Antioxidant and Metal Chelation-Based Therapies in the Treatment of Prion Disease

Abstract
:1. Introduction
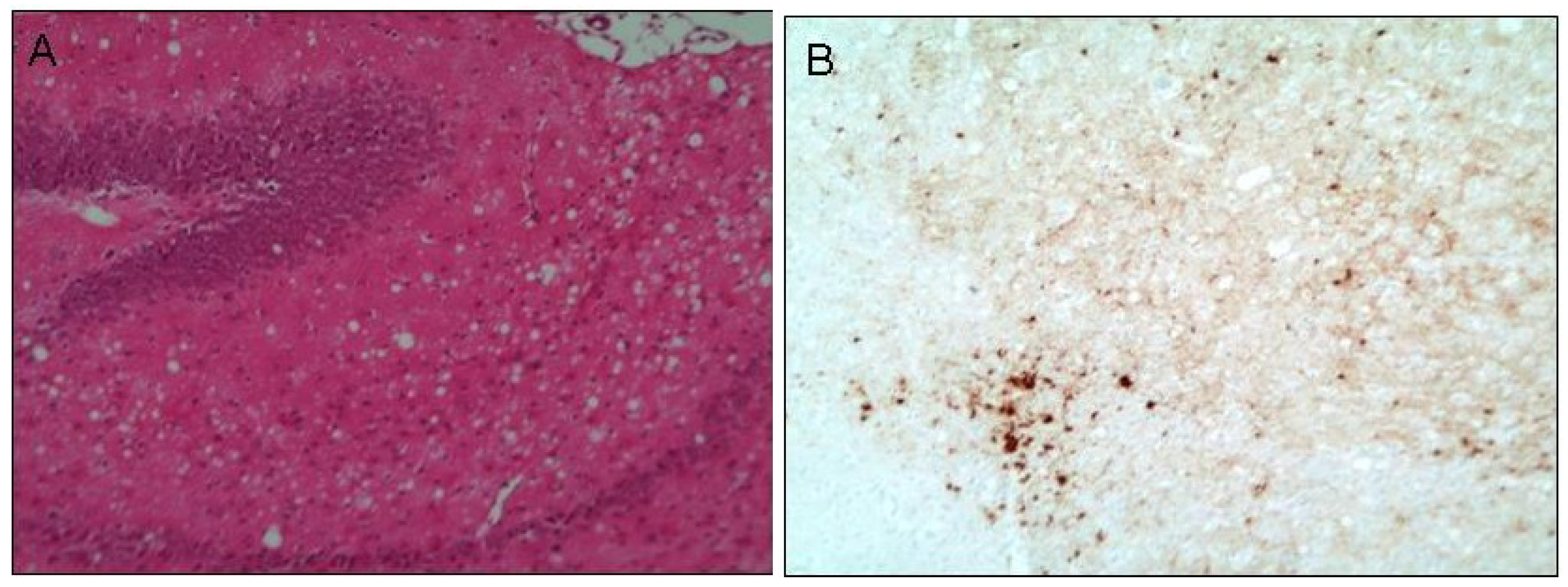
2. Oxidative Stress
3. Reactive Transition Metal Ions

{kind=link}
{kind=link}
{kind=link}
| Reactive Oxygen Species | Reactive Nitrogen Species | |
|---|---|---|
| Radicals | Alkoxyl RO Hydroperoxyl HO2 Hydroxyl OH Peroxyl RO2 Superoxide O2− | Nitric oxide NO Nitrogen dioxide NO2 |
| Closed shell molecules | Hydrogen peroxide H2O2 Hypochlorous acid HOCl Ozone O3 Singlet dioxygen 1O2 *Peroxynitrite OONO− | Dinitrogen trioxide N2O3 Nitronium ion NO2+ Nitrosyl cation NO+ Nitrous acid HNO2 Nitroxyl anion NO− Nitryl chloride NO2Cl * Peroxynitrite OONO− |
4. Superoxide Dismutase
Enzyme-Cu1+ + O2− + 2 H+ → Enzyme-Cu1+ + H2O2
2 O2− + 2 H+ → H2O2 + O2 Net SOD reaction
Enzyme-Mn2+ + O2− + 2 H+ → Enzyme-Mn3+ + H2O2
2 O2− + 2 H+ → H2O2 + O2 Net SOD reaction
5. Fenton and Haber-Weiss Reactions
Mn+ + H2O2 → Mn+1 + OH− + OH
O2− + H2O2 → O2 + OH− + OH Net Haber-Weiss reaction
6. Prion Protein Possesses Antioxidant Activity
7. Oxidative Stress Contributes to Prion Pathogenesis
8. Antioxidant Therapy Combating Models of Prion Disease
9. Transition Metals Contribute to Neurodegeneration
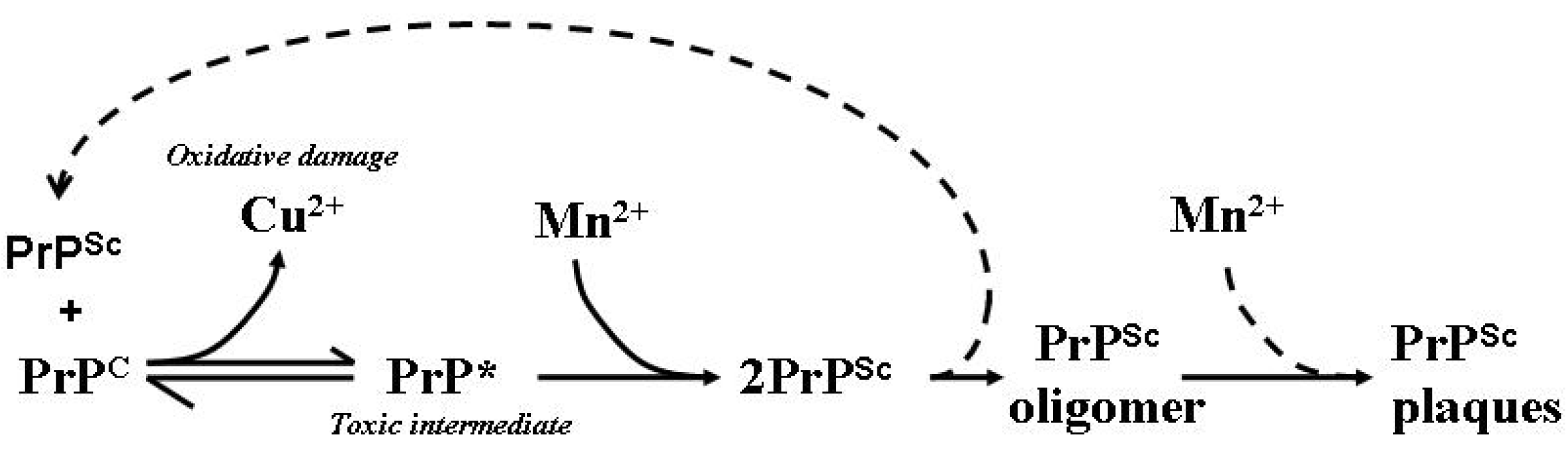
10. Transition Metal Participation in Prion Disease

11. Prion Protein Aggregation in Prion Disease
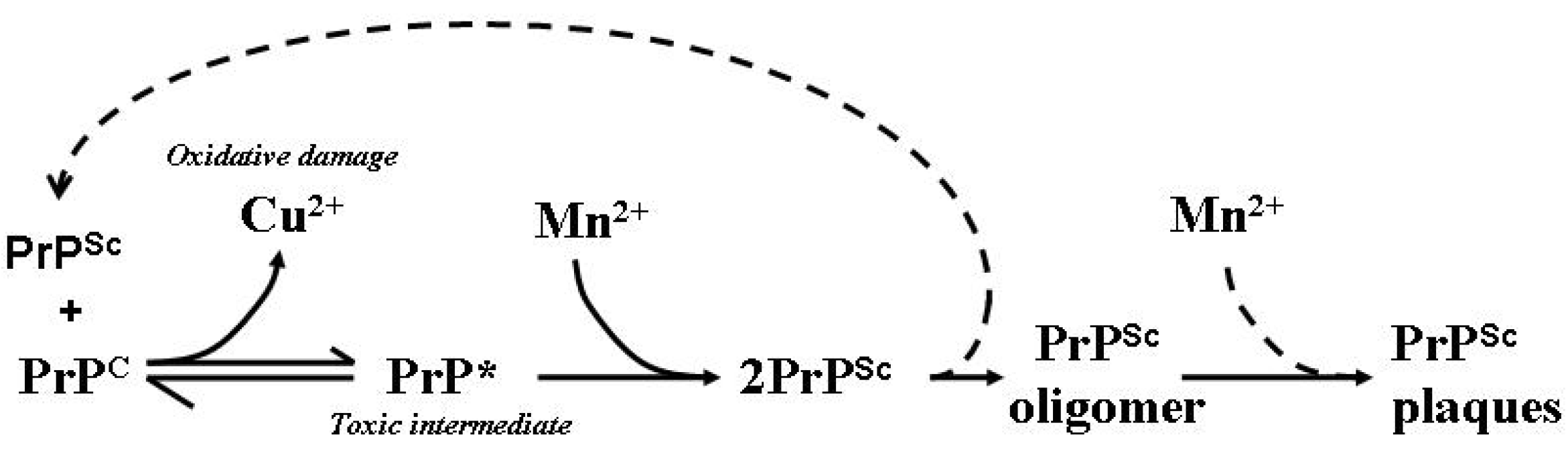
12. Therapeutic Manipulation of CNS Metals in Prion Disease
13. Conclusions
Acknowledgments
Conflicts of Interest
References
- Zucconi, G.G.; Cipriani, S.; Scattoni, R.; Balgkouranidou, I.; Hawkins, D.P.; Ragnarsdottir, K.V. Copper deficiency elicits glial and neuronal response typical of neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2007, 33, 212–225. [Google Scholar] [CrossRef]
- Melov, S.; Doctrow, S.R.; Schneider, J.A.; Haberson, J.; Patel, M.; Coskun, P.E.; Huffman, K.; Wallace, D.C.; Malfroy, B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. J. Neurosci. 2001, 21, 8348–8353. [Google Scholar] [PubMed]
- Collins, S.J.; Lawson, V.A.; Masters, C.L. Transmissible spongiform encephalopathies. Lancet 2004, 363, 51–61. [Google Scholar] [CrossRef]
- Knight, R.; Brazier, M.; Collins, S. Prions: A challenge for Science, Medicine and Public Health Systems. In Contributions to Microbiology; Rabenau, H.F., Cinatl, J., Doerr, H.W., Eds.; Karger: Basel, Switzerland, 2004; Volume 11. [Google Scholar]
- Goldfarb, L.G.; Petersen, R.B.; Tabaton, M.; Brown, P.; LeBlanc, A.C.; Montagna, P.; Cortelli, P.; Julien, J.; Vital, C.; Pendelbury, W.W.; et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: Disease phenotype determined by a DNA polymorphism. Science 1992, 258, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Walsh, D.J.; Piro, J.R.; Wang, F.; Wang, X.; Ma, J.; Rees, J.R.; Supattapone, S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. USA 2012, 109, E1938–E1946. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, X.; Yuan, C.-G.; Ma, J. Generating a Prion with Bacterially Expressed Recombinant Prion Protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef]
