

Mitochondrial Dysfunction, Oxidative Stress, and Therapeutic Strategies in Diabetes, Obesity, and Cardiovascular Disease
 , ,
, ,
Abstract
:1. Introduction
2. Mitochondrion Structure and Function
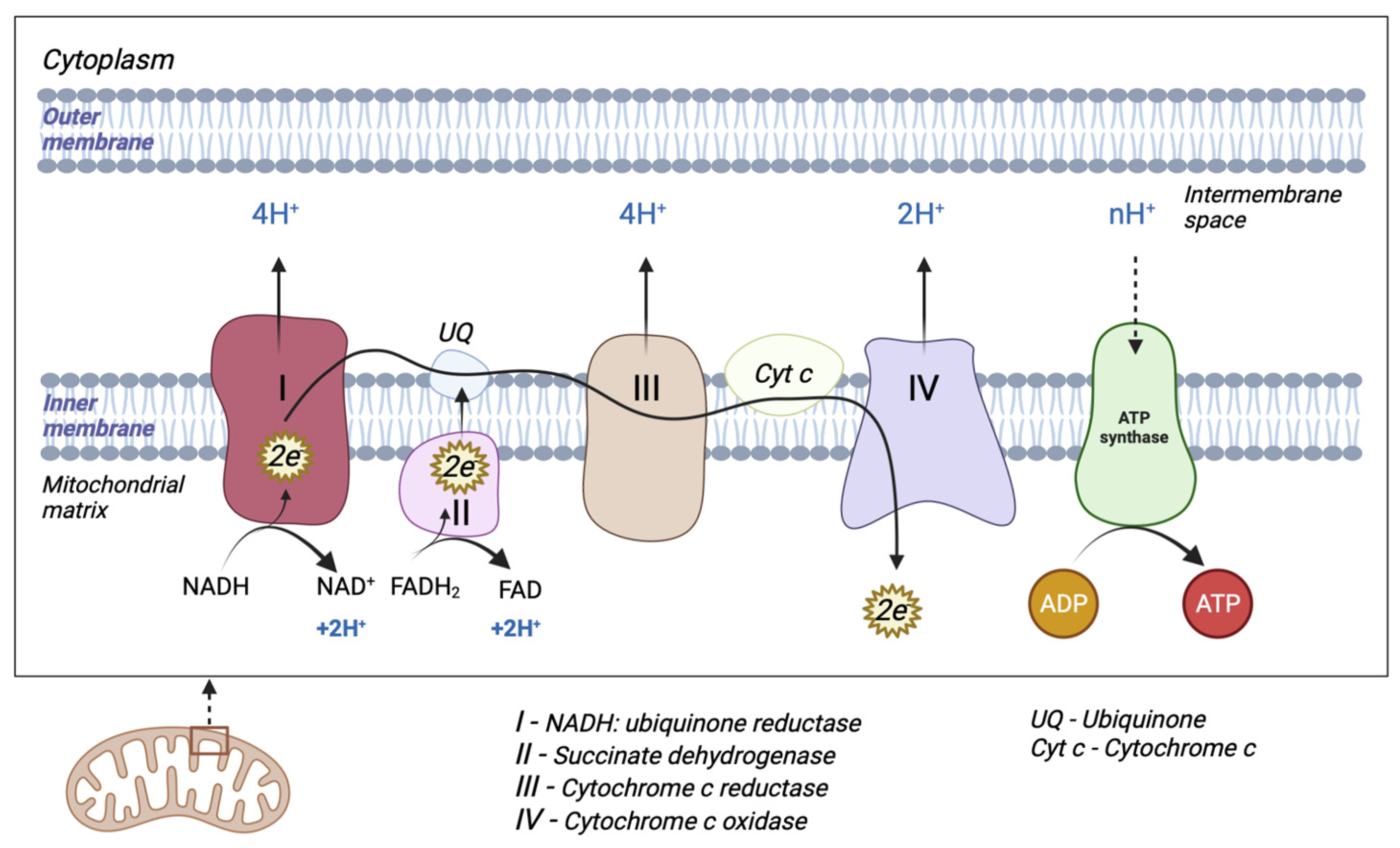
2.1. The Electron Transport Chain
2.2. Mitochondrial DNA Structure
2.3. Mitochondrial Biogenesis and Dynamics
2.4. Mitophagy
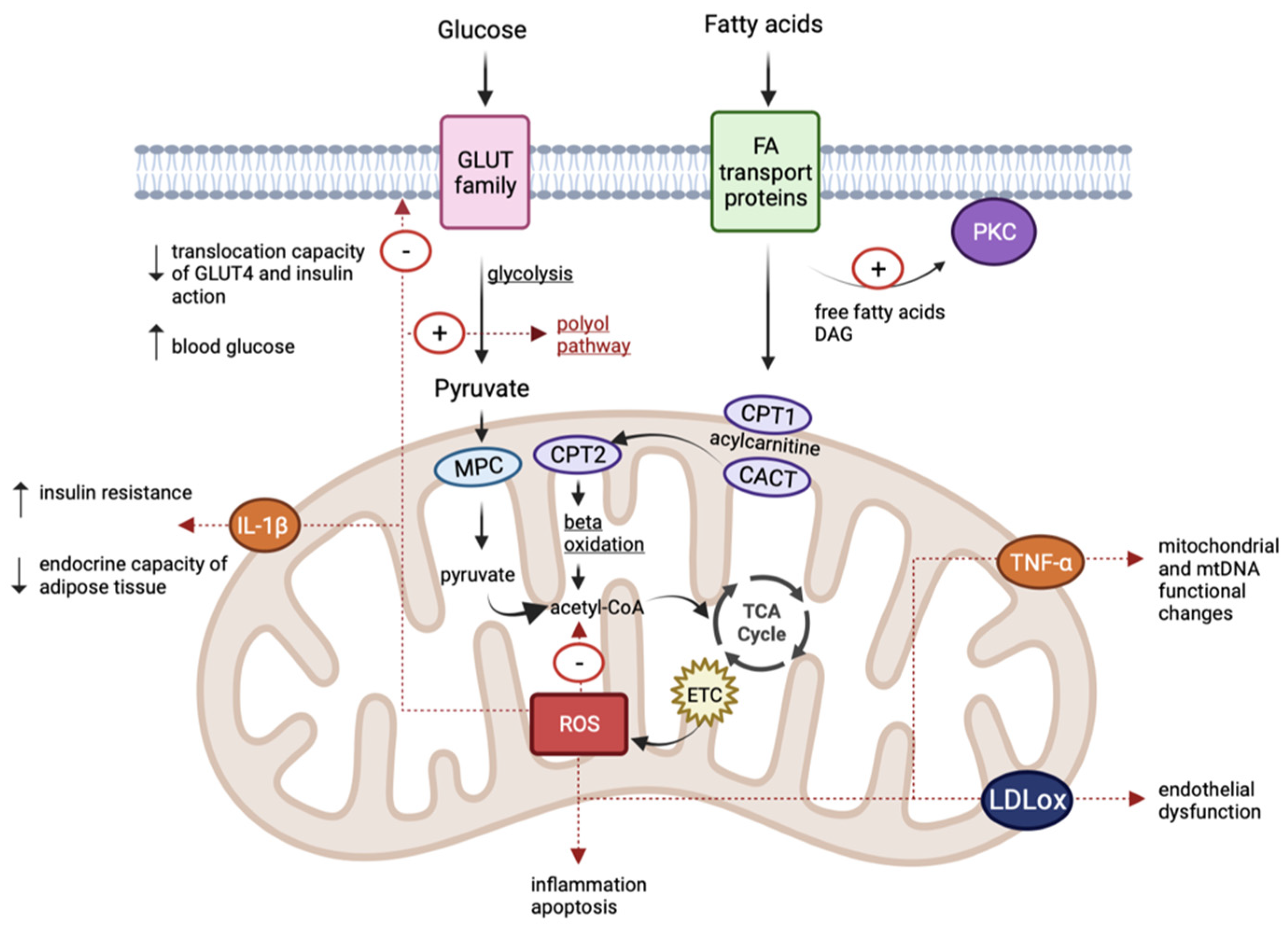
3. Oxidative Stress and Mitochondrial Dysfunctions

{kind=link}
{kind=link}
| ROS/RNS and Promoters of Free Radicals | Antioxidants System | Positive Impacts of Free Radicals | Negative Impacts of Free Radicals | Ref. |
| Superoxide radical anion (O2•−) Hydrogen peroxide (H2O2) Monoamine oxidase (MAO) Singlet oxygen(1O2) Hydroxyl radical (OH•) Hydroperoxyl radical (HOO•) Nitric oxide (NO•) | Superoxide dismutase (SOD): Mn-SOD, Cu/Zn-SOD Catalase Glutathione peroxidase Thioredoxin peroxidase NAD/NADP transhydrogenase Cytochrome c oxidase Vitamin E UQH2 | Signalling pathways and cell structures synthesis (within fibroblasts, endothelial cells, vascular smooth muscle cells, cardiac myocytes) Immune system activity rise Induction of mitogenic response Vasodilation Angiogenesis Wound healing | Lipid peroxidation Damage to cell membranes and lipoproteins Cytotoxic and mutagenic compounds Conformational modifications of proteins DNA lesions Loss of epigenetic information Hypertension Atherosclerosis | [66,67] |
| Disease | Biomarkers | Mechanism of action and effects of oxidative stress | ||
| Diabetes | ↑malondialdehyde ↑8-isoprostane ↑4-hydroxynonenal ↑glycated haemoglobin ↑advanced oxidation protein products ↑protein carbonyls ↓glutathione ↓superoxide dismutase ↓catalase | Lipid peroxidation Protein oxidation Decreased insulin activity Hyperglycaemia Stimulation of the polyol pathway Stimulation of glucose autoxidation Increase in advanced glycosylation end products mtDNA and proteins conformational modifications | [23,68,68,69,70,71] | |
| Obesity | ↑tumour necrosis factor-α ↑nuclear factor-κB ↑interleukin-1β ↑interleukin 6 ↑plasminogen activator inhibitor 1 ↓superoxide dismutase ↓catalase ↓vitamin A ↓vitamin E ↓vitamin C | Excess of pro-inflammatory cytokines and expression of adhesion molecules and growth factors Depleted antioxidant levels Increase in free fatty acids Thrombosis and insulin resistance | [72] | |
| Cardiovascular Disease | ↑oxidized low-density lipoprotein ↑tumour necrosis factor-α ↑nuclear factor-κB ↑interleukin-1β ↑interleukin 6 ↑8-Hydroxyl-2′-deoxyguanosine ↑myeloperoxidase ↑F2-isoprostanes ↑biopyrrins ↓vitamin C ↓glutathione peroxidase 1 ↓total antioxidant status | Endothelial dysfunction Inflammation in blood vessels Atherosclerosis Hypertension Cardiac hypertrophy Cardiomyocytes apoptosis Oxidative damage in DNA Lipid peroxidation | [73,74,75,76,77,78,79,80] | |
4. Insulin Resistance, Diabetes and Mitochondrial Dysfunctions
5. Obesity and Mitochondrial Dysfunctions
6. Cardiovascular Disease and Mitochondrial Dysfunctions
7. Pharmacological Strategies and Lifestyle Interventions in Mitochondrial Dysfunctions
8. Antioxidants
9. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| OXPHOS | oxidative phosphorylation |
| mtDNA | mitochondrial DNA |
| ROS | reactive oxygen species |
| MS | metabolic syndrome |
| T2DM | type II diabetes mellitus |
| NADH | reduced nicotinamide adenine dinucleotide |
| FADH2 | reduced flavin adenine dinucleotide |
| TCA | tricarboxylic acid |
| ETC | electron transport chain |
| UQ | ubiquinone |
| UQH2 | ubiquinol |
| Q- | semi-quinone radical ion Q- |
| Hs | heavy strand |
| Ls | light strand |
| NCR | non-coding region |
| D-loop | displacement loop |
| OH | origin of heavy-strand synthesis |
| HSP | heavy-strand promoter |
| LSP | light-strand promoter |
| TLR9 | toll-like receptor 9 |
| TOM | translocase of the outer mitochondrial membrane |
| TIM | translocase of the inner mitochondrial membrane |
| Mfn | mitofusins |
| OPA1 | optic atrophy 1 protein |
