
Lactation Affects Isolated Mitochondria and Its Fatty Acid Composition but Has No Effect on Tissue Protein Oxidation, Lipid Peroxidation or DNA-Damage in Laboratory Mice

 , and
, and
Abstract
:1. Introduction
2. Experimental Section
2.1. Animals and Housing
2.2. Isolation of Liver and Heart Mitochondria
2.3. Measurement of Mitochondrial Respiration Rates
2.4. Fatty Acid Composition of Isolated Mitochondria
2.5. Measurements of DNA Damage
2.6. Measurement of Protein Oxidation
2.7. Measurement of Lipid Peroxidation
2.8. Statistical Analysis
2.9. Ethics
3. Results
3.1. Mitochondrial Respiration
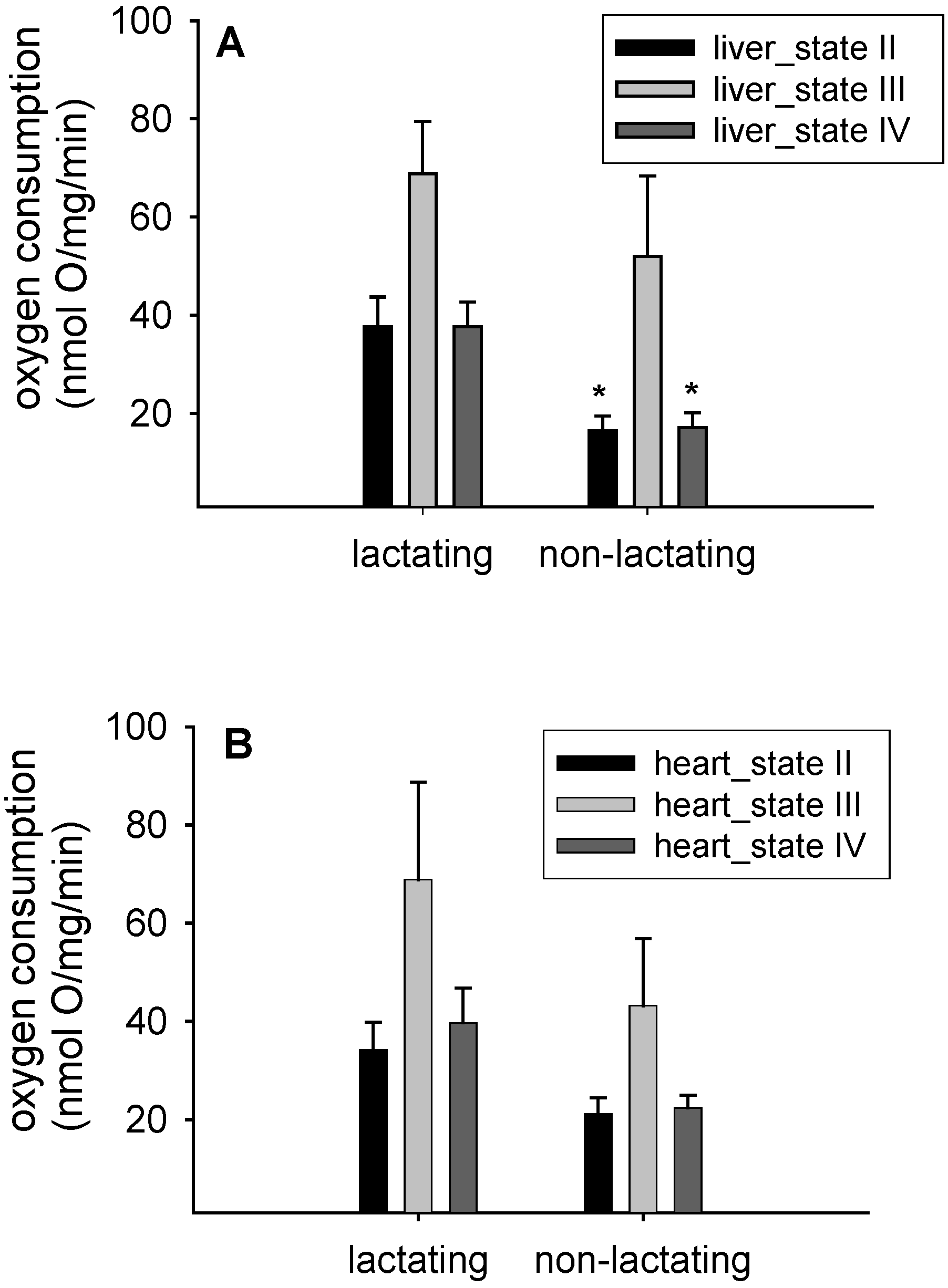

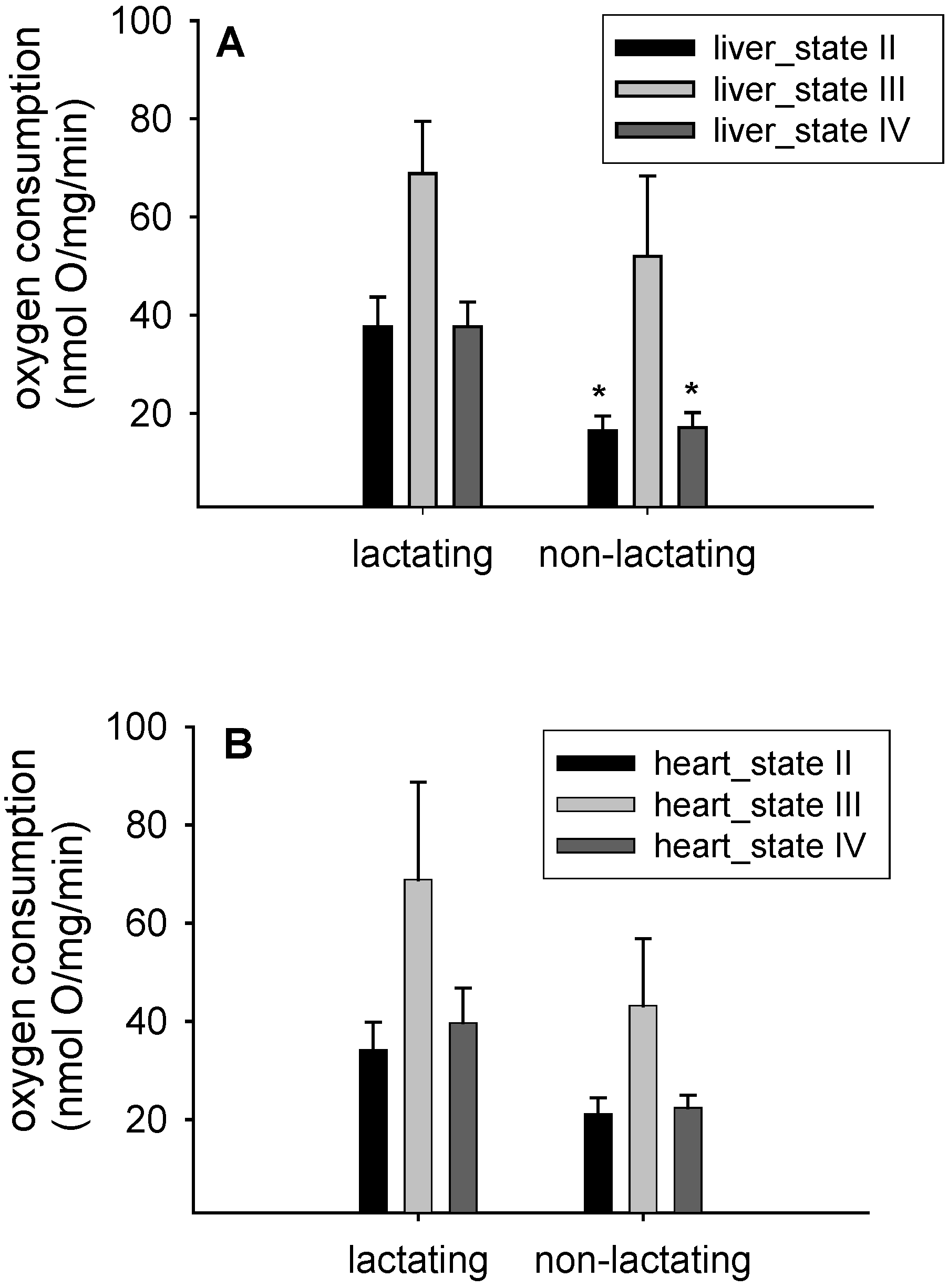{kind=link}
{kind=link}
| Lactating Mice | Non-Lactating Mice | ||
|---|---|---|---|
| Respiratory control ratio (nmol·O/mg/min) | Liver mitochondria | 1.87 ± 1.2 | 3.75 ± 0.98 |
| Heart mitochondria | 1.9 ± 0.78 | 1.96 ± 1.3 | |