- Perry, G.; Nunomura, A.; Hirai, K.; Zhu, X.; Perez, M.; Avila, J.; Castellani, R.J.; Atwood, C.S.; Aliev, G.; Sayre, L.M.; et al. Is oxidative damage the fundamental pathogenic mechanism of Alzheimer’s and other neurodegenerative diseases? Free Radic. Biol. Med. 2002, 33, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Howard, B.J.; LaFontaine, M.A. Brain oxidative stress in animal models of accelerated aging and the age-related neurodegenerative disorders, Alzheimer’s disease and Huntington’s disease. Curr. Med. Chem. 2001, 8, 815–828. [Google Scholar] [CrossRef]
- Liu, D.; Wen, J.; Liu, J.; Li, L. The roles of free radicals in amyotrophic lateral sclerosis: Reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB J. 1999, 13, 2318–2328. [Google Scholar] [PubMed]
- Barnham, K.J.; Cappai, R.; Beyreuther, K.; Masters, C.L.; Hill, A.F. Delineating common molecular mechanisms in Alzheimer’s and prion diseases. Trends Biochem. Sci. 2006, 31, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Park, K.J.; Choi, E.K.; Kim, J.; Carp, R.I.; Wisniewski, H.M.; Kim, Y.S. Expression of inducible nitric oxide synthase in the brains of scrapie-infected mice. J. Neurovirol. 1998, 4, 445–450. [Google Scholar] [CrossRef]
- Choi, S.I.; Ju, W.K.; Choi, E.K.; Kim, J.; Lea, H.Z.; Carp, R.I.; Wisniewski, H.M.; Kim, Y.S. Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 K scrapie agent. Acta Neuropathol. (Berl.) 1998, 96, 279–286. [Google Scholar] [CrossRef]
- Guentchev, M.; Voigtlander, T.; Haberler, C.; Groschup, M.H.; Budka, H. Evidence for oxidative stress in experimental prion disease. Neurobiol. Dis. 2000, 7, 270–273. [Google Scholar] [CrossRef]
- Guentchev, M.; Siedlak, S.L.; Jarius, C.; Tagliavini, F.; Castellani, R.J.; Perry, G.; Smith, M.A.; Budka, H. Oxidative damage to nucleic acids in human prion disease. Neurobiol. Dis. 2002, 9, 275–281. [Google Scholar] [CrossRef]
- Andreoletti, O.; Levavasseur, E.; Uro-Coste, E.; Tabouret, G.; Sarradin, P.; Delisle, M.B.; Berthon, P.; Salvayre, R.; Schelcher, F.; Negre-Salvayre, A. Astrocytes accumulate 4-hydroxynonenal adducts in murine scrapie and human creutzfeldt-jakob disease. Neurobiol. Dis. 2002, 11, 386–393. [Google Scholar] [CrossRef]
- Brazier, M.W.; Lewis, V.; Ciccotosto, G.D.; Klug, G.M.; Lawson, V.A.; Cappai, R.; Ironside, J.W.; Masters, C.L.; Hill, A.F.; White, A.R.; et al. Correlative studies support lipid peroxidation is linked to PrP(res) propagation as an early primary pathogenic event in prion disease. Brain Res. Bull. 2006, 68, 346–354. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- International Programme on Chemical Safety. Environmental Health Criteria No. 200: Copper; World Health Organization: Geneva, Switzerland, 1998. [Google Scholar]
- Stern, B.R. Essentiality and toxicity in copper health risk assessment: Overview, update and regulatory considerations. J. Toxicol. Environ. Health Part A 2010, 73, 114–127. [Google Scholar] [CrossRef]
- Macomber, L.; Imlay, J.A. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 8344–8349. [Google Scholar] [CrossRef]
- Telianidis, J.; Hung, Y.H.; Materia, S.; Fontaine, S.L. Role of the P-Type ATPases, ATP7A and ATP7B in brain copper homeostasis. Front. Aging Neurosci. 2013, 5, 44. [Google Scholar] [PubMed]
- Stern, B.R.; Solioz, M.; Krewski, D.; Aggett, P.; Aw, T.C.; Baker, S.; Crump, K.; Dourson, M.; Haber, L.; Hertzberg, R.; et al. Copper and Human Health: Biochemistry, Genetics, and Strategies for Modeling Dose-Response Relationships. J. Toxicol. Environ. Health Part B 2007, 10, 157–222. [Google Scholar] [CrossRef]
- Margulis, L. Origin of Eukaryotic Cells; Yale University Press: New Haven, CT, USA, 1970. [Google Scholar]
