Oxidative Stress Is Associated with Neuroinflammation in Animal Models of HIV-1 Tat Neurotoxicity

Abstract
:1. Introduction
2. Material and Methods
2.1. Animals
2.2. Reagents
2.3. Antibodies
2.4. Vector Production
2.5. Experimental Design
2.5.1. Tat Injection
2.5.2. Injection of SV(Tat)
2.5.3. Challenge with Tat after Administration of SV(GPx1) or/and SV(SOD1)
2.6. In Vivo Injection of Tat and Vectors
2.7. Procedure for Harvesting the Tissue
2.8. Immunocytochemistry
2.9. Staining of Neurons Using NeuroTrace
2.10. TUNEL Assay
2.11. Morphometry
2.12. Measurement of Malondialdehyde
2.13. Statistical Analysis
3. Results
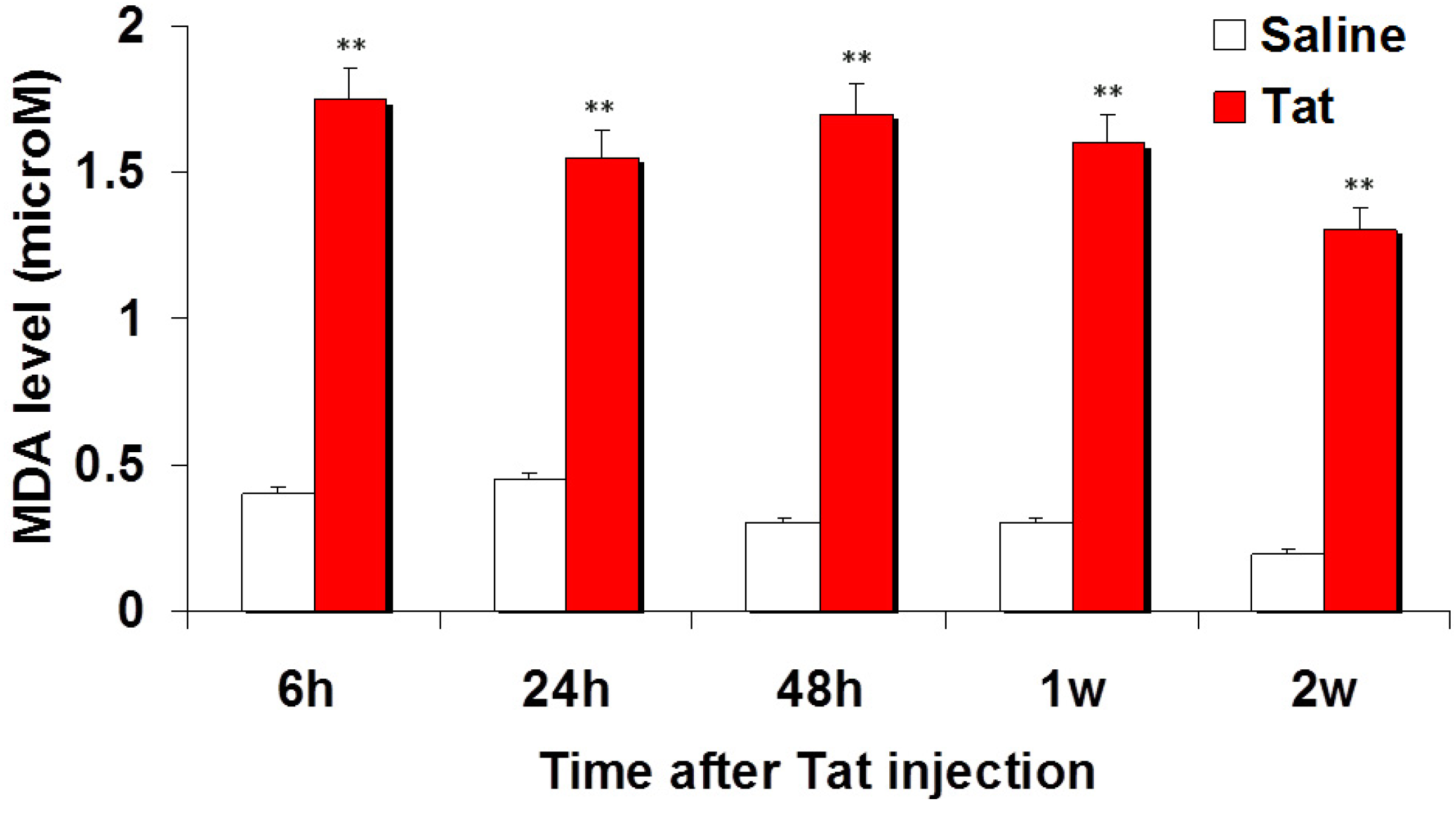
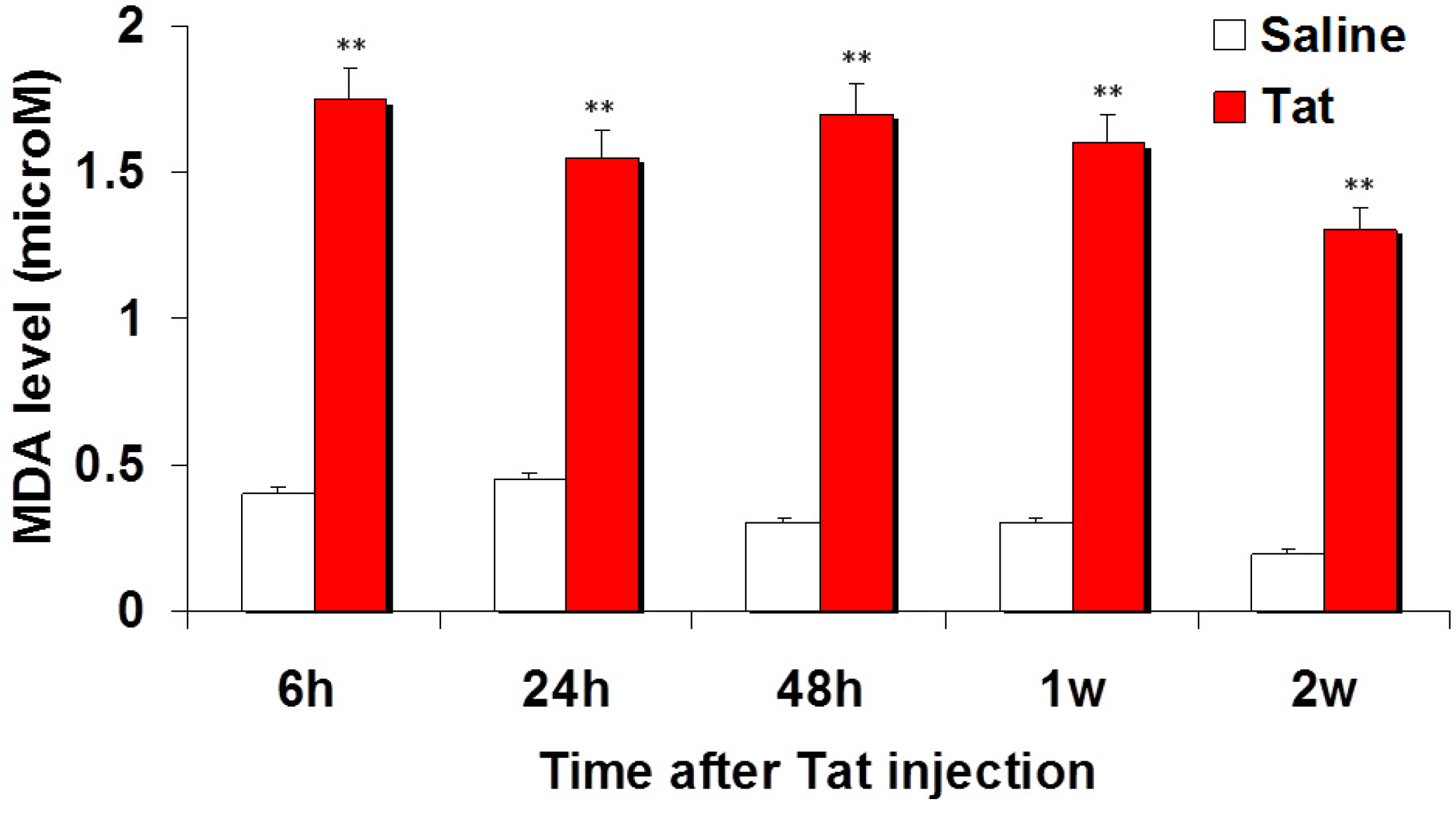
3.1. Injection of Tat Induces Oxidative Damage
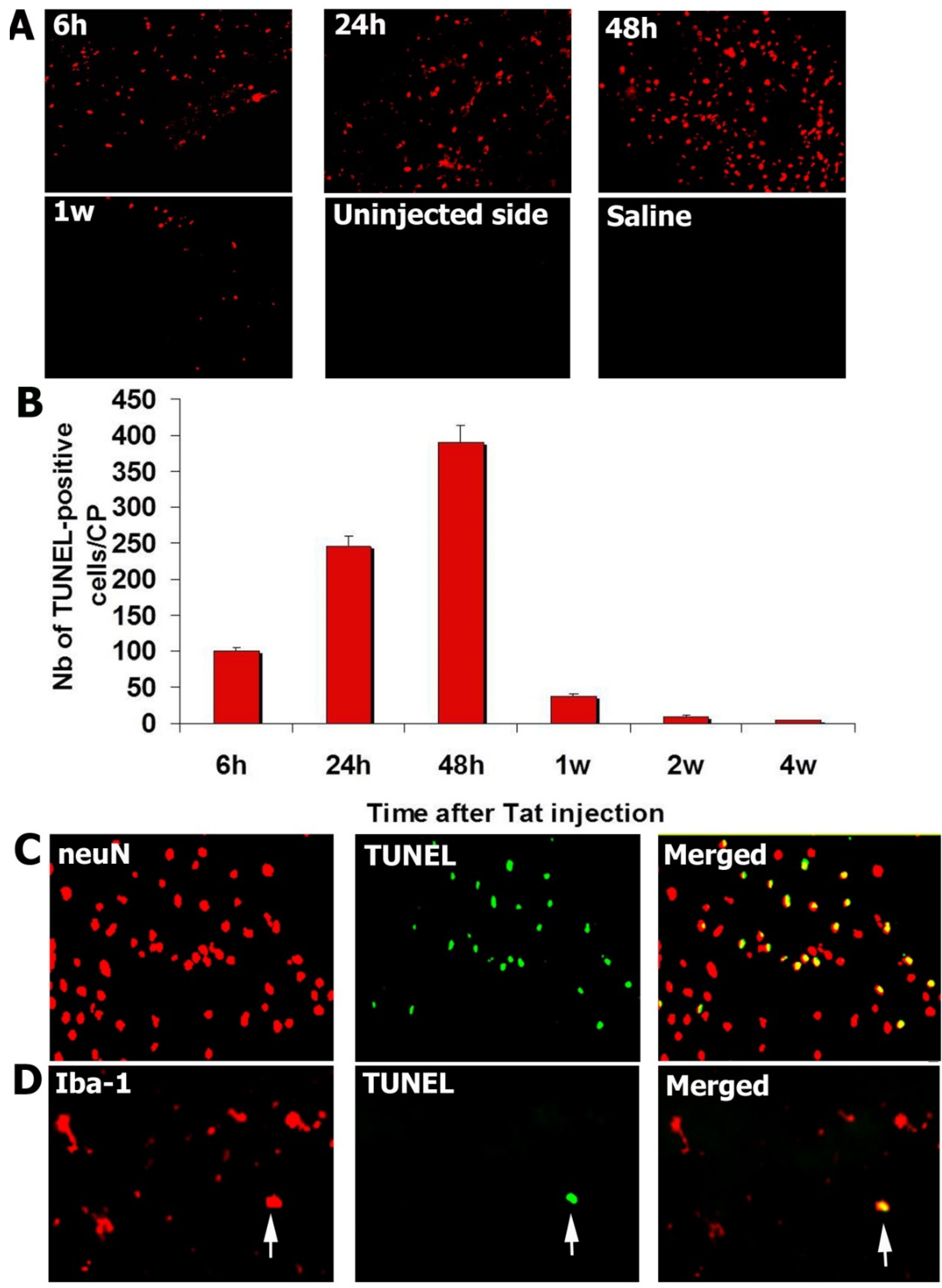
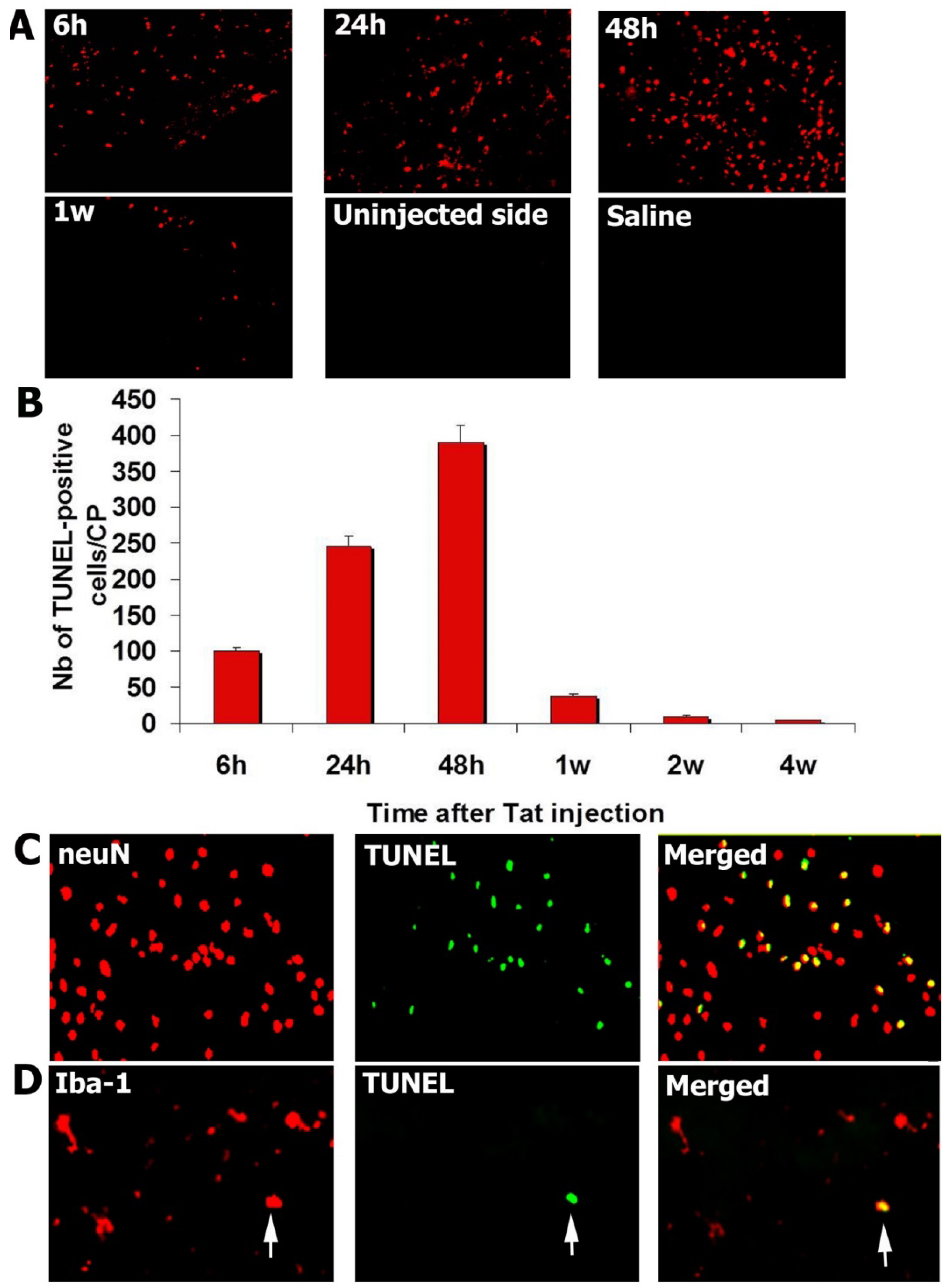
3.2. Injection of Tat into the CP Elicits Apoptosis
3.3. Tat-Induced Apoptotic Cells Are Mainly Neurons
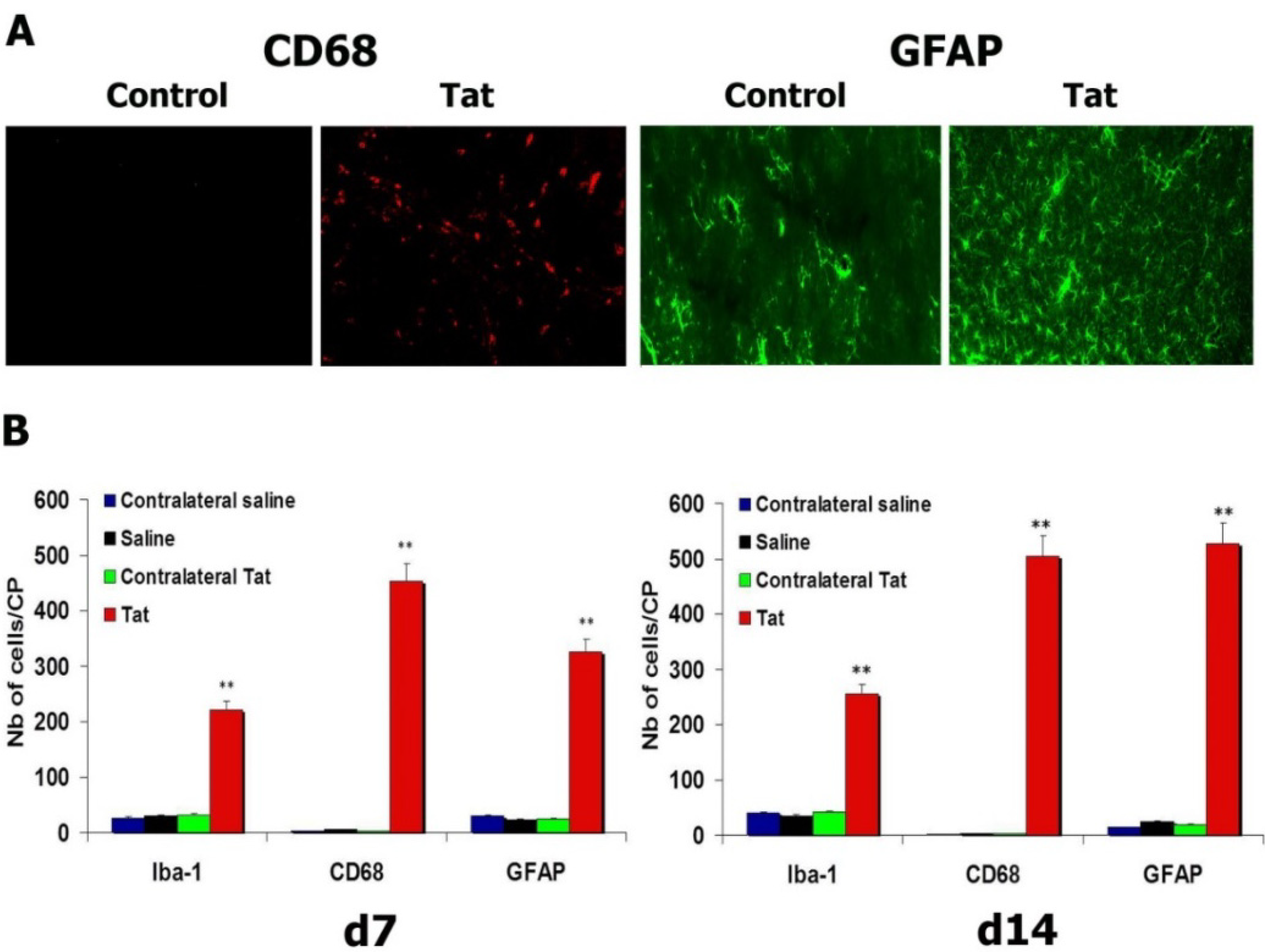
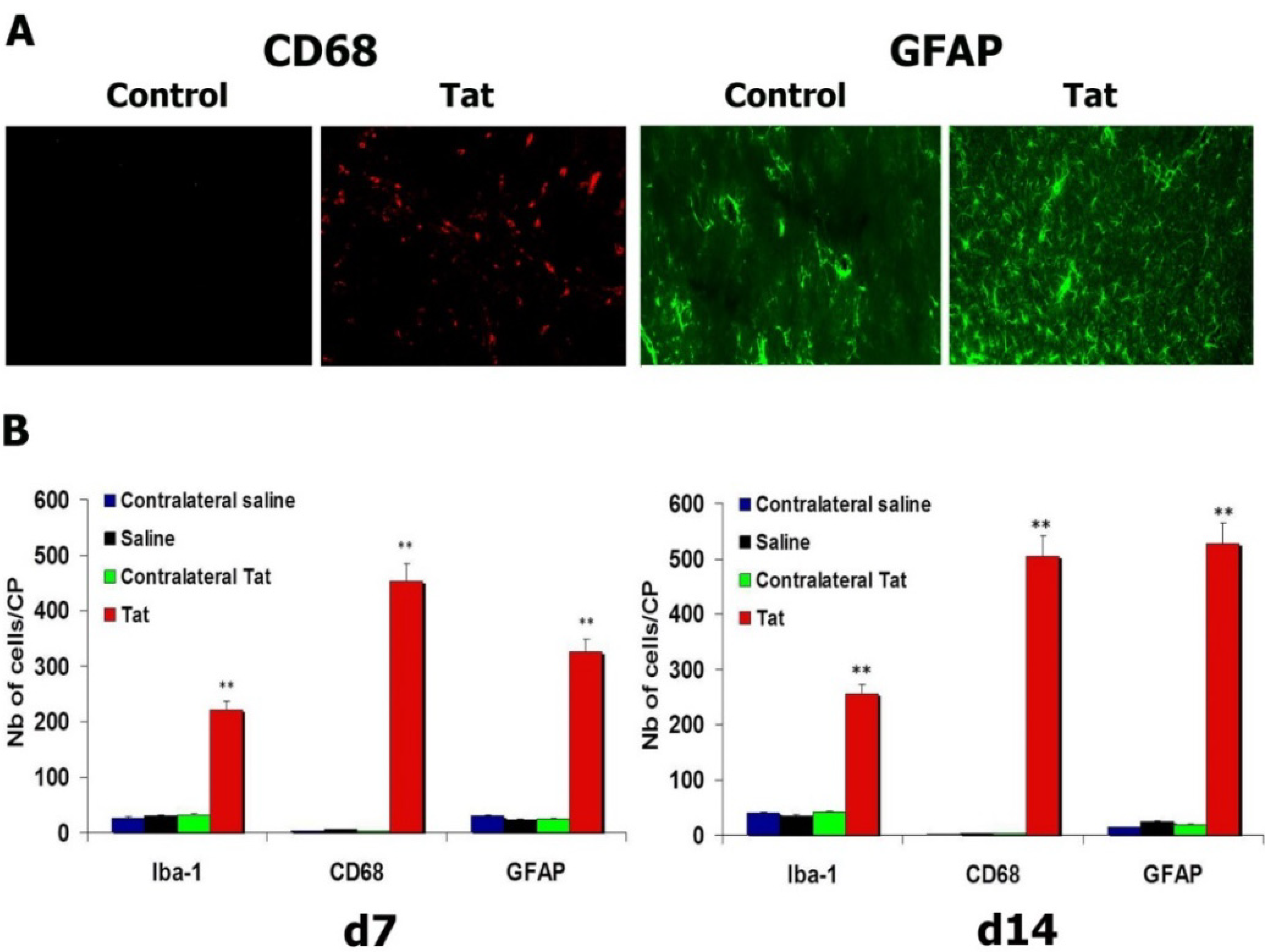
3.4. HIV-1 Tat Triggers Increases in Microglial Cells and Astrocytes
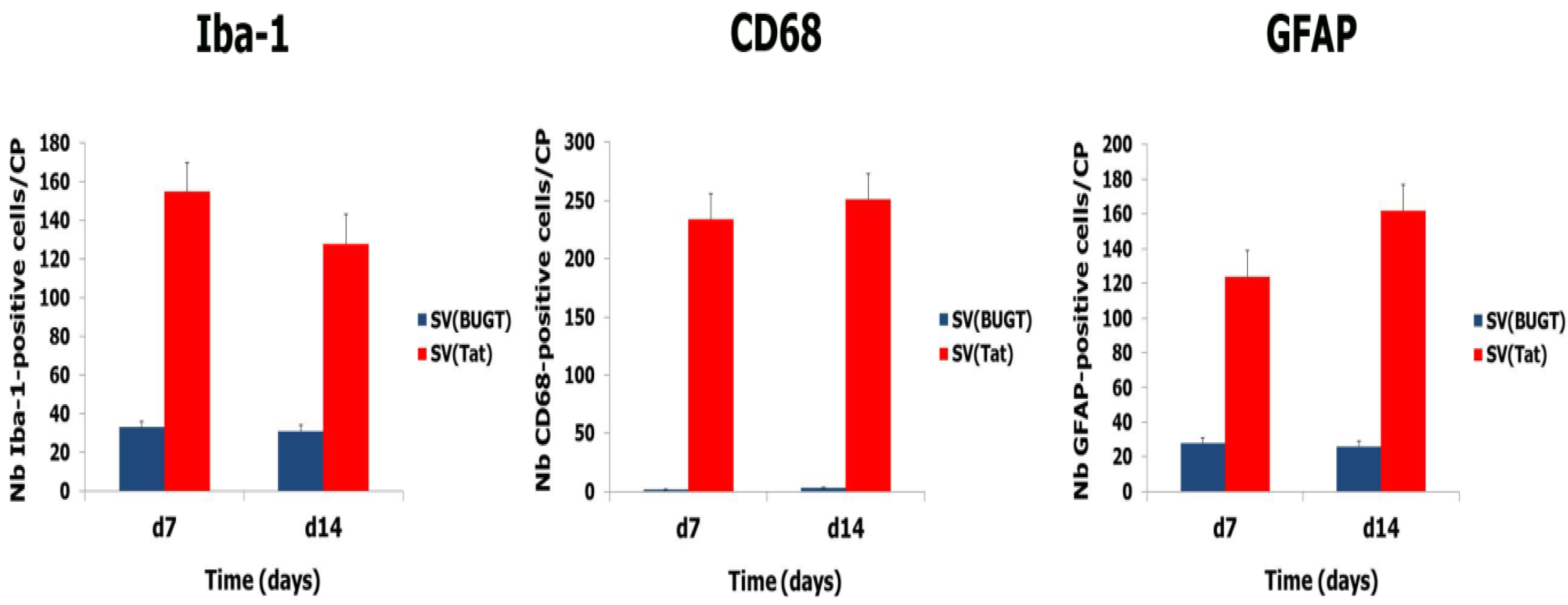
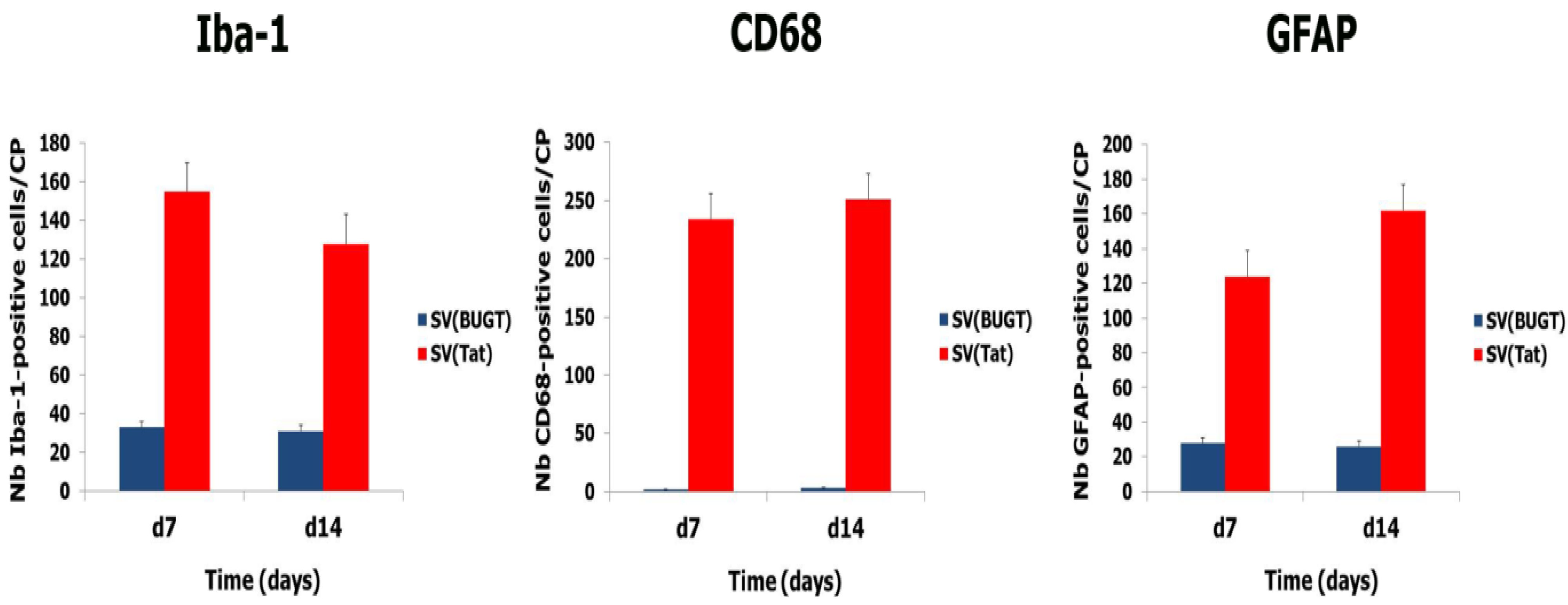
3.5. Transgene Expression of Tat by Intra-CP Injection of SV(Tat) Induces Oxidative Stress, Apoptosis and Neuroinflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Days/Tat-Induced Abnormalities | Nb Tat-Positive Cells/Area | Nb TUNEL-Positive Cells/Area | Neuron Loss (% Compared to Contralateral Area) | MDA (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SV(BUGT) | SV(Tat) | P | SV(BUGT) | SV(Tat) | P | SV(BUGT) | SV(Tat) | P | SV(BUGT) | SV(Tat) | P | |
| d7 | - | 39.4 ± 4.2 | <0.01 | 2.1 ± 1.8 | 42.7 ± 4.8 | <0.01 | 1.9 ± 0.1 | 26.7 ± 3.2 | <0.01 | NA | NA | - |
| d14 | - | 48.7 ± 3.8 | <0.01 | - | 47.2 ± 4.5 | <0.01 | - | 30.8 ± 2.9 | <0.01 | 0.4 ± 0.03 | 1.4 ± 0.1 | <0.01 |
| d28 | - | 28.5 ± 3.4 | <0.01 | - | 24.6 ± 3.1 | <0.01 | - | 34.3 ± 3.6 | <0.01 | NA | NA | - |
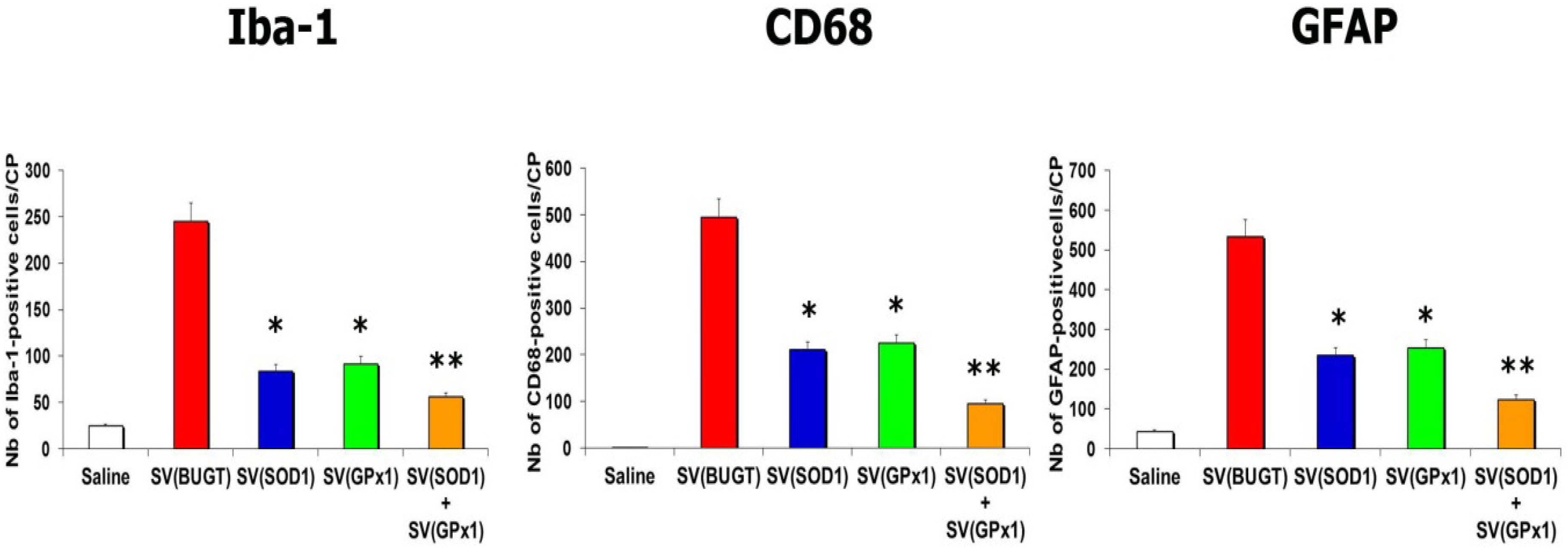
3.6. Overexpression of Antioxidant Enzymes Protects against Tat-Induced Lesions
4. Discussion
| Vector | Saline | SV(BUGT) | SV(SOD1) | SV(GPx1) | SV(SOD1) + SV(GPx1) |
|---|---|---|---|---|---|
| MDA level (μM) | 0.38 ± 0.02 | 1.66 ± 1.5 | 0.83 ± 0.1 | 0.42 ± 0.05 | 0.38 ± 0.04 |
| P value | 0.01 | - | 0.05 | 0.01 | 0.01 |
| Nb of TUNEL-positive cells/CP | 2 ± 0.01 | 382 ± 41 | 232 ± 28 | 255 ± 28 | 52 ± 6 |
| P value | 0.01 | - | 0.05 | 0.05 | 0.01 |

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Broughton, B.R.S.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 11, e331–e339. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995, 38, 357–366. [Google Scholar] [CrossRef]
- Reddy, P.H.; Reddy, T.P. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr. Alzheimer Res. 2011, 8, 393–409. [Google Scholar] [CrossRef]