| Protein concentration (mg/mL) | Liver mitochondria | 77.6 ± 9.64 | 75.4 ± 5.53 |
| Heart mitochondria | 20.2 ± 1.9 | 26.8 ± 2.4 |
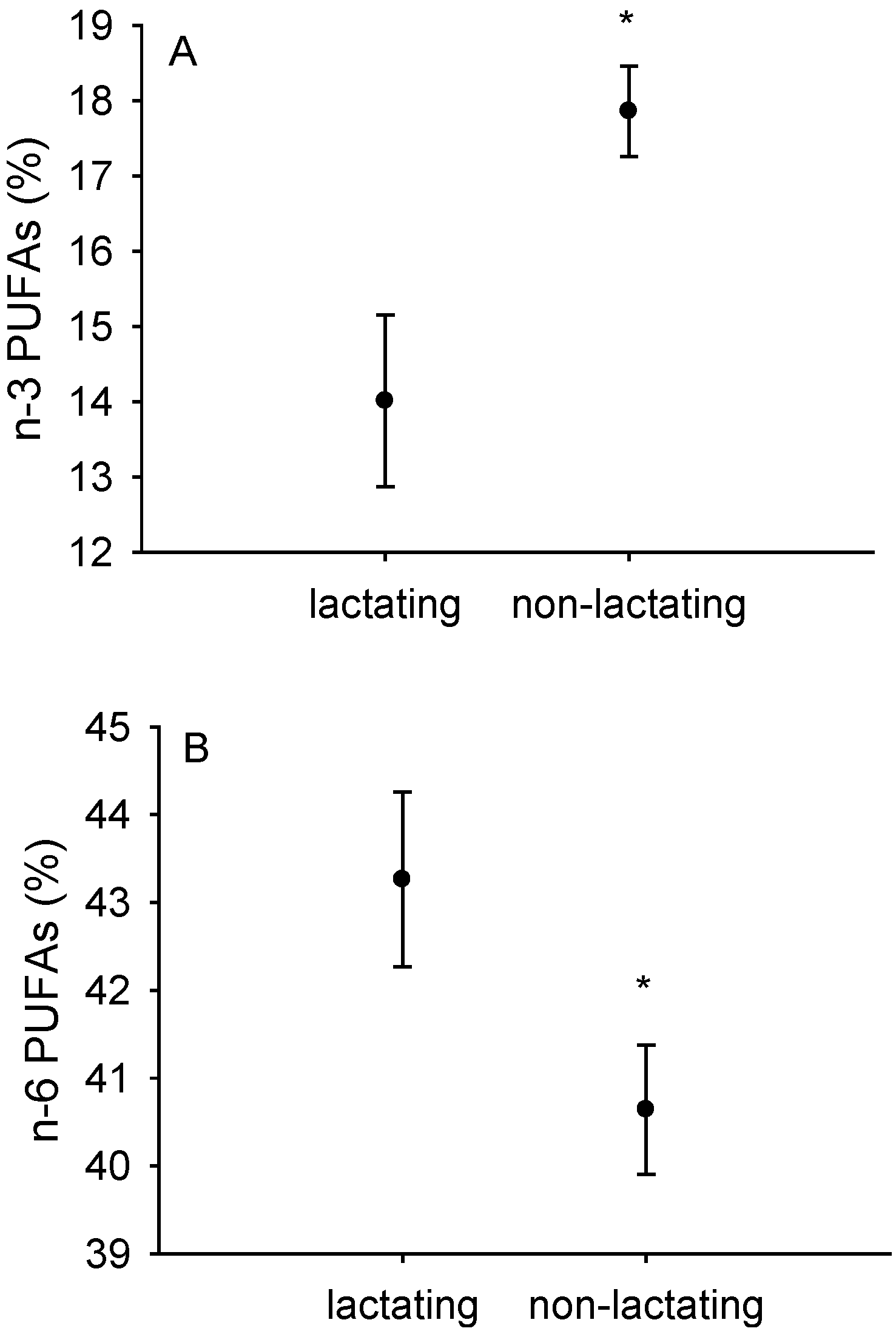
3.2. Fatty Acid Composition of Isolated Liver and Heart Mitochondria
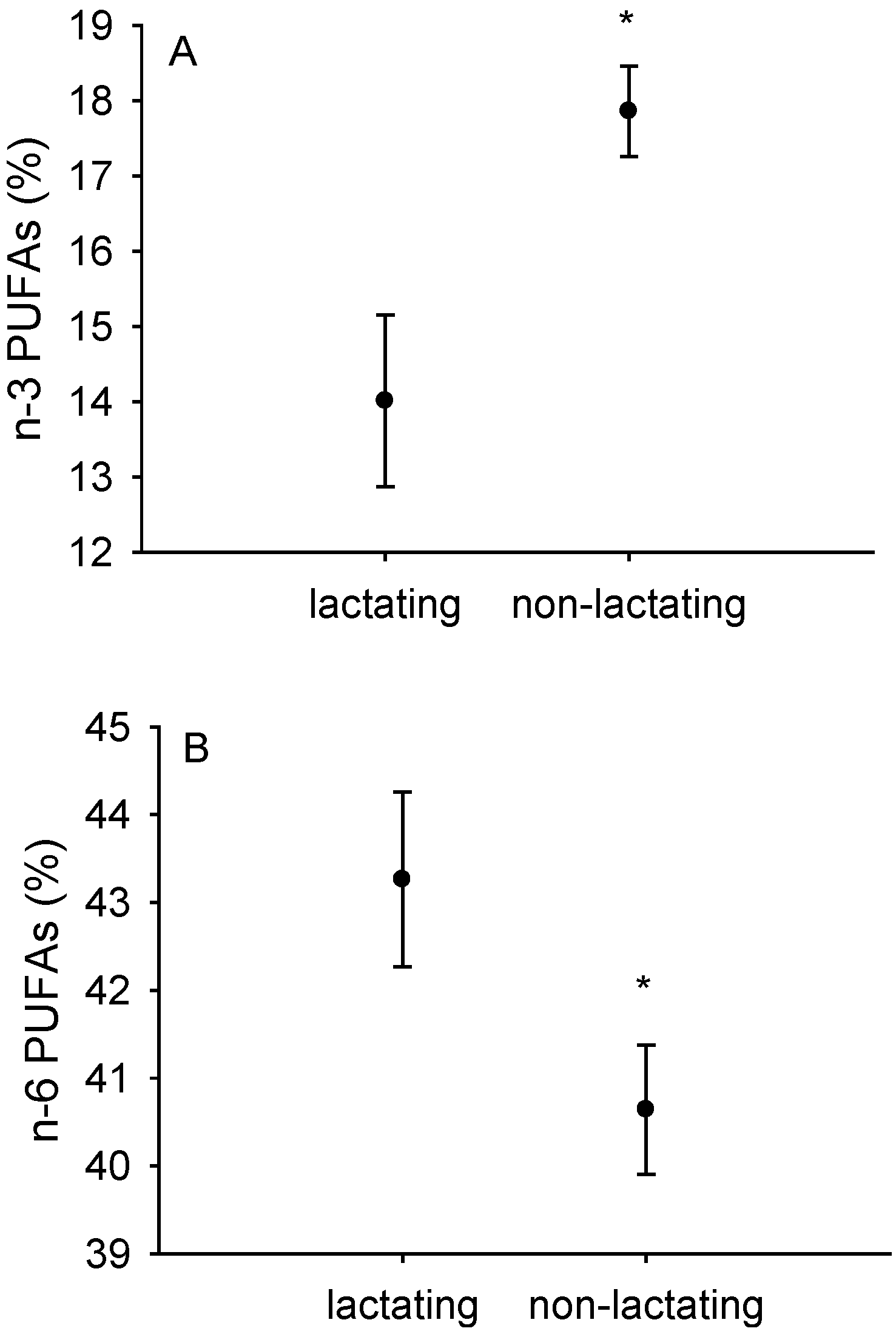
| Liver Mitochondria | Heart Mitochondria | |||
|---|---|---|---|---|
| L | C | L | C | |
| N | 12 | 10 | 12 | 10 |
| C 14:0 | 0.06 ± 0.003 | 0.04 ± 0.0003 | 0.3 ± 0.02 | 0.3 ± 0.003 |
| C 15:0 | 0.03 ± 0.002 | 0.04 ± 0.002 | 0.05 ± 0.003 | 0.05 ± 0.003 |
| C 16:0 | 20.9 ± 0.64 | 19.8 ± 0.4 | 18.2 ± 0.4 | 19.3 ± 1.01 |
| C 17:0 | 0.3 ± 0.01 | 0.3 ± 0.02 | 0.3 ± 0.003 | 0.3 ± 0.01 |
| C 18:0 | 15.4 ± 0.84 | 16.7 ± 0.7 | 24.3 ± 0.09 | 25.1 ± 0.3 |
| C 16:1n-7 | 0.4 ± 0.03 | 0.3 ± 0.03 | 0.6 ± 0.03 | 0.5 ± 0.05 |
| C 18:1n-9 | 5.7 ± 0.32 | 4.4 ± 0.5 | 8.9 ± 0.3 | 5.9 ± 0.4 |
| C 18:2n-6 | 17.2 ± 0.03 | 16.3 ± 0.3 | 19.4 ± 1.3 | 11.6 ± 0.3 |
| C 18:3n-3 | 0.09 ± 0.01 | 0.08 ± 0.01 | 0.13 ± 0.01 | 0.12 ± 0.09 |