- Lang, B.F.; Gray, M.W.; Burger, G. Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 1999, 33, 351–397. [Google Scholar] [CrossRef]
- Moura, I.; Pauleta, S.R.; Moura, J.J. Enzymatic activity mastered by altering metal coordination spheres. J. Biol. Inorg. Chem. 2008, 13, 1185–1195. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncola, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Biologically relevant metal ion-dependent hydroxyl radical generation. An update. FEBS Lett. 1992, 307, 108–112. [Google Scholar] [CrossRef]
- Gaeta, A.; Hider, R.C. The crucial role of metal ions in neurodegeneration: The basis for a promising therapeutic strategy. Br. J. Pharmacol. 2005, 146, 1041–1059. [Google Scholar] [PubMed]
- Rana, A.; Gnaneswari, D.; Bansal, S.; Kundu, B. Prion metal interaction: Is prion pathogenesis a cause or a consequence of metal imbalance? Chem. Biol. Interact. 2009, 181, 282–291. [Google Scholar] [CrossRef]
- Nadal, R.C.; Abdelraheim, S.R.; Brazier, M.W.; Rigby, S.E.; Brown, D.R.; Viles, J.H. The prion protein does not redox silence Cu2+, but is a sacrificial quencher of hydroxyl radicals. Free Radic. Biol. Med. 2007, 42, 79–89. [Google Scholar] [CrossRef]
- Davies, P.; Marken, F.; Salter, S.; Brown, D.R. Thermodynamic and voltammetric characterisation of the metal binding to the prion protein: Insights into pH dependence and redox chemistry. Biochemistry 2009, 48, 2610–2619. [Google Scholar] [CrossRef]
- Hutter, G.; Heppner, F.L.; Aguzzi, A. No superoxide dismutase activity of cellular prion protein in vivo. Biol. Chem. 2003, 384, 1279–1285. [Google Scholar] [PubMed]
- Jones, S.; Batchelor, M.; Bhelt, D.; Clarke, A.R.; Collinge, J.; Jackson, G.S. Recombinant prion protein does not possess SOD-1 activity. Biochem. J. 2005, 392, 309–312. [Google Scholar] [CrossRef]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [PubMed]
- Wong, B.S.; Pan, T.; Liu, T.; Li, R.; Gambetti, P.; Sy, M.S. Differential contribution of superoxide dismutase activity by prion protein in vivo. Biochem. Biophys. Res. Commun. 2000, 273, 136–139. [Google Scholar] [CrossRef]
- Brown, D.R.; Schmidt, B.; Kretzschmar, H.A. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature 1996, 380, 345–347. [Google Scholar] [CrossRef]
- White, A.R.; Collins, S.J.; Maher, F.; Jobling, M.F.; Stewart, L.R.; Thyer, J.M.; Beyreuther, K.; Masters, C.L.; Cappai, R. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am. J. Pathol. 1999, 155, 1723–1730. [Google Scholar] [CrossRef]
- Choi, C.J.; Anantharam, V.; Saetveit, N.J.; Houk, R.; Kanthasamy, A.; Kanthasamy, A.G. Normal cellular prion protein protects against manganese-induced oxidative stress and apoptotic cell death. Toxicol. Sci. 2007, 98, 495–509. [Google Scholar] [CrossRef]
- Brown, D.R.; Schmidt, B.; Kretzschmar, H.A. Effects of copper on survival of prion protein knockout neurons and glia. J. Neurochem. 1998, 70, 1686–1693. [Google Scholar] [PubMed]
- Wong, B.S.; Brown, D.R.; Pan, T.; Whiteman, M.; Liu, T.; Bu, X.; Li, R.; Gambetti, P.; Olesik, J.; Rubenstein, R.; et al. Oxidative impairment in scrapie-infected mice is associated with brain metals perturbations and altered antioxidant activities. J. Neurochem. 2001, 79, 689–698. [Google Scholar] [PubMed]
- Brown, D.R. Neurodegeneration and oxidative stress: Prion disease results from loss of antioxidant defence. Folia Neuropathol. 2005, 43, 229–243. [Google Scholar] [PubMed]