- Smith, M.A.; Sayre, L.M.; Monnier, V.M.; Perry, G. Radical ageing in Alzheimer’s disease. Trends Neurosci. 1995, 18, 172–176. [Google Scholar] [CrossRef]
- Cao, W.; Carney, J.M.; Duchon, A.; Floyd, R.A.; Chevion, M. Oxygen free radicals involvement in ischemia and reperfusion of the brain injury to brain. Neurosci. Lett. 1998, 88, 233–238. [Google Scholar]
- Montoliu, C.; Valles, S.; Renau-Piqueras, J.; Guerri, C. Ethanol-induced oxygen radical formation and lipid peroxidation in rat brain: Effect of chronic alcohol consumption. J. Neurochem. 1994, 63, 1855–1862. [Google Scholar] [PubMed]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A.; Markesberry, W.R. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef]
- Nelson, P.T.; Soma, L.A.; Lavi, E. Microglia in diseases of the central nervous system. Ann. Med. 2002, 34, 491–500. [Google Scholar] [CrossRef]
- Liu, B.; Hong, J.S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.J.; Zhou, C.; Gravanis, I.; Rogove, A.D.; Wu, Y.P.; Bogenhagen, D.F.; Tsirka, S.E. Proteolytic activation of monocyte chemoattractant protein-1 by plasmin underlies excitotoxic neurodegeneration in mice. J. Neurosci. 2007, 27, 1738–1745. [Google Scholar] [CrossRef]
- Ekdahl, C.T.; Claasen, J.H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef]
- Cowell, R.M.; Xu, H.; Galasso, J.M.; Silverstein, F.S. Hypoxic-ischemic injury induces macrophage inflammatory protein-1 alpha expression in immature rat brain. Stroke 2002, 33, 795–801. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Anti-inflammatory drugs in the fight against Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1996, 777, 213–220. [Google Scholar] [CrossRef]
- Perry, V.H.; Newwan, T.A.; Cunningham, C. The impact of systemic infection on the progression of neurodegenerative disease. Nat. Rev. Neurosci. 2003, 4, 103–112. [Google Scholar] [CrossRef]
- McArthur, J.C.; Brew, B.J.; Nath, A. Neurological complications of HIV infection. Lancet Neurol. 2005, 4, 543–555. [Google Scholar] [CrossRef]
- Nath, A.; Sacktor, N. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Curr. Opin. Neurol. 2006, 19, 358–361. [Google Scholar] [CrossRef]
- Ances, B.M.; Ellis, R.J. Dementia and neurocognitive disorders due to HIV-1 infection. Semin. Neurol. 2007, 27, 86–92. [Google Scholar] [CrossRef]
- Mattson, M.P.; Haughey, N.J.; Nath, A. Cell death in HIV dementia. Cell Death Diff. 2005, 12, 893–904. [Google Scholar] [CrossRef]
- Kaul, M.; Lipton, S.A. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc. Natl. Acad. Sci. USA 1999, 96, 8212–8216. [Google Scholar] [CrossRef]
- Kaul, M.; Garden, G.W.; Lipton, S.A. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 2001, 19, 988–994. [Google Scholar] [CrossRef]