| C 20:4n-6 | 26.1 ± 0.04 | 24.4 ± 0.5 | 10.3 ± 0.4 | 6.1 ± 0.24 |
| C 20:5n-3 | 0.69 ± 0.04 | 0.49 ± 0.04 | 0.48 ± 0.06 | 0.24 ± 0.0003 |
| C 22:5n-3 | 0.7 ± 0.03 | 0.5 ± 0.02 | 1.5 ± 0.06 | 1.1 ± 0.22 |
| C 22:6n-3 | 12.5 ± 0.4 | 16.8 ± 0.2 | 15.9 ± 0.6 | 29.7 ± 1.6 |
3.3. DNA Damage
| Lactating Mice | Non-Lactating Mice | |
|---|---|---|
| Tail DNA (%) | 78.9 ± 2.6 | 68.93 ± 2.7 |
| Tail Moment (%) | 133.5 ± 9.3 | 128.4 ± 7.8 |
| Olive Tail Moment (%) | 64.3 ± 4.3 | 59.3 ± 3.04 |
Protein Oxidation
| Brain | Kidney | Lung | Spleen Lymphocytes | |
|---|---|---|---|---|
| Lactating L | ||||
| Protein carbonyls (nmol/mg) | 2.71 ± 0.3 | 1.99 ± 0.4 | 1.96 ± 0.3 | 3.75 ± 0.4 |
| TBARS (nmol/mg) | 45.81 ± 5.2 | 15.498 ± 2.6 | 52.795 ± 5.7 | 47.635 ± 7.7 |
| Non reproducing controls C | ||||
| Protein carbonyls (nmol/mg) | 3.12 ± 0.5 | 2.39 ± 0.5 | 1.71 ± 0.3 | 2.49 ± 0.1 |
| TBARS (nmol/mg) | 53.98 ± 14.4 | 23.17 ± 3.9 | 50.54 ± 4.6 | 36.63 ± 5.3 |