- Brazier, M.W.; Davies, P.; Player, E.; Marken, F.; Viles, J.H.; Brown, D.R. Manganese binding to the prion protein. J. Biol. Chem. 2008, 283, 12831–12839. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Clive, C.; Haswell, S. Antioxidant activity related to copper binding native prion protein. J. Neurochem. 2001, 76, 69–76. [Google Scholar] [CrossRef]
- Thackray, A.M.; Knight, R.; Haswell, S.J.; Bujdoso, R.; Brown, D.R. Metal imbalance and compromised antioxidant function are early changes in prion disease. Biochem. J. 2002, 362, 253–258. [Google Scholar] [CrossRef]
- Miele, G.; Jeffrey, M.; Turnbull, D.; Manson, J.; Clinton, M. Ablation of cellular prion protein expression affects mitochondrial numbers and morphology. Biochem. Biophys. Res. Commun. 2002, 291, 372–377. [Google Scholar] [CrossRef]
- Hachiya, N.S.; Yamada, M.; Watanabe, K.; Jozuka, A.; Ohkubo, T.; Sano, K.; Takeuchi, Y.; Kozuka, Y.; Sakasegawa, Y.; Kaneko, K. Mitochondrial localization of cellular prion protein (PrPC) invokes neuronal apoptosis in aged transgenic mice overexpressing PrPC. Neurosci. Lett. 2005, 374, 98–103. [Google Scholar] [CrossRef]
- Lee, D.W.; Sohn, H.O.; Lim, H.B.; Lee, Y.G.; Kim, Y.S.; Carp, R.I.; Wisniewski, H.M. Alteration of free radical metabolism in the brain of mice infected with scrapie agent. Free Radic. Res. 1999, 30, 499–507. [Google Scholar] [CrossRef]
- Wong, B.S.; Chen, S.G.; Colucci, M.; Xie, Z.; Pan, T.; Liu, T.; Li, R.; Gambetti, P.; Sy, M.S.; Brown, D.R. Aberrant metal binding by prion protein in human prion disease. J. Neurochem. 2001, 78, 1400–1408. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.B. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Petersen, R.B.; Siedlak, S.L.; Lee, H.G.; Kim, Y.S.; Nunomura, A.; Tagliavini, F.; Ghetti, B.; Cras, P.; Moreira, P.I.; Castellani, R.J.; et al. Redox metals and oxidative abnormalities in human prion diseases. Acta Neuropathol. (Berl.) 2005, 110, 232–238. [Google Scholar] [CrossRef]
- Turnbull, S.; Tabner, B.J.; Brown, D.R.; Allsop, D. Quinacrine acts as an antioxidant and reduces the toxicity of the prion peptide PrP106–126. Neuroreport 2003, 14, 1743–1745. [Google Scholar] [CrossRef] [PubMed]
- Doh-Ura, K.; Iwaki, T.; Caughey, B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J. Virol. 2000, 74, 4894–4897. [Google Scholar] [CrossRef]
- Korth, C.; May, B.C.; Cohen, F.; Prusiner, S.B. Acridine and phenothiazine derivatives as phamacotherapeutics for prion disease. Proc. Natl. Acad. Sci. USA 2001, 98, 9836–9841. [Google Scholar] [CrossRef]
- Collins, S.J.; Lewis, V.; Brazier, M.; Hill, A.F.; Fletcher, A.; Masters, C.L. Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann. Neurol. 2002, 52, 503–506. [Google Scholar] [CrossRef]
- Collinge, J.; Gorham, M.; Hudson, F.; Kennedy, A.; Keogh, G.; Pal, S.; Rossor, M.; Rudge, P.; Siddique, D.; Spyer, M.; et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): A patient-preference trial. Lancet Neurol. 2009, 8, 334–344. [Google Scholar] [CrossRef]
- Baudry, M.; Etienne, S.; Bruce, A.; Palucki, M.; Jacobsen, E.; Malfroy, B. Salen-manganese complexes are superoxide dismutase-mimics. Biochem. Biophys. Res. Commun. 1993, 192, 964–968. [Google Scholar] [CrossRef]
- Melov, S.; Ravenscroft, J.; Malik, S.; Gill, M.S.; Walker, D.W.; Clayton, P.E.; Wallace, D.C.; Malfroy, B.; Doctrow, S.R.; Lithgow, G.J. Extension of life-span with superoxide dismutase/catalase mimetics. Science 2000, 289, 1567–1569. [Google Scholar] [CrossRef]
