Scholar]
- Anedda, A.; Lopez-Bernardo, E.; Acosta-Iborra, B.; Saadeh Suleiman, M.; Landazuri, M.O.; Cadenas, S. The transcription factor Nrf2 promotes survival by enhancing the expression of uncoupling protein 3 under conditions of oxidative stress. Free Radic. Biol. Med. 2013, 61, 395–407. [Google Scholar] [CrossRef]
- Callahan, L.A.; Nethery, D.; Stofan, D.; DiMarco, A.; Supinski, G. Free radical-induced contractile protein dysfunction in endotoxin-induced sepsis. Am. J. Respir. Cell Mol. Biol. 2001, 24, 210–217. [Google Scholar] [CrossRef]
- Narasimhan, M.; Hong, J.; Atieno, N.; Muthusamy, V.R.; Davidson, C.J.; Abu-Rmaileh, N.; Richardson, R.S.; Gomes, A.V.; Hoidal, J.R.; Rajasekaran, N.S. Nrf2 deficiency promotes apoptosis and impairs PAX7/MyoD expression in aging skeletal muscle cells. Free Radic. Biol. Med. 2014, 71, 402–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenz, T.; Diaz, F.; Hernandez, D.; Moraes, C.T. Endurance exercise is protective for mice with mitochondrial myopathy. J. Appl. Physiol. 2009, 106, 1712–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iversen, N.; Krustrup, P.; Rasmussen, H.N.; Rasmussen, U.F.; Saltin, B.; Pilegaard, H. Mitochondrial biogenesis and angiogenesis in skeletal muscle of the elderly. Exp. Gerontol. 2011, 46, 670–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballmann, C.; McGinnis, G.; Peters, B.; Slivka, D.; Cuddy, J.; Hailes, W.; Dumke, C.; Ruby, B.; Quindry, J. Exercise-induced oxidative stress and hypoxic exercise recovery. Eur. J. Appl. Physiol. 2014, 114, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Gutierrez, C.; Sepulveda, C.; Caballero, J.; Palomo, I.; Fuentes, E. Study of the interactions between Edaglitazone and Ciglitazone with PPARgamma and their antiplatelet profile. Life Sci. 2017, 186, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, D.L.; Silva, L.A.; Tromm, C.B.; da Rosa, G.L.; Silveira, P.C.; de Souza, C.T.; Latini, A.; Pinho, R.A. Impact of different resistance training protocols on muscular oxidative stress parameters. Appl. Physiol. Nutr. Metab. 2012, 37, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Iavicoli, I.; Calabrese, V. Hormesis: Its impact on medicine and health. Hum. Exp. Toxicol. 2013, 32, 120–152. [Google Scholar] [CrossRef]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr Opin Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.L.; Gomez-Cabrera, M.C.; Vina, J. Exercise and hormesis: Activation of cellular antioxidant signaling pathway. Ann. N. Y. Acad. Sci. 2006, 1067, 425–435. [Google Scholar] [CrossRef]
- Lawler, J.M.; Rodriguez, D.A.; Hord, J.M. Mitochondria in the middle: Exercise preconditioning protection of striated muscle. J. Physiol. 2016, 594, 5161–5183. [Google Scholar] [CrossRef] [PubMed]
- Wafi, A.M.; Hong, J.; Rudebush, T.L.; Yu, L.; Hackfort, B.; Wang, H.; Schultz, H.D.; Zucker, I.H.; Gao, L. Curcumin improves exercise performance of mice with coronary artery ligation-induced HFrEF: Nrf2 and antioxidant mechanisms in skeletal muscle. J. Appl. Physiol. 2019, 126, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.G.R.; Hawke, T.J.P.D. From matrices to mitochondria: Emerging roles and regulation of the striated muscle cytoskeleton. Am. J. Physiol. Cell Physiol. 2019, 316, C655–C656. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.H.; Schachner, D.; Zimmermann, K.; Dirsch, V.M. Glucose availability is a decisive factor for Nrf2-mediated gene expression. Redox Biol. 2013, 1, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef]