3.4. Lipid Peroxidation (TBARS)
4. Discussion
4.1. Mitochondrial Metabolism and Fatty Acid Composition in Lactation
4.2. Oxidative Stress and Lactation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brand, M.D. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp. Gerontol. 2000, 35, 811–820. [Google Scholar] [CrossRef]
- Speakman, J.R.; Talbot, D.A.; Selman, C.; Snart, S.; McLaren, J.S.; Redman, P.; Krol, E.; Jackson, D.M.; Johnson, M.S.; Brand, M.D. Uncoupled and surviving: Individual mice with high metabolism have greater mitochondrial uncoupling and live longer. Aging Cell 2004, 3, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Gittleman, J.L.; Thompson, S.D. Energy allocation in mammalian reproduction. Am. Zool. 1988, 28, 863–875. [Google Scholar] [CrossRef]
- Speakman, J.R.; Król, E. Maximal heat dissipation capacity and hyperthermia risk: Neglected key factors in the ecology of endotherms. J. Anim. Ecol. 2010, 79, 726–746. [Google Scholar] [CrossRef] [PubMed]
- Król, E.; Martin, S.A.; Huhtaniemi, I.T.; Douglas, A.; Speakman, J.R. Negative correlation between milk production and brown adipose tissue gene expression in lactating mice. J. Exp. Biol. 2011, 214, 4160–4170. [Google Scholar] [CrossRef] [PubMed]
- Gamo, Y.; Troup, C.; Mitchell, S.E.; Hambly, C.; Vaanholt, L.M.; Speakman, J.R. Limits to sustained energy intake. XX. Body temperatures and physical activity of female mice during lactation. J. Exp. Biol. 2013, 216, 3751–3761. [Google Scholar] [CrossRef] [PubMed]
- Pichaud, N.; Garratt, M.; Ballard, J.W.; Brooks, R.C. Physiological adaptations to reproduction. II. Mitochondrial adjustments in livers of lactating mice. J. Exp. Biol. 2013, 216, 2889–2895. [Google Scholar] [CrossRef] [PubMed]
- Valencak, T.G.; Ruf, T. Phospholipid composition and longevity: Lessons from Ames dwarf mice. AGE 2013, 35, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Kühnel, H.; Velde, K.; Dadak, A. Immunomodulation during and after castration under inhalation anaesthetic without genotoxic effects on equine lymphocytes. Res. Vet. Sci. 2012, 92, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, S.J.; Agurell, E. Aspects of design and statistical analysis in the Comet assay. Mutagenesis 2003, 18, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Fagan, J.M.; Sleczka, B.G.; Sohar, I. Quantitation of oxidative damage to tissue proteins. Int. J. Biochem. Cell Biol. 1999, 31, 751–757. [Google Scholar] [CrossRef]
- Vincent, A.M.; McLean, L.L.; Backus, C.; Feldman, E.L. Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. FASEB J. 2005, 19, 638–640. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Maechler, M.; Bolker, B.; Walker, S. lme4: Linear Mixed-Effects Models Using Eigen and S4. R Package version 1.1–7. Available online: http://CRAN.R-project.org/package=lme4 (accessed on 29 October 2015).