- Carillon, J.; Rouanet, J.M.; Cristol, J.P.; Brion, R. Superoxide Dismutase Administration, a Potential Therapy Against Oxidative Stress Related Diseases: Several Routes of Supplementation and Proposal of an Original Mechanism of Action. Pharm. Res. 2013, 30, 2718–2728. [Google Scholar] [CrossRef]
- Brazier, M.W.; Doctrow, S.R.; Masters, C.L.; Collins, S.J. A manganese-superoxide dismutase/catalase mimetic extends survival in a mouse model of human prion disease. Free Radic. Biol. Med. 2008, 45, 184–192. [Google Scholar] [CrossRef]
- Bowman, A.B.; Kwakye, G.F.; Herrero Hernández, E.; Aschner, M. Role of manganese in neurodegenerative diseases. J. Trace Elem. Med. Biol. 2011, 25, 191–203. [Google Scholar] [CrossRef]
- Herrero Hernandez, E.; Discalzi, G.; Valentini, C.; Venturi, F.; Chiò, A.; Carmellino, C.; Rossi, L.; Sacchetti, A.; Pira, E. Follow-up of patients affected by manganese-induced Parkinsonism after treatment with CaNaEDTA. Neurotoxicology 2006, 27, 333–339. [Google Scholar] [CrossRef]
- Allsop, D.; Mayes, J.; Moore, S.; Masad, A.; Tabner, B.J. Metal-dependent generation of reactive oxygen species from amyloid proteins implicated in neurodegenerative disease. Biochem. Soc. Trans. 2008, 36, 1293–1298. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Scott, M.R.; DeArmond, S.J.; Cohen, F.E. Prion protein biology. Cell 1998, 93, 337–348. [Google Scholar] [CrossRef]
- Linden, R.; Martins, V.R.; Prado, M.A.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the prion protein. Physiol. Rev. 2008, 88, 673–728. [Google Scholar] [CrossRef]
- Younan, N.D.; Sarell, C.J.; Davies, P.; Brown, D.R.; Viles, J.H. The cellular prion protein trapsAlzheimer’s Aβ in an oligomeric form and disassembles amyloid fibers. FASEB J. 2013, 27, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Lesné, S.E. The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes inAlzheimer’s disease. J. Neurosci. 2012, 32, 16857–16871. [Google Scholar] [CrossRef]
- Kudo, W.; Petersen, R.B.; Lee, H.G. Cellular prion protein andAlzheimerdisease: Link to oligomeric amyloid-β and neuronal cell death. Prion 2013, 7, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef]
- Viles, J.H.; Klewpatinond, M.; Nadal, R.C. Copperand the structural biology of the prion protein. Biochem. Soc. Trans. 2008, 36, 1288–1292. [Google Scholar] [CrossRef]
- Gaggelli, E.; Bernardi, F.; Molteni, E.; Pogni, R.; Valensin, D.; Valensin, G.; Remelli, M.; Luczkowski, M.; Kozlowski, H. Interaction of the human prion protein PrP(106–126) sequence with copper (II), manganese (II) and zinc (II): NMR and EPR studies. J. Am. Chem. Soc. 2005, 127, 996–1006. [Google Scholar] [CrossRef]
- Davies, P.; Brown, D.R. The chemistry of copper binding to PrP: Is there sufficient evidence to elucidate a role for copper in protein function? Biochem. J. 2008, 410, 237–244. [Google Scholar] [CrossRef]
- Jackson, G.S.; Murray, I.; Hosszu, L.L.; Gibbs, N.; Waltho, J.P.; Clarke, A.R.; Collinge, J. Location and properties of metal-binding sites on the human prion protein. Proc. Natl. Acad. Sci. USA 2001, 98, 8531–8535. [Google Scholar] [CrossRef]
- Kozlowski, H.; Luczkowski, M.; Remelli, M. Prion proteins and copper ions. Biological and chemical controversies. Dalton Trans. 2010, 39, 6371–6385. [Google Scholar] [CrossRef]