- Van de Bovenkamp, M.; Nottet, H.S.; Pereira, C.F. Interactions of human immunodeficiencyvirus-1 proteins with neurons: Possible role in the development of human immunodeficiencyvirus-1 associated dementia. Eur. J. Clin. Investig. 2002, 32, 619–627. [Google Scholar] [CrossRef]
- Bansal, A.K.; Mactutus, C.F.; Nath, A.; Maragos, W.; Hauser, K.F.; Booze, R.M. Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res. 2000, 879, 42–49. [Google Scholar] [CrossRef]
- Agrawal, L.; Louboutin, J.P.; Reyes, B.A.S.; van Bockstaele, E.J.; Strayer, D.S. Antioxidant enzyme gene delivery to protect from HIV-1 gp120-induced neuronal apoptosis. Gene Ther. 2006, 13, 1645–1656. [Google Scholar] [CrossRef]
- Eugenin, E.A.; D’Aversa, T.G.; Lopez, L.; Calderon, T.M.; Berman, J.W. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J. Neurochem. 2003, 85, 1299–1311. [Google Scholar] [CrossRef]
- Ghezzi, S.; Noolan, D.M.; Aluigi, M.G.; Vallanti, G.; Cota, M.; Benelli, R.; Morini, M.; Reeves, J.D.; Vicenzi, E.; Poli, G.; et al. Inhibition of CXCR-3-dependent HIV-1 infection by extracellular HIV-1 Tat. Biochem. Biophys. Res. Commun. 2000, 270, 992–996. [Google Scholar] [CrossRef]
- Bonavia, R.; Bajetto, A.; Barbero, S.; Albini, A.; Noonan, D.M.; Schettini, G. HIV-1 Tat causes apoptosis death and calcium homeostasis alterations in rat neurons. Biochem. Biophys. Res. Commun. 2001, 288, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Nath, A.; Mattson, M.P. HIV-1 tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J. Neurochem. 2001, 78, 457–467. [Google Scholar] [CrossRef]
- Norman, J.P.; Perry, S.W.; Reynolds, H.M.; Kiebala, M.; de Mesy Bentley, K.L.; Trejo, M.; Volsky, D.J.; Maggirwar, S.B.; Dewhurst, S.; Masliah, E. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS One 2008, 3, e3731. [Google Scholar] [CrossRef]
- Nath, A.; Haughey, N.J.; Jones, M.; Anderson, C.; Bell, J.E.; Geiger, J.D. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: Protection by memantine. Ann. Neurol. 2000, 47, 186–194. [Google Scholar] [CrossRef]
- Kruman, L.L.; Nath, A.; Mattson, M.P. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp. Neurol. 1998, 154, 276–288. [Google Scholar] [CrossRef]
- Magnuson, D.S.; Knudsen, B.E.; Geiger, J.D.; Brownstone, R.M.; Nath, A. Human immunodeficiency virus type 1 Tat activates non-N-methyl-o-aspartate excitatory amino receptors and causes neurotoxicity. Ann. Neurol. 1995, 37, 373–380. [Google Scholar] [CrossRef]
- Bonfoco, E.; Krainc, D.; Ankarcrona, M.; Nicotera, P.; Lipton, S.A. Apoptosis and necrosis: Two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc. Natl. Acad. Sci. USA 1995, 92, 7162–7166. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Olafson, K.; del Bigio, M.R.; Peeling, A.; Nath, A. Intraventricular injection of human immunodeficiency virus type 1 (HIV-1) tat protein causes inflammation, gliosis, apoptosis, and ventricular enlargment. J. Neuropathol. Exp. Neurol. 1998, 57, 563–570. [Google Scholar] [CrossRef]