- Valencak, T.G.; Tataruch, F.; Ruf, T. Peak energy turnover in lactating European hares: The role of fat reserves. J. Exp. Biol. 2009, 212, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Valencak, T.G.; Wright, P.; Weir, A.; Mitchell, S.E.; Vaanholt, L.M.; Hambly, C.; Król, E.; Speakman, J.R. Limits to sustained energy intake. XXI. Effect of exposing the mother, but not her pups, to a cold environment during lactation in mice. J. Exp. Biol. 2013, 216, 4326–4333. [Google Scholar] [CrossRef] [PubMed]
- Naya, D.E.; Karasov, W.H.; Bozinovic, F. Phenotypic plasticity in laboratory mice and rats: A meta-analysis of current ideas on gut size flexibility. Evol. Ecol. Res. 2007, 9, 1363–1374. [Google Scholar]
- Gutgesell, A.; Ringseis, R.; Schmidt, E.; Brandsch, C.; Stangl, G.I.; Eder, K. Downregulation of peroxisome proliferator-activated receptor α and its coactivators in liver and skeletal muscle mediates the metabolic adaptations during lactation in mice. J. Mol. Endocrinol. 2009, 43, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Valencak, T.G.; Ruf, T. N-3 polyunsaturated fatty acids impair lifespan but have no role for metabolism. Aging Cell 2007, 6, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, A.J.; Faulks, S.C.; Harper, J.M.; Miller, R.A.; Buffenstein, R. Extended longevity of wild-derived mice is associated with peroxidation-resistant membranes. Mech. Ageing Dev. 2006, 127, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Harper, J.M.; Dysko, R.C.; Durkee, S.J.; Austad, S.N. Longer life spans and delayed maturation in wild-derived mice. Exp. Biol. Med. 2002, 227, 500–508. [Google Scholar]
- Garratt, M.; Vasilaki, A.; Stockley, P.; McArdle, F.; Jackson, M.; Hurst, J.L. Is oxidative stress a physiological cost of reproduction? An experimental test in house mice. Proc. R. Soc. B 2011, 278, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R.; Garratt, M. Oxidative stress as a cost of reproduction: Beyond the simplistic trade-off model. Bioessays 2014, 36, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, P.; Metcalfe, N.B.; Torres, R. Oxidative stress as a mediator of life history trade-offs: Mechanisms, measurements and interpretation. Ecol. Lett. 2009, 12, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Brzęk, P.; Książek, A.; Ołdakowski, Ł.; Konarzewski, M. High basal metabolic rate does not elevate oxidative stress during reproduction in laboratory mice. J. Exp. Biol. 2014, 217, 1504–1509. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Lin, J.T.; Zheng, W.H.; Cao, J.; Zhao, Z.J. Energy intake, oxidative stress and antioxidant in mice during lactation. Zool. Res. 2015, 36, 95–102. [Google Scholar] [PubMed]
- Garratt, M.; Pichaud, N.; King, E.D.A.; Brooks, R.C. Physiological adaptations to reproduction. I. Experimentally increasing litter size enhances aspects of antioxidant defence but does not cause oxidative damage in mice. J. Exp. Biol. 2013, 216, 2879–2888. [Google Scholar] [CrossRef] [PubMed]
- Blount, J.D.; Vitikainen, E.I.; Stott, I.; Cant, M.A. Oxidative shielding and the cost of reproduction. Biol. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valencak, T.G.; Raith, J.; Staniek, K.; Gille, L.; Strasser, A. Lactation Affects Isolated Mitochondria and Its Fatty Acid Composition but Has No Effect on Tissue Protein Oxidation, Lipid Peroxidation or DNA-Damage in Laboratory Mice. Antioxidants 2016, 5, 2. https://doi.org/10.3390/antiox5010002
Valencak TG, Raith J, Staniek K, Gille L, Strasser A. Lactation Affects Isolated Mitochondria and Its Fatty Acid Composition but Has No Effect on Tissue Protein Oxidation, Lipid Peroxidation or DNA-Damage in Laboratory Mice. Antioxidants. 2016; 5(1):2. https://doi.org/10.3390/antiox5010002
Chicago/Turabian StyleValencak, Teresa G., Johannes Raith, Katrin Staniek, Lars Gille, and Alois Strasser. 2016. "Lactation Affects Isolated Mitochondria and Its Fatty Acid Composition but Has No Effect on Tissue Protein Oxidation, Lipid Peroxidation or DNA-Damage in Laboratory Mice" Antioxidants 5, no. 1: 2. https://doi.org/10.3390/antiox5010002