| Drp1 | dynamin-related protein 1 |
| Fis1 | fission protein |
| PGC-1α | proliferator-activated receptor gamma coactivator-1α |
| NRF | nuclear respiratory factor |
| TFAmt | mitochondrial transcription factor |
| AMPK | AMP-activated protein kinase |
| SIRT1 | silent information regulator 1 |
| O2•− | superoxide radical anions |
| H2O2 | hydrogen peroxide |
| MAO | monoamine oxidase |
| 1O2 | singlet oxygen |
| OH• | hydroxyl radical |
| HOO• | hydroperoxyl radical |
| NO• | nitric oxide |
| RNS | reactive nitrogen species |
| NOS | nitric oxide synthases |
| GLUT4 | glucose transporter 4 |
| EXT | exostosin |
| NAC | N-acetylcysteine |
| COQ10 | coenzyme Q10 |
| MitoQ | mitoquinone |
| MitoE | mitovitamin E |
| TPP+ | triphenylphosphonium cation |
References
- Friedman, J.R.; Nunnari, J. Mitochondrial Form and Function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Shadel, G.S. Expression and Maintenance of Mitochondrial DNA: New Insights into Human Disease Pathology. Am. J. Pathol. 2008, 172, 1445–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nass, M.M. The Circularity of Mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1966, 56, 1215–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brailoiu, E.; Shipsky, M.M.; Yan, G.; Abood, M.E.; Brailoiu, G.C. Mechanisms of Modulation of Brain Microvascular Endothelial Cells Function by Thrombin. Brain Res. 2017, 1657, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B. Reactive Species and Antioxidants. Redox Biology Is a Fundamental Theme of Aerobic Life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Holbrook, N.J. Oxidants, Oxidative Stress and the Biology of Ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Grundy, S.M. Metabolic Syndrome Pandemic. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, E.L.; VonWald, T.; Hansen, K. The Metabolic Syndrome. S D Med. 2015, 24–28. [Google Scholar] [PubMed]
- Huang, G.T.-J.; Gronthos, S.; Shi, S. Mesenchymal Stem Cells Derived from Dental Tissues vs. Those from Other Sources: Their Biology and Role in Regenerative Medicine. J. Dent. Res. 2009, 88, 792–806. [Google Scholar] [CrossRef]
- Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration. Cardiovascular Disease, Chronic Kidney Disease, and Diabetes Mortality Burden of Cardiometabolic Risk Factors from 1980 to 2010: A Comparative Risk Assessment. Lancet Diabetes Endocrinol. 2014, 2, 634–647. [Google Scholar] [CrossRef]
- Sherratt, H.S. Mitochondria: Structure and Function. Rev. Neurol. 1991, 147, 417–430. [Google Scholar]
- Chen, X.J.; Butow, R.A. The Organization and Inheritance of the Mitochondrial Genome. Nat. Rev. Genet. 2005, 6, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallner, G.; Sindelar, P.J. Regulation of Ubiquinone Metabolism. Free Radic. Biol. Med. 2000, 29, 285–294. [Google Scholar] [CrossRef]
- Ahmad, M.; Wolberg, A.; Kahwaji, C.I. Biochemistry, Electron Transport Chain. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial Metabolism of Reactive Oxygen Species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and Organization of the Human Mitochondrial Genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and Revision of the Cambridge Reference Sequence for Human Mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.-X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Shokolenko, I.; Alexeyev, M. Mitochondrial DNA: Consensuses and Controversies. DNA 2022, 2, 131–148. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and Reactive Oxygen Species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Yapa, N.M.B.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial Dynamics in Health and Disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef]
- Araiso, Y.; Imai, K.; Endo, T. Role of the TOM Complex in Protein Import into Mitochondria: Structural Views. Annu. Rev. Biochem. 2022, 91, 679–703. [Google Scholar] [CrossRef]