- Askenov, M.Y.; Hasselrot, U.; Bansal, A.K.; Wu, G.; Nath, A.; Anderson, C.; Mactutus, C.F.; Booze, R.M. Oxidative damage induced by the injection of HIV-1 Tat protein in the rat striatum. Neurosci. Lett. 2001, 305, 5–8. [Google Scholar] [CrossRef]
- Askenov, M.Y.; Hasselrot, U.; Wu, G.; Nath, A.; Anderson, C.; Mactutus, C.F.; Booze, R.M. Temporal relationship between HIV-1 Tat-induced neuronal degeneration, OX-42 immunoreactivity, reactive astrocytosis, and protein oxidation in the rat striatum. Brain Res. 2003, 987, 1–9. [Google Scholar] [CrossRef]
- Agrawal, L.; Louboutin, J.P.; Strayer, D.S. Preventing HIV-1 Tat-induced neuronal apoptosis using antioxidant enzymes: Mechanistic and therapeutic implications. Virology 2007, 363, 462–472. [Google Scholar] [CrossRef]
- Strayer, D.S. Gene therapy using SV40-derived vectors: What does the future hold? J. Cell. Physiol. 1999, 181, 375–384. [Google Scholar] [CrossRef]
- McKee, H.J.; Strayer, D.S. Immune responses against SIV envelope glycoprotein, using recombinant SV40 as a vaccine delivery vector. Vaccine 2002, 20, 3613–3625. [Google Scholar] [CrossRef]
- Strayer, D.S.; Kondo, R.; Milano, J.; Duan, L.X. Use of SV40-based vectors to transduce foreign genes to normal human peripheral blood mononuclear cells. Gene Ther. 1997, 4, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Strayer, D.S.; Lamothe, M.; Wei, D.; Milano, J.; Kondo, R. Generation of Recombinant SV40 Vectors for Gene Transfer. In Methods in Molecular Biology, SV40 Protocols; Raptis, L., Ed.; Humana Press: Totowa, NJ, USA, 2001; Volume 165, pp. 103–117. [Google Scholar]
- Sauter, B.V.; Parashar, B.; Chowdhury, N.R.; Kadakol, A.; Ilan, Y.; Singh, H.; Milano, J.; Strayer, D.S.; Chowdhury, J.R. A replication-deficient rSV40 mediates liver-directed gene transfer and a long-term amelioration of jaundice in gunn rats. Gastroenterology 2000, 119, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Theodore, S.; Cass, W.A.; Maragos, W.F. Methamphetamine and human immunodeficiency virus protein Tat synergize to destroy dopaminergic terminals in the rat striatum. Neuroscience 2006, 137, 925–935. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: New York, NY, USA, 1986. [Google Scholar]
- Rouger, K.; Louboutin, J.P.; Villanova, M.; Cherel, Y.; Fardeau, M. X-linked vacuolated myopathy: TNF-alpha and IFN-gamma expression in muscle fibers with MHC class I on sarcolemma. Am. J. Pathol. 2001, 158, 355–359. [Google Scholar] [CrossRef]
- Nikonov, A.A.; Finger, T.E.; Caprio, J. Beyond the olfactory bulb: An odotopic map in the forebrain. Proc. Natl. Acad. Sci. USA 2005, 102, 18688–18693. [Google Scholar] [CrossRef]
- Louboutin, J.P.; Liu, B.; Reyes, B.A.S.; van Bockstaele, E.J.; Strayer, D.S. Rat bone marrow progenitor cells transduced in situ by rSV40 vectors differentiate into multiple CNS cell lineages. Stem Cells 2006, 24, 2801–2809. [Google Scholar] [CrossRef]