- Aronoff-Spencer, E.; Burns, C.S.; Avdievich, N.I.; Gerfen, G.J.; Peisach, J.; Antholine, W.E.; Ball, H.L.; Cohen, F.E.; Prusiner, S.B.; Millhauser, G.L. Identification of the Cu2+ binding sites in the N-terminal domain of the prion protein by EPR and CD spectroscopy. Biochemistry 2000, 39, 13760–13771. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.S.; Aronoff-Spencer, E.; Legname, G.; Prusiner, S.B.; Antholine, W.E.; Gerfen, G.J.; Peisach, J.; Millhauser, G.L. Copper coordination in the full-length, recombinant prion protein. Biochemistry 2003, 42, 6794–6803. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Walter, E.D.; Newell, D.J.; Jackson, P.J.; Aronoff-Spencer, E.; Peisach, J.; Gerfen, G.J.; Bennett, B.; Antholine, W.E.; Millhauser, G.L. The octarepeat domain of the prion protein binds Cu(II) with three distinct coordination modes at pH 7.4. J. Am. Chem. Soc. 2005, 127, 12647–12656. [Google Scholar] [CrossRef]
- Jones, C.E.; Klewpatinond, M.; Abdelraheim, S.R.; Brown, D.R.; Viles, J.H. Probing copper2+ binding to the prion protein using diamagnetic nickel2+ and 1H NMR: The unstructured N terminus facilitates the coordination of six copper2+ ions at physiological concentrations. J. Mol. Biol. 2005, 346, 1393–1407. [Google Scholar] [CrossRef]
- Treiber, C.; Thompsett, A.R.; Pipkorn, R.; Brown, D.R.; Multhaup, G. Real-time kinetics of discontinuous and highly conformational metal-ion binding sites of prion protein. J. Biol. Inorg. Chem. 2007, 12, 711–720. [Google Scholar] [CrossRef]
- Qin, K.; Yang, D.S.; Yang, Y.; Chishti, M.A.; Meng, L.J.; Kretzschmar, H.A.; Yip, C.M.; Fraser, P.E.; Westaway, D. Copper(II)-induced conformational changes and protease resistance in recombinant and cellular PrP. Effect of protein age and deamidation. J. Biol. Chem. 2000, 275, 19121–19131. [Google Scholar] [CrossRef]
- Quaglio, E.; Chiesa, R.; Harris, D.A. Copper converts the cellular prion protein into a protease-resistant species that is distinct from the scrapie isoform. J. Biol. Chem. 2001, 276, 11432–11438. [Google Scholar] [CrossRef]
- Bocharova, O.V.; Breydo, L.; Salnikov, V.V.; Baskakov, I.V. Copper(II) inhibits in vitro conversion of prion protein into amyloid fibrils. Biochemistry 2005, 44, 6776–6787. [Google Scholar] [CrossRef]
- Brazier, M.W.; Volitakis, I.; Kvasnicka, M.; White, A.R.; Underwood, J.R.; Green, J.E.; Han, S.; Hill, A.F.; Masters, C.L.; Collins, S.J. Manganese chelation therapy extends survival in a mouse model of M1000 prion disease. J. Neurochem. 2010, 114, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Anantharam, V.; Jin, H.; Witte, T.; Houk, R.; Kanthasamy, A.; Kanthasamy, A.G. Infectious prion protein alters manganese transport and neurotoxicity in a cell culture model of priondisease. Neurotoxicology 2011, 32, 554–562. [Google Scholar] [CrossRef]
- Pauly, P.C.; Harris, D.A. Copper stimulates endocytosis of the prion protein. J. Biol. Chem. 1998, 273, 33107–33110. [Google Scholar] [CrossRef]
- Brown, L.R.; Harris, D.A. Copper and zinc cause delivery of the prion protein from the plasma membrane to a subset of early endosomes and the Golgi. J. Neurochem. 2003, 87, 353–363. [Google Scholar] [CrossRef]
- Brown, D.R. Brain proteins that mind metals: A neurodegenerative perspective. Dalton Trans. 2009, 21, 4069–4076. [Google Scholar] [CrossRef]