- Tang, K.; Zhao, Y.; Li, H.; Zhu, M.; Li, W.; Liu, W.; Zhu, G.; Xu, D.; Peng, W.; Xu, Y.-W. Translocase of Inner Membrane 50 Functions as a Novel Protective Regulator of Pathological Cardiac Hypertrophy. J. Am. Heart Assoc. 2017, 6, e004346. [Google Scholar] [CrossRef]
- Bauer, M.F.; Hofmann, S.; Neupert, W.; Brunner, M. Protein Translocation into Mitochondria: The Role of TIM Complexes. Trends Cell Biol. 2000, 10, 25–31. [Google Scholar] [CrossRef] [PubMed]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of Mitochondrial Fission and Fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Kaikini, A.A.; Kanchan, D.M.; Nerurkar, U.N.; Sathaye, S. Targeting Mitochondrial Dysfunction for the Treatment of Diabetic Complications: Pharmacological Interventions through Natural Products. Pharmacogn. Rev. 2017, 11, 128–135. [Google Scholar] [CrossRef] [Green Version]
- Popov, L.-D. Mitochondrial Biogenesis: An Update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular Definitions of Autophagy and Related Processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Live to Die Another Way: Modes of Programmed Cell Death and the Signals Emanating from Dying Cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-Consumption: The Interplay of Autophagy and Apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Wang, Y.; Li, B.; Shen, K.; Li, Q.; Ni, Y.; Huang, L. Mitophagy in Carcinogenesis, Drug Resistance and Anticancer Therapeutics. Cancer Cell Int. 2021, 21, 350. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Iorio, R.; Celenza, G.; Petricca, S. Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis. Cells 2021, 11, 30. [Google Scholar] [CrossRef]
- Kim, S.-J.; Ahn, D.-G.; Syed, G.H.; Siddiqui, A. The Essential Role of Mitochondrial Dynamics in Antiviral Immunity. Mitochondrion 2018, 41, 21–27. [Google Scholar] [CrossRef]
- Mitophagy Regulated by the PINK1-Parkin Pathway|IntechOpen. Available online: https://www.intechopen.com/chapters/49196 (accessed on 26 January 2023).
- Killackey, S.A.; Philpott, D.J.; Girardin, S.E. Mitophagy Pathways in Health and Disease. J. Cell Biol. 2020, 219, e202004029. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Fa, W.H.; Tian, C.R.; Yuan, C.S.; Jie, N. Mitophagy and Mitochondrial Dynamics in Type 2 Diabetes Mellitus Treatment. Aging 2022, 14, 2902–2919. [Google Scholar] [CrossRef]
- Riquelme, J.A.; Chavez, M.N.; Mondaca-Ruff, D.; Bustamante, M.; Vicencio, J.M.; Quest, A.F.G.; Lavandero, S. Therapeutic Targeting of Autophagy in Myocardial Infarction and Heart Failure. Expert Rev. Cardiovasc. Ther. 2016, 14, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Liu, F. Mitochondrial Stress: A Bridge between Mitochondrial Dysfunction and Metabolic Diseases? Cell Signal. 2011, 23, 1528–1533. [Google Scholar] [CrossRef] [Green Version]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial Dysfunction and Molecular Pathways of Disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef]
- Miller, D.M.; Buettner, G.R.; Aust, S.D. Transition Metals as Catalysts of “Autoxidation” Reactions. Free Radic. Biol. Med. 1990, 8, 95–108. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free Radicals and Antioxidants in Normal Physiological Functions and Human Disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative Stress: The Mitochondria-Dependent and Mitochondria-Independent Pathways of Apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef] [PubMed]
- Weisiger, R.A.; Fridovich, I. Superoxide Dismutase. Organelle Specificity. J. Biol. Chem. 1973, 248, 3582–3592. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, A. Antioxidant Function of Thioredoxin and Glutaredoxin Systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef]
- Nordberg, J.; Arnér, E.S. Reactive Oxygen Species, Antioxidants, and the Mammalian Thioredoxin System. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial Oxidative Stress: Implications for Cell Death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Kakhlon, O.; Manning, H.; Breuer, W.; Melamed-Book, N.; Lu, C.; Cortopassi, G.; Munnich, A.; Cabantchik, Z.I. Cell Functions Impaired by Frataxin Deficiency Are Restored by Drug-Mediated Iron Relocation. Blood 2008, 112, 5219–5227. [Google Scholar] [CrossRef]