- Louboutin, J.P.; Reyes, B.A.S.; Agrawal, L.; van Bockstaele, E.J.; Strayer, D.S. Strategies for CNS-directed gene delivery: In vivo gene transfer to the brain using SV40-derived vectors. Gene Ther. 2007, 14, 939–949. [Google Scholar] [CrossRef]
- Louboutin, J.P.; Chekmasova, A.; Marusich, E.; Agrawal, L.; Strayer, D.S. Role of CCR5 and its ligands in the control of vascular inflammation and leukocyte recruitment required for acute excitotoxic seizure induction and neural damage. FASEB J. 2011, 25, 737–753. [Google Scholar] [CrossRef]
- Louboutin, J.P.; Chekmasova, A.A.; Marusich, E.; Chowdury, D.; Strayer, D.S. Efficient CNS gene delivery by intravenous injection. Nat. Meth. 2010, 7, 905–907. [Google Scholar] [CrossRef]
- Mandel, R.J.; Rendahl, K.G.; Spratt, S.K.; Snyder, R.O.; Cohen, L.K.; Leff, S.E. Characterization of intrastriatal recombinant adeno-associated virus-mediated gene transfer of human tyrosine hydroxylase and human GTP-cyclohydrolase I in a rat model of Parkinson’s disease. J. Neurosci. 1998, 18, 4271–4284. [Google Scholar] [PubMed]
- Louboutin, J.P.; Agrawal, L.; Reyes, B.A.S.; van Bockstaele, E.J.; Strayer, D.S. A rat model of human immunodeficiency virus 1 encephalopathy using envelope glycoprotein gp120 expression delivered by SV40 vectors. J. Neuropathol. Exp. Neurol. 2009, 68, 456–473. [Google Scholar] [CrossRef] [PubMed]
- McArthur, J.C.; Hoover, D.R.; Bacellar, H.; Miller, E.N.; Cohen, B.A.; Becker, J.T.; Graham, N.M.; McArthur, J.H.; Selnes, O.A.; Jacobson, L.P.; et al. Dementia in AIDS patients: Incidence and risk factors. Multicenter AIDS cohort study. Neurology 1993, 43, 2245–2252. [Google Scholar] [CrossRef]
- Major, E.O.; Rausch, D.; Marra, C.; Clifford, D. HIV-associated dementia. Science 2000, 288, 440–442. [Google Scholar] [PubMed]
- Koutsilieri, E.; Sopper, S.; Scheller, C.; ter Meulen, V.; Riederer, P. Parkinsonism in HIV dementia. J. Neural Transm. 2002, 109, 767–775. [Google Scholar] [CrossRef]
- Antinori, A.; Arendt, G.; Becker, J.T.; Brew, B.J.; Byrd, D.A.; Cherner, M.; Clifford, D.B.; Cinque, P.; Epstein, L.G.; Gookin, K.; et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007, 69, 1789–1799. [Google Scholar] [CrossRef]
- Woods, S.P.; Moore, D.J.; Weber, E.; Grant, I. Cognitive neuropsychology of HIV-associated neurocognitive disorders. Neuropsychol. Rev. 2009, 19, 152–168. [Google Scholar] [CrossRef]
- Mollace, V.; Nottet, H.S.; Clayette, P.; Turco, M.C.; Muscoli, C.; Salvemini, D.; Perno, C.F. Oxidative stress and neuroAIDS: Triggers, modulators and novel antioxidants. Trends Neurosci. 2001, 24, 411–416. [Google Scholar] [CrossRef]
- Turchan, J.; Pocernich, C.B.; Gairola, C.; Chauhan, A.; Schifitto, G.; Butterfield, D.A.; Buch, S.; Naravan, O.; Sinai, A.; Geiger, J.; et al. Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology 2003, 60, 307–314. [Google Scholar] [CrossRef]