- Uruno, A.; Yagishita, Y.; Katsuoka, F.; Kitajima, Y.; Nunomiya, A.; Nagatomi, R.; Pi, J.; Biswal, S.S.; Yamamoto, M. Nrf2-Mediated Regulation of Skeletal Muscle Glycogen Metabolism. Mol. Cell. Biol. 2016, 36, 1655–1672. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, A.; Kitteringham, N.R.; Jenkins, R.E.; Goldring, C.; Higgins, L.; Yamamoto, M.; Hayes, J.; Park, B.K. Analysis of the role of Nrf2 in the expression of liver proteins in mice using two-dimensional gel-based proteomics. Pharmacol. Rep. PR 2012, 64, 680–697. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Baird, L.; Holmstrom, K.M.; Meyer, C.J.; Abramov, A.Y. The spatiotemporal regulation of the Keap1-Nrf2 pathway and its importance in cellular bioenergetics. Biochem. Soc. Trans. 2015, 43, 602–610. [Google Scholar] [CrossRef]
- Svensson, M.; Lexell, J.; Deierborg, T. Effects of Physical Exercise on Neuroinflammation, Neuroplasticity, Neurodegeneration, and Behavior: What We Can Learn From Animal Models in Clinical Settings. Neurorehabilit. Neural Repair 2015, 29, 577–589. [Google Scholar] [CrossRef]
- Sallam, N.; Laher, I. Exercise Modulates Oxidative Stress and Inflammation in Aging and Cardiovascular Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 7239639. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Pathway/Enzimatic System | Activity |
|---|---|
| Glutathione synthesis and regeneration: | |
| -GCL: GCLM/GCLC complex | Carrying out glutathione synthesis forms glutamate and cysteine |
| -GPx | Detoxification of H2O2 |
| -GR | Reduction of GSSG to GSH |
| -XCT | Transports cysteine to the cell to be reduced to cysteine from GSH |
| Phase-II detoxifying enzymes: | |
| -HO-1 | Degradation of the heme group gives rise to biliverdin, free iron, and carbon monoxide |
| -UGT | Glucoronidation: conjugation of glucuronic acid |
| -SULT | Sulfonation: the addition of sulfuryl groups donated by 3′-PhosphoAdenosine-5′-PhosphoSulfate (PAPS) to hydroxyl or amine groups |
| Expression of NADPH- producing enzymes: | |
| -G6PD | Synthesis of NADPH in the PPP pathwa |
| -IDH | Synthesis of NADPH in the conversion ofisocytrate into α-ketoglutarate in the KC |
| -ME1 | Synthesis of NADHP in the conversion of pyruvate into malate in the KC |
| Expression of Thioredoxins: | |
| -TXN1 | Their two active cysteine residues can be oxidized for reducing the oxidized thiols of proteins |
| -TXNRD1 | NADPH-dependent can reduce oxidized TXN |
| Detoxification of quinones: | |
| -NQO1 | These compete with the CYP 450 reductases and convert quinones into more stable molecules (quinoles) |
| -AKR | |
| GCL: glutamate-cysteine ligase | SULT´s: sulfotransferases |
| GCLM: glutamate-cysteine ligase modifier subunit | G6PD: glucose-6-phosphate dehydrogenase |
| GCLC: glutamate-cysteine ligase catalytic subunit | PPP: pentose phosphate pathway |
| GST: glutathione S-transferases | NADPH: nicotinamine dinucleotide phophate |
| GR: glutathione reductase | IDH: isocitrate dehydrogenase |
| XCT: cystine/glutamate transporter | ME1: malic enzyme 1 |
| GSSG: oxidized glutathione | KC: Kreb’s cycle |
| GSH: reducedglutathione | TXN1: thioredoxine 1 |
| GPx: glutathioneperoxidases | TXNRD1: thioredoxine reductase 1 |
| HO-1: heme oxygenase-1 | NQO1: N-quinone oxido reductase 1 |