- De Grey, A.D.N.J. HO2*: The Forgotten Radical. DNA Cell Biol. 2002, 21, 251–257. [Google Scholar] [CrossRef]
- Migliaccio, E.; Giorgio, M.; Pelicci, P.G. Apoptosis and Aging: Role of P66Shc Redox Protein. Antioxid. Redox Signal. 2006, 8, 600–608. [Google Scholar] [CrossRef]
- Cosso, R.G.; Turim, J.; Nantes, I.L.; Almeida, A.M.; Mascio, P.D.; Vercesi, A.E. Mitochondrial Permeability Transition Induced by Chemically Generated Singlet Oxygen. J. Bioenerg. Biomembr. 2002, 34, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Nantes, I.L.; Faljoni-Alario, A.; Vercesi, A.E.; Santos, K.E.; Bechara, E.J. Liposome Effect on the Cytochrome C-Catalyzed Peroxidation of Carbonyl Substrates to Triplet Species. Free Radic. Biol. Med. 1998, 25, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Cassina, A.; Hodara, R. Nitric Oxide and Peroxynitrite Interactions with Mitochondria. Biol. Chem. 2002, 383, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative Stress in Chronic Kidney Disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.A.P.; Romagna, C.D.; Pereira, J.L.; Souza-Pinto, N.C. The Role of Mitochondrial DNA Damage in the Citotoxicity of Reactive Oxygen Species. J. Bioenerg. Biomembr. 2011, 43, 25–29. [Google Scholar] [CrossRef]
- Barber, R.G.; Grenier, Z.A.; Burkhead, J.L. Copper Toxicity Is Not Just Oxidative Damage: Zinc Systems and Insight from Wilson Disease. Biomedicines 2021, 9, 316. [Google Scholar] [CrossRef]
- Chen, P.; Bornhorst, J.; Aschner, M. Manganese Metabolism in Humans. Front. Biosci. 2018, 23, 1655–1679. [Google Scholar] [CrossRef] [Green Version]
- Wolff, N.A.; Ghio, A.J.; Garrick, L.M.; Garrick, M.D.; Zhao, L.; Fenton, R.A.; Thévenod, F. Evidence for Mitochondrial Localization of Divalent Metal Transporter 1 (DMT1). FASEB J. 2014, 28, 2134–2145. [Google Scholar] [CrossRef]
- Mezzaroba, L.; Alfieri, D.F.; Colado Simão, A.N.; Vissoci Reiche, E.M. The Role of Zinc, Copper, Manganese and Iron in Neurodegenerative Diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef]
- Menon, A.V.; Chang, J.; Kim, J. Mechanisms of Divalent Metal Toxicity in Affective Disorders. Toxicology 2016, 339, 58–72. [Google Scholar] [CrossRef] [Green Version]
- Krauss, S.; Zhang, C.-Y.; Lowell, B.B. The Mitochondrial Uncoupling-Protein Homologues. Nat. Rev. Mol. Cell. Biol. 2005, 6, 248–261. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jîtcă, G.; Ősz, B.E.; Tero-Vescan, A.; Miklos, A.P.; Rusz, C.-M.; Bătrînu, M.-G.; Vari, C.E. Positive Aspects of Oxidative Stress at Different Levels of the Human Body: A Review. Antioxidants 2022, 11, 572. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, B.K.; Pandey, K.B.; Abidi, A.B.; Rizvi, S.I. Markers of Oxidative Stress during Diabetes Mellitus. J. Biomark. 2013, 2013, 378790. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I.H. Molecular Mechanisms of ROS Production and Oxidative Stress in Diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Ding, X.-W.; Robinson, M.; Li, R.; Aldhowayan, H.; Geetha, T.; Babu, J.R. Mitochondrial Dysfunction and Beneficial Effects of Mitochondria-Targeted Small Peptide SS-31 in Diabetes Mellitus and Alzheimer’s Disease. Pharmacol. Res. 2021, 171, 105783. [Google Scholar] [CrossRef] [PubMed]
- Leguisamo, N.M.; Lehnen, A.M.; Machado, U.F.; Okamoto, M.M.; Markoski, M.M.; Pinto, G.H.; Schaan, B.D. GLUT4 Content Decreases along with Insulin Resistance and High Levels of Inflammatory Markers in Rats with Metabolic Syndrome. Cardiovasc. Diabetol. 2012, 11, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative Stress in Obesity: A Critical Component in Human Diseases. Int. J. Mol. Sci. 2014, 16, 378–400. [Google Scholar] [CrossRef] [Green Version]