- Steiner, J.; Haughey, N.; Li, W.; Venkatesan, A.; Anderson, C.; Reid, R.; Malpica, T.; Pocernich, C.; Butterfield, D.A.; Nath, A. Oxidative stress and therapeutic approaches in HIV dementia. Antioxid. Redox Signal. 2006, 8, 2089–2100. [Google Scholar] [CrossRef]
- Haughey, N.J.; Cutler, R.G.; Tamara, A.; McArthur, J.C.; Vargas, D.L.; Pardo, C.A.; Turchan, J.; Nath, A.; Mattson, M.P. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann. Neurol. 2004, 5, 257–267. [Google Scholar]
- Cutler, R.G.; Haughey, N.J.; Tammara, A.; McArthur, J.C.; Nath, A.; Reid, R.; Vargas, D.L.; Pardo, C.A.; Mattson, M.P. Dysregulation of sphingolipids and sterol metabolism by ApoE4 in HIV dementia. Neurology 2004, 63, 626–630. [Google Scholar] [CrossRef]
- Agrawal, L.; Louboutin, J.P.; Reyes, B.A.S.; van Bockstaele, E.J.; Strayer, D.S. HIV-1 Tat neurotoxicity: A model of acute and chronic exposure, and neuroprotection by gene delivery by antioxidant enzymes. Neurobiol. Dis. 2012, 45, 657–670. [Google Scholar] [CrossRef]
- Kruman, I.; Bruce-Keller, A.J.; Bredesen, D.; Waq, G.; Mattson, M.P. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J. Neurosci. 1997, 17, 5089–5100. [Google Scholar] [PubMed]
- Bruce-Keller, A.J.; Li, Y.J.; Lovell, M.A.; Kraemer, P.J.; Gary, D.S.; Brown, R.R.; Markesbery, W.R.; Mattson, M.P. 4-Hydroxynonenal, a product of lipid peroxidation, damages cholinergic neurons and impairs visuospatial memory in rats. J. Neuropathol. Exp. Neurol. 1998, 57, 257–267. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Barger, S.W.; Moss, N.I.; Pham, J.T.; Keller, J.N.; Nath, A. Proinflammatory and pro-oxidant properties of Tat in a microglial cell line: Attenuation by 17β-estradiol. J. Neurochem. 2001, 78, 1315–1324. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Gomez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of microglial proliferation during chronic degeneration. J. Neurosci. 2013, 33, 2481–2493. [Google Scholar] [CrossRef]
- Li, T.; Pang, S.; Yu, Y.; Wu, X.; Guo, J.; Zhang, S. Proliferation of parenchymal microglia is the main source of microgliosis after ischaemic stroke. Brain 2013, 136, 3578–3588. [Google Scholar] [CrossRef]
- Nosheny, R.L.; Bachis, A.; Acquas, E.; Mocchetti, I. Human immunodeficiency virus type 1 glycoprotein gp120 reduces the levels of brain-derived neurotrophic factor in vivo: Potential implication for neuronal cell death. Eur. J. Neurosci. 2004, 20, 2857–2864. [Google Scholar] [CrossRef]
- Louboutin, J.P.; Agrawal, L.; Reyes, B.A.S.; van Bockstaele, E.J.; Strayer, D.S. Protecting neurons from HIV-1 gp120 induced oxidant stress using both localized intracerebral and generalized intraventricular administration of antioxidant enzymes delivered by SV40 derived vectors. Gene Ther. 2007, 14, 1650–1661. [Google Scholar] [CrossRef]