- Kralovicova, S.; Fontaine, S.N.; Alderton, A.; Alderman, J.; Ragnarsdottir, K.V.; Collins, S.J.; Brown, D.R. The effects of prion protein expression on metal metabolism. Mol. Cell. Neurosci. 2009, 41, 135–147. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Petrukhin, K.; Chernov, I.; Pellequer, J.L.; Wasco, W.; Ross, B.; Romano, D.M.; Parano, E.; Pavone, L.; Brzustowicz, L.M.; et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 1993, 5, 344–350. [Google Scholar] [CrossRef]
- Menkes, J.H.; Alter, M.; Steigleder, G.K.; Weakley, D.R.; Sung, J.H. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics 1962, 29, 764–779. [Google Scholar] [PubMed]
- Pandey, K.; Snyder, J.P.; Liotta, D.C.; Musaev, D.G. Computational studies of transition metal selectivity of octapeptide repeat region of prion protein (PrP). J. Phys. Chem. 2010, 114, 1127–1135. [Google Scholar] [CrossRef]
- Hesketh, S.; Sassoon, J.; Knight, R.; Brown, D.R. Elevated manganese levels in blood and CNS in human prion disease. Mol. Cell. Neurosci. 2008, 37, 590–598. [Google Scholar] [CrossRef]
- Hesketh, S.; Sassoon, J.; Knight, R.; Hopkins, J.; Brown, D.R. Elevated manganese levels in blood and central nervous system occur before onset of clinical signs in scrapie and bovine spongiform encephalopathy. J. Anim. Sci. 2007, 85, 1596–1609. [Google Scholar] [CrossRef]
- Brown, D.R.; Hafiz, F.; Glasssmith, L.L.; Wong, B.S.; Jones, I.M.; Clive, C.; Haswell, S.J. Consequences of manganese replacement of copper for prion protein function and proteinase resistance. EMBO J. 2000, 19, 1180–1186. [Google Scholar] [CrossRef]
- Giese, A.; Levin, J.; Bertsch, U.; Kretzschmar, H. Effect of metal ions on de novo aggregation of full-length prion protein. Biochem. Biophys. Res. Commun. 2004, 320, 1240–1246. [Google Scholar] [CrossRef]
- Levin, J.; Bertsch, U.; Kretzschmar, H.; Giese, A. Single particle analysis of manganese-induced prion protein aggregates. Biochem. Biophys. Res. Commun. 2005, 329, 1200–1207. [Google Scholar] [CrossRef]
- Zhu, F.; Davies, P.; Thompsett, A.R.; Kelly, S.M.; Tranter, G.E.; Hecht, L.; Isaacs, N.W.; Brown, D.R.; Barron, L.D. Raman optical activity and circular dichroism reveal dramatic differences in the influence of divalent copper and manganese ions on prion protein folding. Biochemistry 2008, 47, 2510–2517. [Google Scholar] [CrossRef]
- Tsenkova, R.N.; Iordanova, I.K.; Toyoda, K.; Brown, D.R. Prion protein fate governed by metal binding. Biochem. Biophys. Res. Commun. 2004, 325, 1005–1012. [Google Scholar] [CrossRef]
- Kim, N.H.; Choi, J.K.; Jeong, B.H.; Kim, J.I.; Kwon, M.S.; Carp, R.I.; Kim, Y.S. Effect of transition metals (Mn, Cu, Fe) and deoxycholic acid (DA) on the conversion of PrPC to PrPres. FASEB J. 2005, 19, 783–785. [Google Scholar] [PubMed]
- Li, X.; Dong, C.; Wang, G.; Zhou, R.M.; Shi, Q.; Tian, C.; Gao, C.; Mei, G.Y.; Chen, C.; Xu, K.; et al. Manganese induces changes of the biochemical characteristics of the recombinant wild-type and mutant PrPs. Med. Microbiol. Immunol. 2009, 198, 239–245. [Google Scholar] [CrossRef]
- Stefureac, R.I.; Madampage, C.A.; Andrievskaia, O.; Lee, J.S. Nanopore analysis of the interaction of metal ions with prion proteins and peptides. Biochem. Cell Biol. 2010, 88, 347–358. [Google Scholar] [CrossRef]
- Jobling, M.F.; Huang, X.; Stewart, L.R.; Barnham, K.J.; Curtain, C.; Volitakis, I.; Perugini, M.; White, A.R.; Cherny, R.A.; Masters, C.L.; et al. Copper and zinc binding modulates the aggregation and neurotoxic properties of the prion peptide PrP106–126. Biochemistry 2001, 40, 8073–8084. [Google Scholar] [CrossRef]