- Stephens, J.W.; Gable, D.R.; Hurel, S.J.; Miller, G.J.; Cooper, J.A.; Humphries, S.E. Increased Plasma Markers of Oxidative Stress Are Associated with Coronary Heart Disease in Males with Diabetes Mellitus and with 10-Year Risk in a Prospective Sample of Males. Clin. Chem. 2006, 52, 446–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, Y.; Nakamura, K.; Kimura, H.; Nishii, N.; Watanabe, A.; Banba, K.; Miura, A.; Nagase, S.; Sakuragi, S.; Kusano, K.F.; et al. Elevated Levels of Oxidative DNA Damage in Serum and Myocardium of Patients With Heart Failure. Circ. J. 2006, 70, 1001–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vita, J.A.; Keaney, J.F.; Raby, K.E.; Morrow, J.D.; Freedman, J.E.; Lynch, S.; Koulouris, S.N.; Hankin, B.R.; Frei, B. Low Plasma Ascorbic Acid Independently Predicts the Presence of an Unstable Coronary Syndrome. J. Am. Coll. Cardiol. 1998, 31, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Polidori, M.C.; Praticó, D.; Savino, K.; Rokach, J.; Stahl, W.; Mecocci, P. Increased F2 Isoprostane Plasma Levels in Patients with Congestive Heart Failure Are Correlated with Antioxidant Status and Disease Severity. J. Card. Fail. 2004, 10, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, S.; Rupprecht, H.J.; Bickel, C.; Torzewski, M.; Hafner, G.; Tiret, L.; Smieja, M.; Cambien, F.; Meyer, J.; Lackner, K.J.; et al. Glutathione Peroxidase 1 Activity and Cardiovascular Events in Patients with Coronary Artery Disease. N. Engl. J. Med. 2003, 349, 1605–1613. [Google Scholar] [CrossRef] [Green Version]
- Ndrepepa, G.; Braun, S.; Mehilli, J.; von Beckerath, N.; Schömig, A.; Kastrati, A. Myeloperoxidase Level in Patients with Stable Coronary Artery Disease and Acute Coronary Syndromes. Eur. J. Clin. Investig. 2008, 38, 90–96. [Google Scholar] [CrossRef]
- Lahera, V.; de Las Heras, N.; López-Farré, A.; Manucha, W.; Ferder, L. Role of Mitochondrial Dysfunction in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The Role of Mitochondrial Dysfunction in Cardiovascular Disease: A Brief Review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Melmer, A.; Laimer, M. Treatment Goals in Diabetes. Endocr. Dev. 2016, 31, 1–27. [Google Scholar] [CrossRef]
- Turner, N.; Heilbronn, L.K. Is Mitochondrial Dysfunction a Cause of Insulin Resistance? Trends Endocrinol. Metab. 2008, 19, 324–330. [Google Scholar] [CrossRef]
- Chen, L.; Xu, W.M.; Zhang, D. Association of Abdominal Obesity, Insulin Resistance, and Oxidative Stress in Adipose Tissue in Women with Polycystic Ovary Syndrome. Fertil. Steril. 2014, 102, 1167–1174.e4. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Biswas, S.; Mukherjee, S.; Bandyopadhyay, S.K. Association of Oxidative Stress and Obesity with Insulin Resistance in Type 2 Diabetes Mellitus. Mymensingh Med. J. 2016, 25, 148–152. [Google Scholar]
- Korkmaz, G.G.; Altınoglu, E.; Civelek, S.; Sozer, V.; Erdenen, F.; Tabak, O.; Uzun, H. The Association of Oxidative Stress Markers with Conventional Risk Factors in the Metabolic Syndrome. Metabolism 2013, 62, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Duanmu, X.; Zeng, L.; Liu, B.; Song, Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells 2019, 8, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szendroedi, J.; Phielix, E.; Roden, M. The Role of Mitochondria in Insulin Resistance and Type 2 Diabetes Mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.; Chiou, C.C.; Chang, P.Y.; Wu, J.T. Urinary 8-OHdG: A Marker of Oxidative Stress to DNA and a Risk Factor for Cancer, Atherosclerosis and Diabetics. Clin. Chim. Acta 2004, 339, 1–9. [Google Scholar] [CrossRef]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative Toxicity in Diabetes and Alzheimer’s Disease: Mechanisms behind ROS/RNS Generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef]
- Gastaldi, G.; Giacobino, J.P.; Ruiz, J. Metabolic syndrome, a mitochondrial disease? Rev. Med. Suisse 2008, 4, 1387–1388, 1390–1391. [Google Scholar]
- Rendra, E.; Riabov, V.; Mossel, D.M.; Sevastyanova, T.; Harmsen, M.C.; Kzhyshkowska, J. Reactive Oxygen Species (ROS) in Macrophage Activation and Function in Diabetes. Immunobiology 2019, 224, 242–253. [Google Scholar] [CrossRef]