- Louboutin, J.P.; Agrawal, L.; Reyes, B.A.S.; van Bockstaele, E.J.; Strayer, D.S. HIV-1 gp120 neurotoxicity proximally and at a distance from the point of exposure: Protection by rSV40 delivery of antioxidant enzymes. Neurobiol. Dis. 2009, 34, 462–476. [Google Scholar] [CrossRef]
- Louboutin, J.P.; Reyes, B.A.S.; Agrawal, L.; van Bockstaele, E.J.; Strayer, D.S. HIV-1 gp120-induced neuroinflammation: Relationship to neuron loss and protection by rSV40-delivered antioxidant enzymes. Exp. Neurol. 2010, 221, 231–245. [Google Scholar] [CrossRef]
- Chan, P.H.; Schmidley, J.W.; Fishman, R.A.; Longar, S.M. Brain injury, edema, and vascular permeability changes induced by oxygen-derived free radicals. Neurology 1984, 34, 315–320. [Google Scholar] [CrossRef]
- Chan, P.H.; Yang, G.Y.; Carlson, E.; Epstein, C.J. Cold-induced brain edema and infarction are reduced in transgenic mice overexpressing CuZn-superoxide dismutase. Ann. Neurol. 1991, 29, 482–486. [Google Scholar] [CrossRef]
- Wang, H.K.; Park, U.J.; Kim, S.Y.; Lee, J.H.; Kim, S.U.; Gwaq, B.J.; Lee, Y.B. Free radical production in CA1 neurons induces MIP-1alpha expression, microglial recruitment, and delayed neuronal death after transient forebrain ischemia. J. Neurosci. 2008, 28, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Nishi, T.; Maier, C.M.; Hayashi, T.; Saito, A.; Chan, P.H. Superoxide dismutase 1 overexpression reduces MCP-1 and MIP-1 alpha expression after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2005, 25, 1312–1324. [Google Scholar] [CrossRef]
- Shah, A.; Kumar, S.; Simon, S.D.; Singh, D.P.; Kumar, A. HIV gp120- and methamphetamine-mediated oxidative stress induces astrocyte apoptosis via cytochrome P450 2E1. Cell Death Dis. 2013, 4, e850. [Google Scholar] [CrossRef]
- Lee, Y.W.; Hirani, A.A.; Kyprianou, N.; Toborek, M. Human immunodeficiency virus-1Tat protein up-regulates interleukin-6 and interleukin-8 expression in human breast cancer cells. Inflamm. Res. 2005, 54, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Nookala, A.R.; Shah, A.; Noel, R.J.; Kumar, A. HIV-1 Tat-mediated induction of CCL5 in astrocytes involves NF-κB, AP-1, C/EBPα and C/EBPγ transcription factors and JAK, PI3K/Akt and p38 MAPK signaling pathways. PLoS One 2013, 8, e78855. [Google Scholar] [CrossRef]
- El-Hage, N.; Bruce-Keller, A.J.; Yakovleva, T.; Bazov, I.; Bakalkin, G.; Knapp, P.E.; Hauser, K.F. Morphine exacerbates HIV1 Tat-induced cytokine production in astrocytes through convergent effets on ([Ca2+]i, NF-κB trafficking and transcription. PLoS One 2008, 3, e4093. [Google Scholar] [CrossRef] [PubMed]
- Consortium, D. A randomized, double-blind, placebo-controlled trial of deprenyl and thioctic acid in human immunodeficiency virus-associated cognitive impairment: Dana Consortium on the therapy of HIV dementia and related cognitive disorders. Neurology 1998, 50, 645–651. [Google Scholar] [CrossRef]