- Kim, N.H.; Park, S.J.; Jin, J.K.; Kwon, M.S.; Choi, E.K.; Carp, R.I.; Kim, Y.S. Increased ferric iron content and iron-induced oxidative stress in the brains of scrapie-infected mice. Brain Res. 2000, 884, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Mitteregger, G.; Korte, S.; Shakarami, M.; Herms, J.; Kretzschmar, H.A. Role of copper and manganese in prion disease progression. Brain Res. 2009, 1292, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, E.M.; Brown, D.R.; Alim, M.A.; Scholtzova, H.; Carp, R.; Meeker, H.C.; Prelli, F.; Frangione, B.; Wisniewski, T. Copper chelation delays the onset of prion disease. J. Biol. Chem. 2003, 278, 46199–46202. [Google Scholar] [CrossRef]
- Pollera, C.; Lucchini, B.; Formentin, E.; Bareggi, S.; Poli, G.; Ponti, W. Evaluation of anti-prionic activity of clioquinol in an in vivo model (Mesocricetus auratus). Vet. Res. Commun. 2005, 29 (Suppl. S2), 253–255. [Google Scholar] [CrossRef]
- Bareggi, S.R.; Cornelli, U. Clioquinol: Review of its mechanisms of action and clinical uses in neurodegenerative disorders. CNS Neurosci. Ther. 2012, 18, 41–46. [Google Scholar] [CrossRef]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Stepanyan, Z.; Carroll, M.; Guimond, M.P.; Hihi, A.; Hayes, S.; McBride, K.; Hekimi, S. The anti-neurodegeneration drug clioquinol inhibits the aging-associated protein CLK-1. J. Biol. Chem. 2009, 284, 314–323. [Google Scholar] [CrossRef]
- Ponti, W.; Sala, M.; Pollera, C.; Braida, D.; Poli, G.; Bareggi, S. In vivo model for the evaluation of molecules active towards transmissible spongiform encephalopathies. Vet. Res. Commun. 2004, 28, 307–310. [Google Scholar] [CrossRef]
- Brazier, M.W.; Wall, V.; Brazier, B.W.; Masters, C.L.; Collins, S.J. Therapeutic interventions ameliorating prion disease. Exp. Rev. Anti-Infect. Ther. 2009, 7, 83–105. [Google Scholar] [CrossRef]
- Hortells, P.; Monleón, E.; Acín, C.; Vargas, A.; Vasseur, V.; Salomon, A.; Ryffel, B.; Cesbron, J.Y.; Badiola, J.J.; Monzón, M. The effect of metal imbalances on scrapie neurodegeneration. Zoonoses Public Health 2010, 57, 358–366. [Google Scholar] [PubMed]
- Hijazi, N.; Shaked, Y.; Rosenmann, H.; Ben-Hur, T.; Gabizon, R. Copper binding to PrPC may inhibit prion disease propagation. Brain Res. 2003, 993, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Everyedit. Available online: http://www.everyedit.com (accessed on 13 February 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brazier, M.W.; Wedd, A.G.; Collins, S.J. Antioxidant and Metal Chelation-Based Therapies in the Treatment of Prion Disease. Antioxidants 2014, 3, 288-308. https://doi.org/10.3390/antiox3020288
Brazier MW, Wedd AG, Collins SJ. Antioxidant and Metal Chelation-Based Therapies in the Treatment of Prion Disease. Antioxidants. 2014; 3(2):288-308. https://doi.org/10.3390/antiox3020288
Chicago/Turabian StyleBrazier, Marcus W., Anthony G. Wedd, and Steven J. Collins. 2014. "Antioxidant and Metal Chelation-Based Therapies in the Treatment of Prion Disease" Antioxidants 3, no. 2: 288-308. https://doi.org/10.3390/antiox3020288
APA StyleBrazier, M. W., Wedd, A. G., & Collins, S. J. (2014). Antioxidant and Metal Chelation-Based Therapies in the Treatment of Prion Disease. Antioxidants, 3(2), 288-308. https://doi.org/10.3390/antiox3020288