- Darenskaya, M.A.; Kolesnikova, L.I.; Kolesnikov, S.I. Oxidative Stress: Pathogenetic Role in Diabetes Mellitus and Its Complications and Therapeutic Approaches to Correction. Bull. Exp. Biol. Med. 2021, 171, 179–189. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in Skeletal Muscle Mitochondrial Function with Aging in Humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial Dynamics in Type 2 Diabetes: Pathophysiological Implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Zheng, L.D.; Linarelli, L.E.; Liu, L.; Wall, S.S.; Greenawald, M.H.; Seidel, R.W.; Estabrooks, P.A.; Almeida, F.A.; Cheng, Z. Insulin Resistance Is Associated with Epigenetic and Genetic Regulation of Mitochondrial DNA in Obese Humans. Clin. Epigenetics 2015, 7, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, C.; Poulsen, P.; Simonsson, S.; Rönn, T.; Holmkvist, J.; Almgren, P.; Hagert, P.; Nilsson, E.; Mabey, A.G.; Nilsson, P.; et al. Genetic and Epigenetic Factors Are Associated with Expression of Respiratory Chain Component NDUFB6 in Human Skeletal Muscle. J. Clin. Investig. 2007, 117, 3427–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Schmelz, E.M.; Liu, D.; Hulver, M.W. Targeting Mitochondrial Alterations to Prevent Type 2 Diabetes--Evidence from Studies of Dietary Redox-Active Compounds. Mol. Nutr. Food Res. 2014, 58, 1739–1749. [Google Scholar] [CrossRef]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial Dysfunction in Obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Prasun, P. Mitochondrial Dysfunction in Metabolic Syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Xu, L.; Yan, X.; Zhao, Y.; Wang, J.; Liu, B.; Yu, S.; Fu, J.; Liu, Y.; Su, J. Macrophage Polarization Mediated by Mitochondrial Dysfunction Induces Adipose Tissue Inflammation in Obesity. Int. J. Mol. Sci. 2022, 23, 9252. [Google Scholar] [CrossRef]
- Cade, W.T. The Manifold Role of the Mitochondria in Skeletal Muscle Insulin Resistance. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 267–272. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.-C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets Release Mitochondria Serving as Substrate for Bactericidal Group IIA-Secreted Phospholipase A2 to Promote Inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of Mitochondria from Astrocytes to Neurons after Stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brestoff, J.R.; Wilen, C.B.; Moley, J.R.; Li, Y.; Zou, W.; Malvin, N.P.; Rowen, M.N.; Saunders, B.T.; Ma, H.; Mack, M.R.; et al. Intercellular Mitochondria Transfer to Macrophages Regulates White Adipose Tissue Homeostasis and Is Impaired in Obesity. Cell Metab. 2021, 33, 270–282.e8. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial Dysfunction in Type 2 Diabetes Mellitus: An Organ-Based Analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef] [PubMed]
- Kaaman, M.; Sparks, L.M.; van Harmelen, V.; Smith, S.R.; Sjölin, E.; Dahlman, I.; Arner, P. Strong Association between Mitochondrial DNA Copy Number and Lipogenesis in Human White Adipose Tissue. Diabetologia 2007, 50, 2526–2533. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Lee, D.-C.; Im, J.-A.; Lee, J.-W. Mitochondrial DNA Copy Number in Peripheral Blood Is Independently Associated with Visceral Fat Accumulation in Healthy Young Adults. Int. J. Endocrinol. 2014, 2014, 586017. [Google Scholar] [CrossRef] [Green Version]
- Cox, F.F.; Misiou, A.; Vierkant, A.; Ale-Agha, N.; Grandoch, M.; Haendeler, J.; Altschmied, J. Protective Effects of Curcumin in Cardiovascular Diseases-Impact on Oxidative Stress and Mitochondria. Cells 2022, 11, 342. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-Targeted Drugs. Curr. Mol. Pharmacol. 2019, 12, 202–214. [Google Scholar] [CrossRef]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial Dynamics, Mitophagy and Cardiovascular Disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef]