- Sacktor, N.; Schifitto, G.; McDermott, M.P.; Marder, K.; McArthur, J.C.; Kieburtz, K. Transdermal seleginine in HIV-associated cognitive impairment: Pilot, placebo-controlled study. Neurology 2000, 54, 233–235. [Google Scholar] [CrossRef]
- Consortium, D. Safety and tolerability of the antioxidant OPC-14117 in HIV-associated cognitive impairment: The Dana Consortium on the therapy of HIV dementia and related cognitive disorders. Neurology 1997, 49, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Pulliam, L.; Irwin, I.; Kusdra, L.; Rempel, H.; Flitter, W.D.; Garland, W.A. CPI-1189 attenuates effects of suspected neurotoxins associated with AIDS dementia: A possible role for ERK activation. Brain Res. 2001, 893, 95–103. [Google Scholar] [CrossRef]
- Clifford, D.B.; McArthur, J.C.; Schifitto, G.; Kieburtz, K.; McDermott, M.P.; Letendre, S.; Cohen, B.A.; Marder, K.; Ellis, R.J.; Marra, C.M. Neurologic AIDS Research Consortium. A randomized clinical trial of CPI-1189 for HIV-associated cognitive-motor impairment. Neurology 2002, 59, 1568–1573. [Google Scholar] [CrossRef]
- Louboutin, J.P.; Strayer, D.S. HIV-1-Associated Neurocognitive Disorder: Role of Oxidative Stress and Protection by Gene Delivery of Antioxidant Enzymes. In Encephalitis, Encephalomyelitis, Encephalopathies: Symptoms, Causes and Potential Complications; Ruiz, A., Fleming, D., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2013. [Google Scholar]
- Pocernich, C.B.; la Fontaine, M.; Butterfield, D.A. In-vivo glutathione elevation protects against hydroxyl free radical-induced protein oxidation in rat brain. Neurochem. Int. 2000, 36, 185–191. [Google Scholar] [CrossRef]
- Banerjee, A.; Zhanq, X.; Manda, K.R.; Banks, W.A.; Ercal, N. HIV proteins (gp120 and Tat) and methamphetamine in oxidative stress induced damage in the brain: Potential role of the thiol antioxidant N-acetylcysteine amide. Free Radic. Biol. Med. 2010, 48, 1388–1398. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Louboutin, J.-P.; Agrawal, L.; Reyes, B.A.S.; Van Bockstaele, E.J.; Strayer, D.S. Oxidative Stress Is Associated with Neuroinflammation in Animal Models of HIV-1 Tat Neurotoxicity. Antioxidants 2014, 3, 414-438. https://doi.org/10.3390/antiox3020414
Louboutin J-P, Agrawal L, Reyes BAS, Van Bockstaele EJ, Strayer DS. Oxidative Stress Is Associated with Neuroinflammation in Animal Models of HIV-1 Tat Neurotoxicity. Antioxidants. 2014; 3(2):414-438. https://doi.org/10.3390/antiox3020414
Chicago/Turabian StyleLouboutin, Jean-Pierre, Lokesh Agrawal, Beverly A. S. Reyes, Elisabeth J. Van Bockstaele, and David S. Strayer. 2014. "Oxidative Stress Is Associated with Neuroinflammation in Animal Models of HIV-1 Tat Neurotoxicity" Antioxidants 3, no. 2: 414-438. https://doi.org/10.3390/antiox3020414