| UGT: UDP-glucuronosyltransferases | AKR: aldo-ketoreductase |
| KC: Kreb’s cycle |
| Model | Training Protocol | Objective | Results | Reference |
|---|---|---|---|---|
| Male mice Nrf2, WT, and KO aged 3 and 12 months | Free run on wheel for 6–8 weeks. Estimation of revolutions in 24 h and converted into distance (km) | Estimate the role of Nrf2 in biogenesis and mitochondrial content of SkM and the physical performance | Without difference in mitochondrial content ↓mitochondrial respiration, in KO mice: ↑ROS IMF ↓performance, ↑ fatigue. In WT ↑ COX. ET normalized ROS, performance, and respiration in KO | Crilly et al. [117] |
| Male mice 13 weeks of age Nrf2+/+ and Nrf2−/− | AT (5–10 m/min) 3 days prior to administration with SFN ExT on treadmill 5 m/min up to 28 m/min increase every 3 min | Evaluate performance, markers of damage, and OS ExT low conditions of ExT in mice administered SFN pre-treatment | ↑distance covered by Nrf2+/+ SFN, ↓ markers of damage in Nrf2+/+ SFN after the SFN test → protection against muscle damage, regulation of Nrf2, and the antioxidant response. ↓ fatigue due to ↓of OS causing ExT | Oh et al. [118] |
| Young males aged 25 ± 1 years | HIIT Cycling protocol of 30 min Sample taking of blood before and after the session | Determine whether HIIT exercise can more efficiently evaluate Nrf2 than MET in humans | ↑ Nrf2 in HIIT vs. MET ↑ GR activity and response ↑ 8-isoprostanes | Done et al. [119] |
| Male Sprague–Dawley rats aged 20–22 weeks | Exhaustive swimming every day for 3 weeks. After each session, the animals received 20–75 mg of LN or 100 mg of AA | Determine the effect of supplementation with LN on the diminution of fatigue and the modulation of the Nrf2/ARE pathway in a forced swimming model in rats | LN ↑performance resistance exercise normalized metabolic markers. ↓LA and LDH ↑capacity of resistance to the exercise ↑activity of antioxidant enzymes and antioxidant capacity ↓ TNFα, IL-1β, and IL-6 ↑ IL-10 anti-inflammatory in SkM and in liver | Duan et al. [120] |
| Male aged mice ICR/CD-1 | AE at different durations (45, 90, 120, or 150 min) | Evaluate effect of AE on the Ref1/Nrf2 pathway, association with H2O2 and EAS | AE ↑ OS by the Ref1/Nrf2 pathway in time-dependent fashion in linear correlation of the content of H2O2 and the expression of Ref1/Nrf2. ↑GSH and ↑ activity of MnSOD. CuZnSOD not modified | Wang et al. [121] |
| Male mice C57/BL6/SJ aged 15–30 weeks Nrf2+/+ and Nrf2−/− | AT 5 days prior to the study, 5 min (0–9 m/min) 0 degrees of inclination ET included 30–60 min of treadmill running at 10–15 m min−1 at 10% inclination, 4–5 days per week. AE consisted of 1 h of treadmill running at 12 m min, at a 10° inclination | Determine the role of NFE2L2 in AE mitochondrial biogenesis and antioxidant response | ROS and NO regulate the expression of NFE2L2 in SkM cells ↓ NFE2L2 →↓ tolerance to exercise, mitochondrial density, and low SOD activity ↓of markers of mitochondrial biogenesis, citrate synthase, and mtDNA AE, NO, and H2O2→increase of NRF-1 and mtTFA was dependent on NFE2L2 | Merry et al. [122] |