- Suematsu, N.; Tsutsui, H.; Wen, J.; Kang, D.; Ikeuchi, M.; Ide, T.; Hayashidani, S.; Shiomi, T.; Kubota, T.; Hamasaki, N.; et al. Oxidative Stress Mediates Tumor Necrosis Factor-Alpha-Induced Mitochondrial DNA Damage and Dysfunction in Cardiac Myocytes. Circulation 2003, 107, 1418–1423. [Google Scholar] [CrossRef] [Green Version]
- Stoccoro, A.; Coppedè, F. Mitochondrial DNA Methylation and Human Diseases. Int. J. Mol. Sci. 2021, 22, 4594. [Google Scholar] [CrossRef]
- Röckl, K.S.C.; Witczak, C.A.; Goodyear, L.J. Signaling Mechanisms in Skeletal Muscle: Acute Responses and Chronic Adaptations to Exercise. IUBMB Life 2008, 60, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloway, G.P. Mitochondrial Function and Dysfunction in Exercise and Insulin Resistance. Appl. Physiol. Nutr. Metab. 2009, 34, 440–446. [Google Scholar] [CrossRef]
- Joseph, A.-M.; Hood, D.A. Relationships between Exercise, Mitochondrial Biogenesis and Type 2 Diabetes. Med. Sport Sci. 2014, 60, 48–61. [Google Scholar] [CrossRef]
- Jabczyk, M.; Nowak, J.; Hudzik, B.; Zubelewicz-Szkodzińska, B. Curcumin in Metabolic Health and Disease. Nutrients 2021, 13, 4440. [Google Scholar] [CrossRef] [PubMed]
- Toogood, P.L. Mitochondrial Drugs. Curr. Opin. Chem. Biol. 2008, 12, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Milane, L.; Trivedi, M.; Singh, A.; Talekar, M.; Amiji, M. Mitochondrial Biology, Targets, and Drug Delivery. J. Control. Release 2015, 207, 40–58. [Google Scholar] [CrossRef]
- Ishigaki, Y.; Katagiri, H.; Yamada, T.; Ogihara, T.; Imai, J.; Uno, K.; Hasegawa, Y.; Gao, J.; Ishihara, H.; Shimosegawa, T.; et al. Dissipating Excess Energy Stored in the Liver Is a Potential Treatment Strategy for Diabetes Associated with Obesity. Diabetes 2005, 54, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Nübel, T.; Ricquier, D. Respiration under Control of Uncoupling Proteins: Clinical Perspective. Horm. Res. 2006, 65, 300–310. [Google Scholar] [CrossRef]
- Turkmen, K.; Karagoz, A.; Kucuk, A. Sirtuins as Novel Players in the Pathogenesis of Diabetes Mellitus. World J. Diabetes 2014, 5, 894–900. [Google Scholar] [CrossRef]
- Huynh, F.K.; Hershberger, K.A.; Hirschey, M.D. Targeting Sirtuins for the Treatment of Diabetes. Diabetes Manag. 2013, 3, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Kume, S.; Kanasaki, K.; Takeda-Watanabe, A.; Koya, D. Sirtuins as Possible Drug Targets in Type 2 Diabetes. Curr. Drug Targets 2013, 14, 622–636. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Meuer, K.; Suppanz, I.E.; Lingor, P.; Planchamp, V.; Göricke, B.; Fichtner, L.; Braus, G.H.; Dietz, G.P.H.; Jakobs, S.; Bähr, M.; et al. Cyclin-Dependent Kinase 5 Is an Upstream Regulator of Mitochondrial Fission during Neuronal Apoptosis. Cell Death Differ. 2007, 14, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Qvit, N.; Su, Y.-C.; Mochly-Rosen, D. A Novel Drp1 Inhibitor Diminishes Aberrant Mitochondrial Fission and Neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Amanakis, G.; Murphy, E. Cyclophilin D: An Integrator of Mitochondrial Function. Front. Physiol. 2020, 11, 595. [Google Scholar] [CrossRef]
- Mailloux, R.J. Application of Mitochondria-Targeted Pharmaceuticals for the Treatment of Heart Disease. Curr. Pharm. Des. 2016, 22, 4763–4779. [Google Scholar] [CrossRef]
- Silva, F.S.G.; Simoes, R.F.; Couto, R.; Oliveira, P.J. Targeting Mitochondria in Cardiovascular Diseases. Curr. Pharm. Des. 2016, 22, 5698–5717. [Google Scholar] [CrossRef]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.-C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic Inhibition of Mitochondrial Reactive Oxygen Species with Mito-TEMPO Reduces Diabetic Cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Harris, N.; Ren, J.; Han, X. Mitochondria-Targeted Antioxidant Prevents Cardiac Dysfunction Induced by Tafazzin Gene Knockdown in Cardiac Myocytes. Oxid. Med. Cell Longev. 2014, 2014, 654198. [Google Scholar] [CrossRef] [Green Version]