| Male mice aged 20 months Nrf2+/+ and Nrf2−/− | Test of previous resistance ability; 1 week of treadmill running for 10 min; 15–22 m/min; 0–12% inclination. HIES treadmill running for 6 weeks at 20–25 m/min; 12% inclination for 60 min per day | Determine the role of Nrf2 under stress by HIES in atrial cardiomyocyte hypertrophic changes | HIES →↑ markers of the gene expression of hypertrophy of cardiomyocytes (Anf, Bnf, and β-Mhc) in mice Nrf2−/− ↓Gclc, Gsr, and Gstμ, levels of protein of NQO1, Cat, GPX1, GSH in Nrf2−/− after HIES ↑expression of LC3 and ATG7 ↑ ubiquitination of ATG7 →↑ OS | Kumar et al. [123] |
| Male Wistar rats aged 8 weeks | 1 week of adaptation CE: 25 m/min, 45 min/day, 5 days per week during 6 weeks Supplementation of Coenzyme Q10 at doses of 300 mg/kg | Investigate the effect of Coenzyme Q10 or ubiquinone on NFκB, IκB, Nrf2, and HO-1 after CET after 6 weeks of training in sedentary and active rats. | ↓Significant NFκB in muscle, liver, and heart in the group that received Q10 post-training vs. sedentary group. ↑ Nrf2 and HO-1 in muscle, liver, and heart of the active group. ↓plasma triglycerides Without changes in metabolites related with CHO and proteins | Pala et al. [124] |
| Male mice Nrf2+/+ and Nrf2−/− aged 2 months | AE on treadmill; 60 min/day, 14 m/min, 10% inclination, for 2 days. | Determine the impact of the AE in the activation of the Nrf2/ARE pathway and of the EAS system in mouse heart | ↑activation of the Ref1/Nrf2 pathway and of the EAS pathway in mice Nrf2+/+. ↑ OS and ↓ EAS (Cat, NQO1, GCS, GSR, GPx-1, G6PD, GSH) in Nrf2−/− ↑activation of the Ref1/Nrf2 pathway and of the EAS pathway in Nrf2+/+ in old mice vs. Nrf2 AE protects against OS in cardiac muscle in mice | Muthusamy et al. [125] |
| Male mice C57/Bl6/SJ Young (aged ~2 months) and old (aged ≥23 months) Nrf2+/+ and Nrf2−/− | EES: 2 consecutive days on treadmill 90 min/day; 20 m/min; 12% inclination MET: 50 min/day; 10 m/min; 7% inclination min/day for 6 weeks. The protocol included 5 min of ramping at 5 m/min, the speed Increased to 10 m/min/45 min | Evaluate the regulation of Nrf2 depending on age, the antioxidant mechanisms, and redox equilibrium in mouse cardiac muscle Antioxidants under EES and MET conditions | ↑susceptibility in old mice by OS produced by EES Proteins of the ARE antioxidant system ↑ in young mice, with respect to the old mice. ↓ Cat, NQO1 young mice Nrf2−/−, in old mice ↓G6PD, NQO1, cat, HO-1, and GPX1 MET: ↑Nrf2 nuclear and EAS in heart of old mice close to the levels of the young mice | Gounder et al. [34] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vargas-Mendoza, N.; Morales-González, Á.; Madrigal-Santillán, E.O.; Madrigal-Bujaidar, E.; Álvarez-González, I.; García-Melo, L.F.; Anguiano-Robledo, L.; Fregoso-Aguilar, T.; Morales-Gonzalez, J.A. Antioxidant and Adaptative Response Mediated by Nrf2 during Physical Exercise. Antioxidants 2019, 8, 196. https://doi.org/10.3390/antiox8060196
Vargas-Mendoza N, Morales-González Á, Madrigal-Santillán EO, Madrigal-Bujaidar E, Álvarez-González I, García-Melo LF, Anguiano-Robledo L, Fregoso-Aguilar T, Morales-Gonzalez JA. Antioxidant and Adaptative Response Mediated by Nrf2 during Physical Exercise. Antioxidants. 2019; 8(6):196. https://doi.org/10.3390/antiox8060196
Chicago/Turabian StyleVargas-Mendoza, Nancy, Ángel Morales-González, Eduardo Osiris Madrigal-Santillán, Eduardo Madrigal-Bujaidar, Isela Álvarez-González, Luis Fernando García-Melo, Liliana Anguiano-Robledo, Tomás Fregoso-Aguilar, and José A. Morales-Gonzalez. 2019. "Antioxidant and Adaptative Response Mediated by Nrf2 during Physical Exercise" Antioxidants 8, no. 6: 196. https://doi.org/10.3390/antiox8060196