- Yorek, M.A. The Role of Oxidative Stress in Diabetic Vascular and Neural Disease. Free Radic. Res. 2003, 37, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Van Thiel, D.H.; Shah, N.; Mobarhan, S. Nonalcoholic Fatty Liver Disease: Pathogenesis and the Role of Antioxidants. Nutr. Rev. 2002, 60, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Rani, M.; Aggarwal, R.; Vohra, K. Effect of N-Acetylcysteine on Metabolic Profile in Metabolic Syndrome Patients. Metab. Syndr. Relat. Disord. 2020, 18, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Aaseth, J.; Alexander, J.; Alehagen, U. Coenzyme Q10 Supplementation–In Ageing and Disease. Mech. Ageing Dev. 2021, 197, 111521. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Martorell, M.; Arbiser, J.L.; Sureda, A.; Martins, N.; Maurya, P.K.; Sharifi-Rad, M.; Kumar, P.; Sharifi-Rad, J. Antioxidants: Positive or Negative Actors? Biomolecules 2018, 8, 124. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxid. Med. Cell Longev. 2020, 2020, 8837893. [Google Scholar] [CrossRef]
- Yang, J.; Suo, H.; Song, J. Protective Role of Mitoquinone against Impaired Mitochondrial Homeostasis in Metabolic Syndrome. Crit. Rev. Food Sci. Nutr. 2021, 61, 3857–3875. [Google Scholar] [CrossRef]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective Targeting of a Redox-Active Ubiquinone to Mitochondria within Cells: Antioxidant and Antiapoptotic Properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Manczak, M.; Reddy, P.H. Mitochondria-Targeted Molecules MitoQ and SS31 Reduce Mutant Huntingtin-Induced Mitochondrial Toxicity and Synaptic Damage in Huntington’s Disease. Hum. Mol. Genet. 2016, 25, 1739–1753. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Manczak, M.; Shirendeb, U.P.; Reddy, P.H. MitoQ, a Mitochondria-Targeted Antioxidant, Delays Disease Progression and Alleviates Pathogenesis in an Experimental Autoimmune Encephalomyelitis Mouse Model of Multiple Sclerosis. Biochim. Et Biophys. Acta (BBA)–Mol. Basis Dis. 2013, 1832, 2322–2331. [Google Scholar] [CrossRef] [Green Version]
- Powell, R.D.; Swet, J.H.; Kennedy, K.L.; Huynh, T.T.; Murphy, M.P.; Mckillop, I.H.; Evans, S.L. MitoQ Modulates Oxidative Stress and Decreases Inflammation Following Hemorrhage. J. Trauma. Acute Care Surg. 2015, 78, 573–579. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective Targeting of an Antioxidant to Mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef]
- Titova, E.; Shagieva, G.; Ivanova, O.; Domnina, L.; Domninskaya, M.; Strelkova, O.; Khromova, N.; Kopnin, P.; Chernyak, B.; Skulachev, V.; et al. Mitochondria-Targeted Antioxidant SkQ1 Suppresses Fibrosarcoma and Rhabdomyosarcoma Tumour Cell Growth. Cell Cycle 2018, 17, 1797–1811. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Harashima, H. MITO-Porter for Mitochondrial Delivery and Mitochondrial Functional Analysis. Handb. Exp. Pharmacol. 2017, 240, 457–472. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cojocaru, K.-A.; Luchian, I.; Goriuc, A.; Antoci, L.-M.; Ciobanu, C.-G.; Popescu, R.; Vlad, C.-E.; Blaj, M.; Foia, L.G. Mitochondrial Dysfunction, Oxidative Stress, and Therapeutic Strategies in Diabetes, Obesity, and Cardiovascular Disease. Antioxidants 2023, 12, 658. https://doi.org/10.3390/antiox12030658
Cojocaru K-A, Luchian I, Goriuc A, Antoci L-M, Ciobanu C-G, Popescu R, Vlad C-E, Blaj M, Foia LG. Mitochondrial Dysfunction, Oxidative Stress, and Therapeutic Strategies in Diabetes, Obesity, and Cardiovascular Disease. Antioxidants. 2023; 12(3):658. https://doi.org/10.3390/antiox12030658
Chicago/Turabian StyleCojocaru, Karina-Alexandra, Ionut Luchian, Ancuta Goriuc, Lucian-Mihai Antoci, Cristian-Gabriel Ciobanu, Roxana Popescu, Cristiana-Elena Vlad, Mihaela Blaj, and Liliana Georgeta Foia. 2023. "Mitochondrial Dysfunction, Oxidative Stress, and Therapeutic Strategies in Diabetes, Obesity, and Cardiovascular Disease" Antioxidants 12, no. 3: 658. https://doi.org/10.3390/antiox12030658