Expression Comparison of Oil Biosynthesis Genes in Oil Palm Mesocarp Tissue Using Custom Array

Abstract
:1. Introduction
2. Experimental Section
2.1. Experimental Setup and Sampling
2.2. Oil Palm Mesocarp RNA Extraction
2.3. Building the Transcriptome, Custom Design of the Oil Palm Mesocarp Array and Agilent Commercial Array
2.4. Synthesis of cRNA, Microarray Hybridization and Scanning
2.5. Data Extraction, Normalisation and Comparisons
2.6. Quantitative Real-Time PCR
3. Results and Discussion
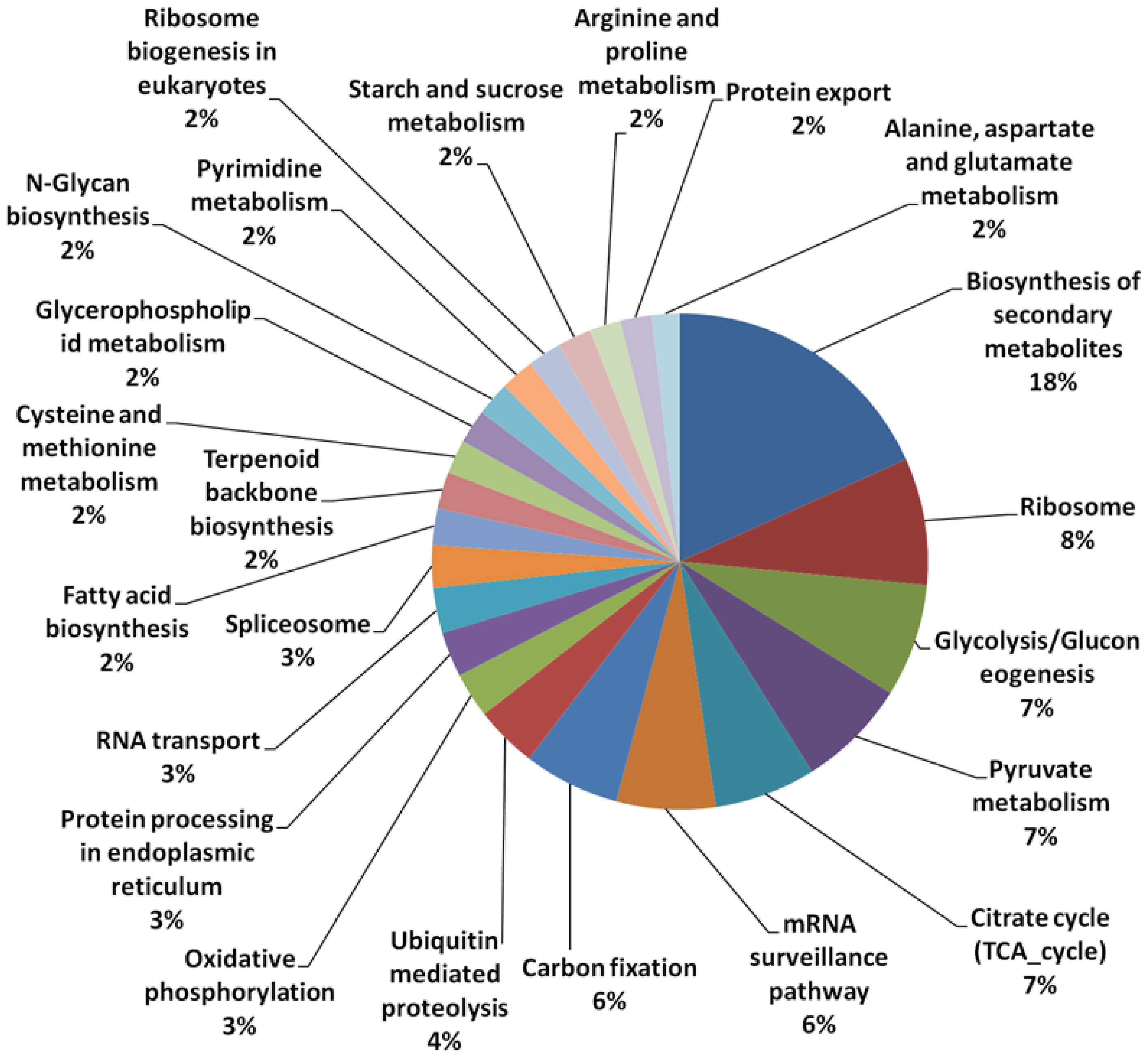
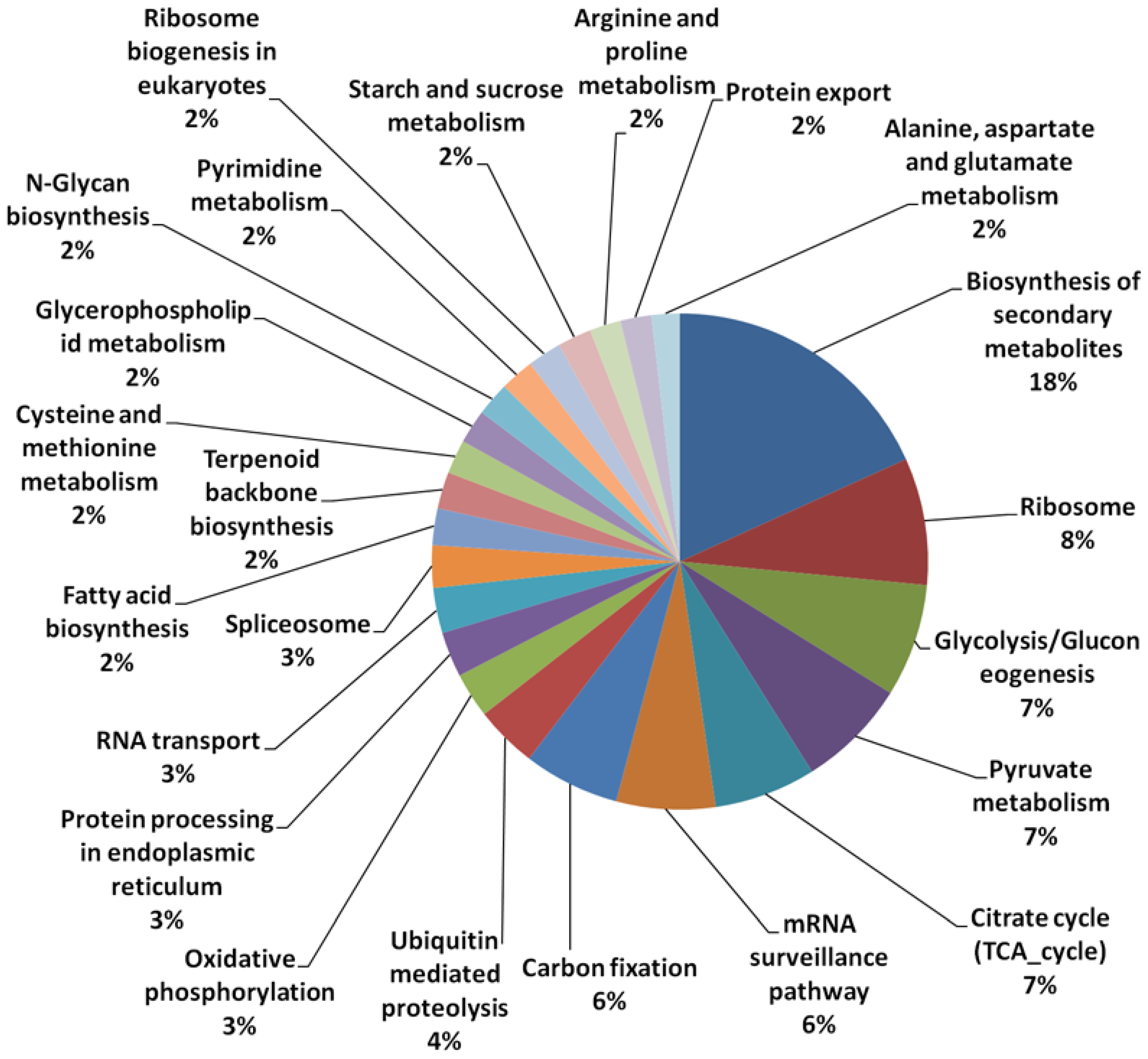
3.1. Array Design and Sequence Annotation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript | Number of Sequences | Percentage (%) |
|---|---|---|
| Transcripts selected for probe design | 31,804 | |
| Transcripts with Uniprot database hits | 15,695 | 49.3 |
| Transcripts with KEGG database hits | 2569 | 8.1 |
| Transcripts with KEGG Orthologs ID (pathways) hits | 624 | 1.9 |
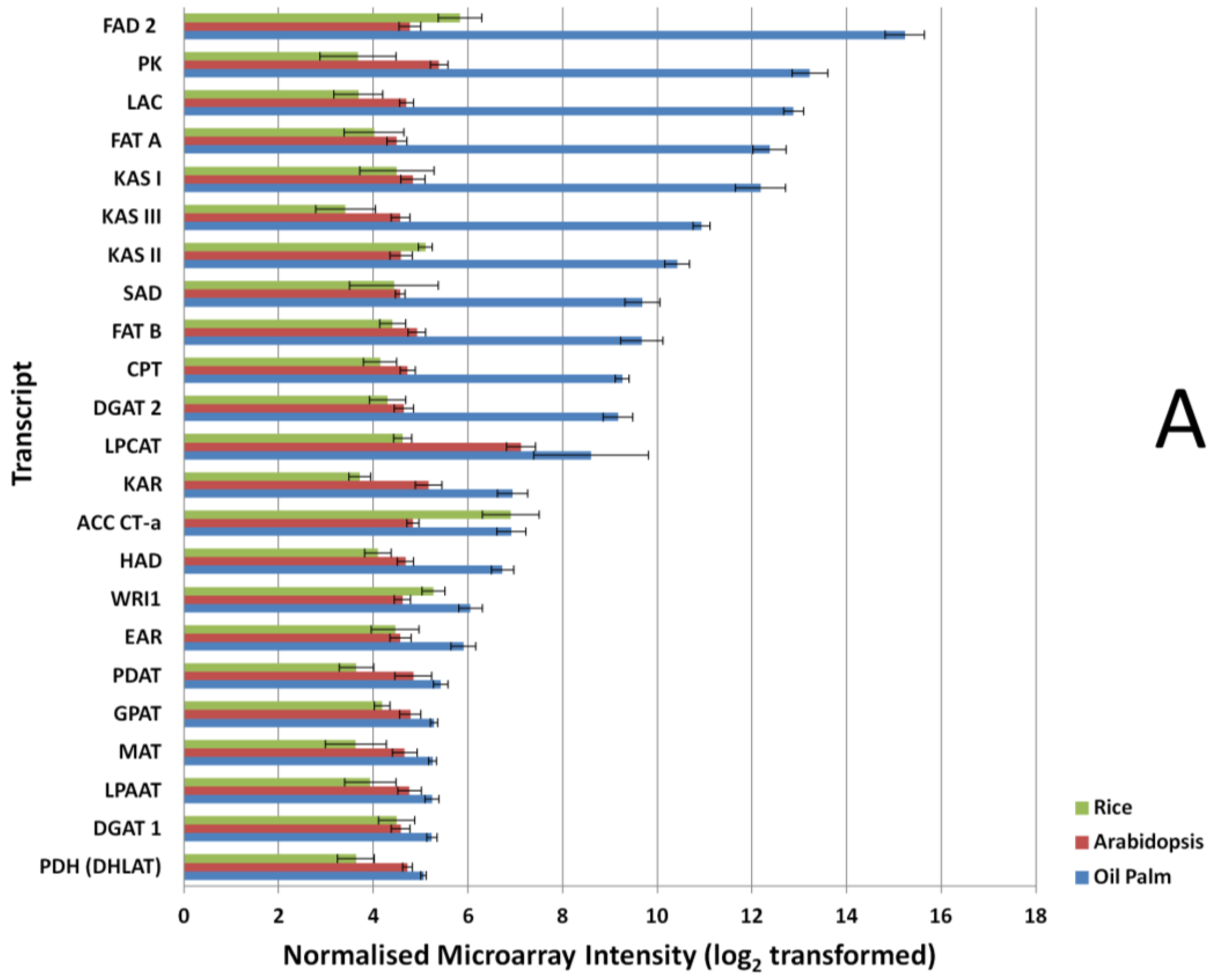
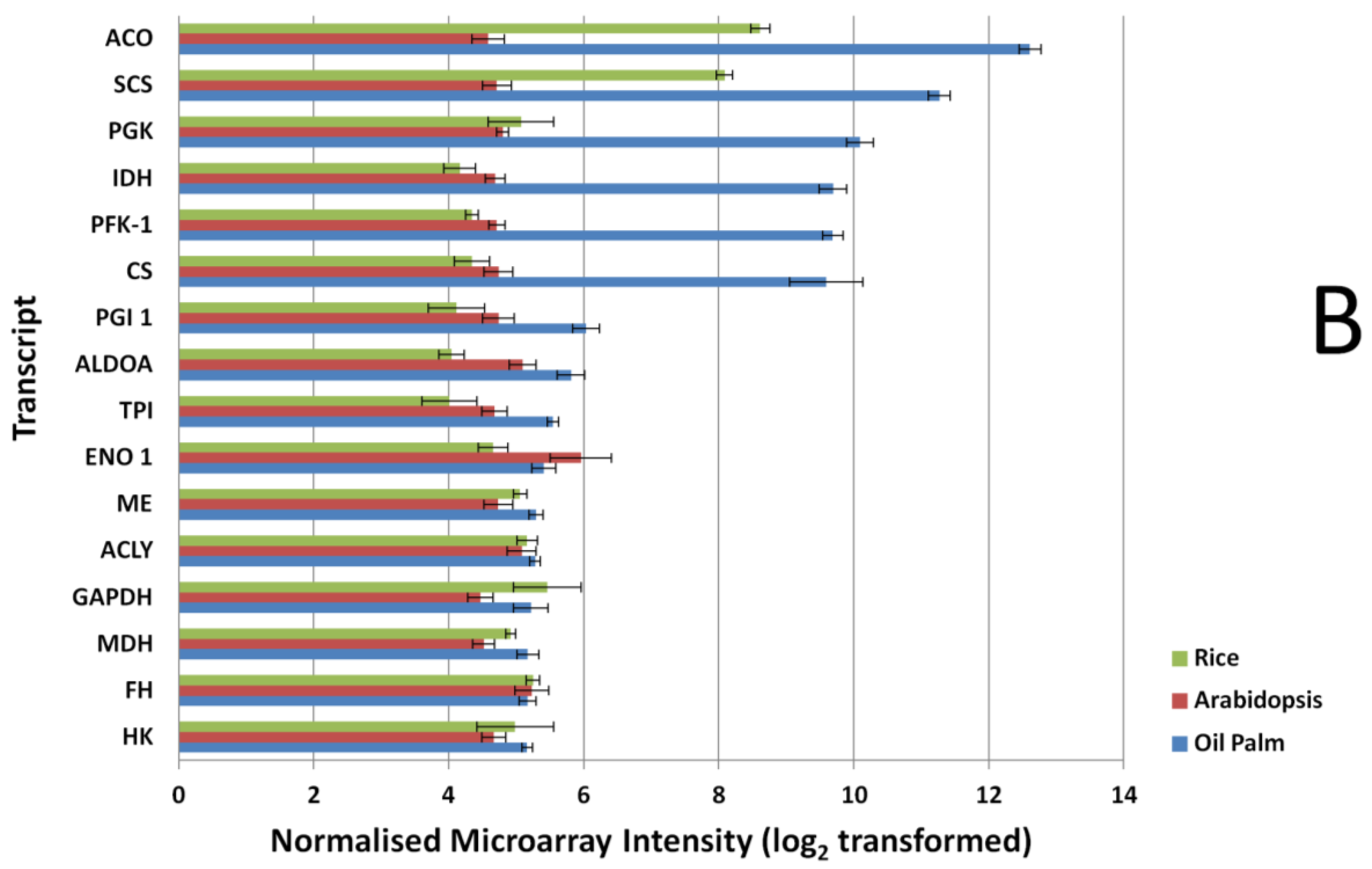
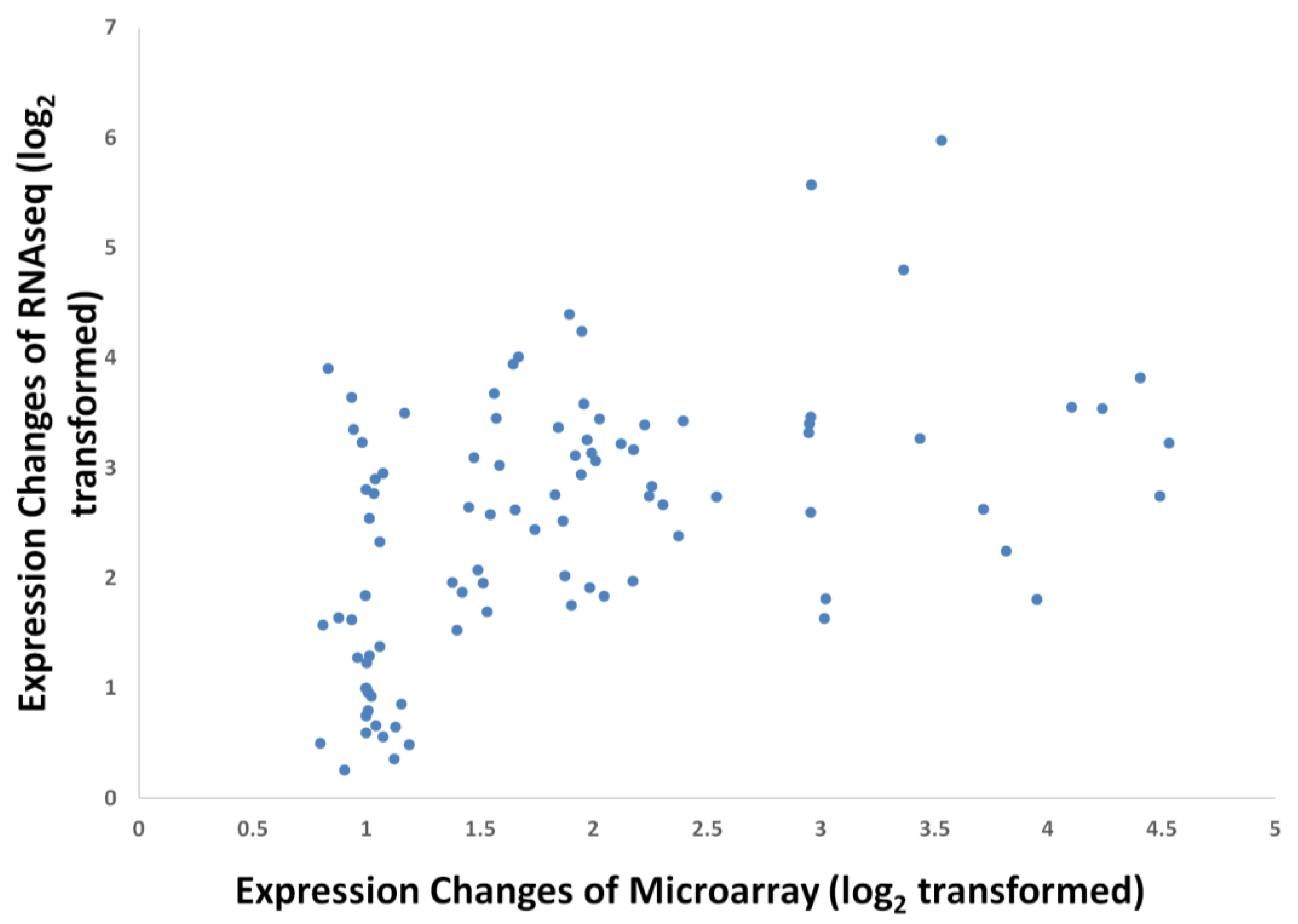
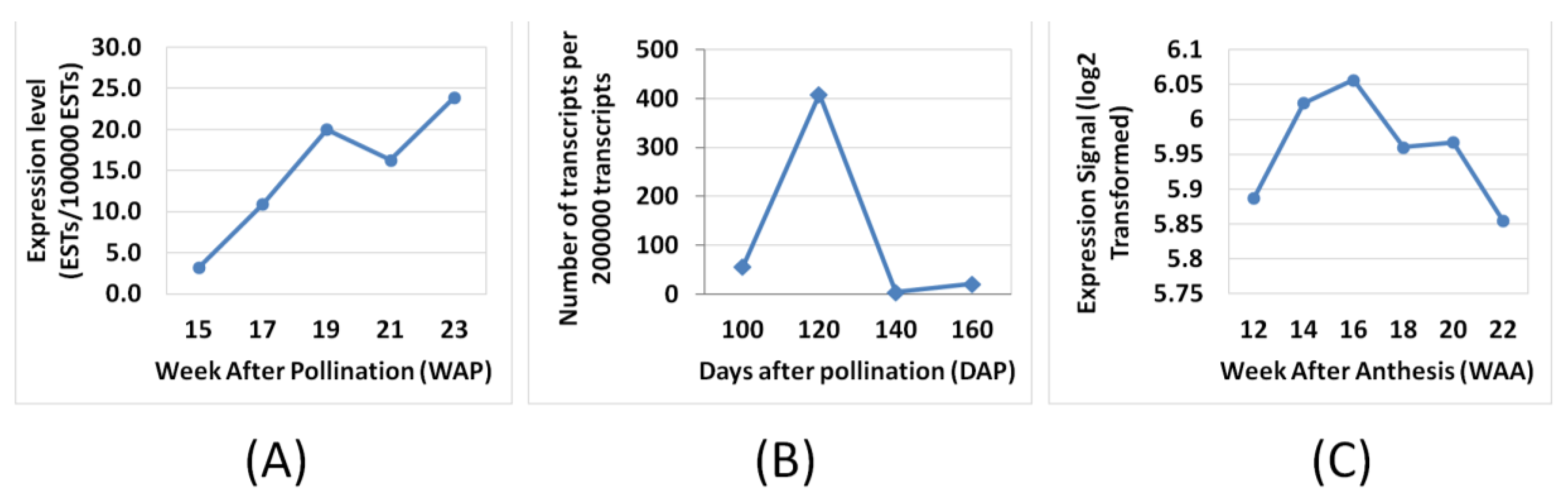
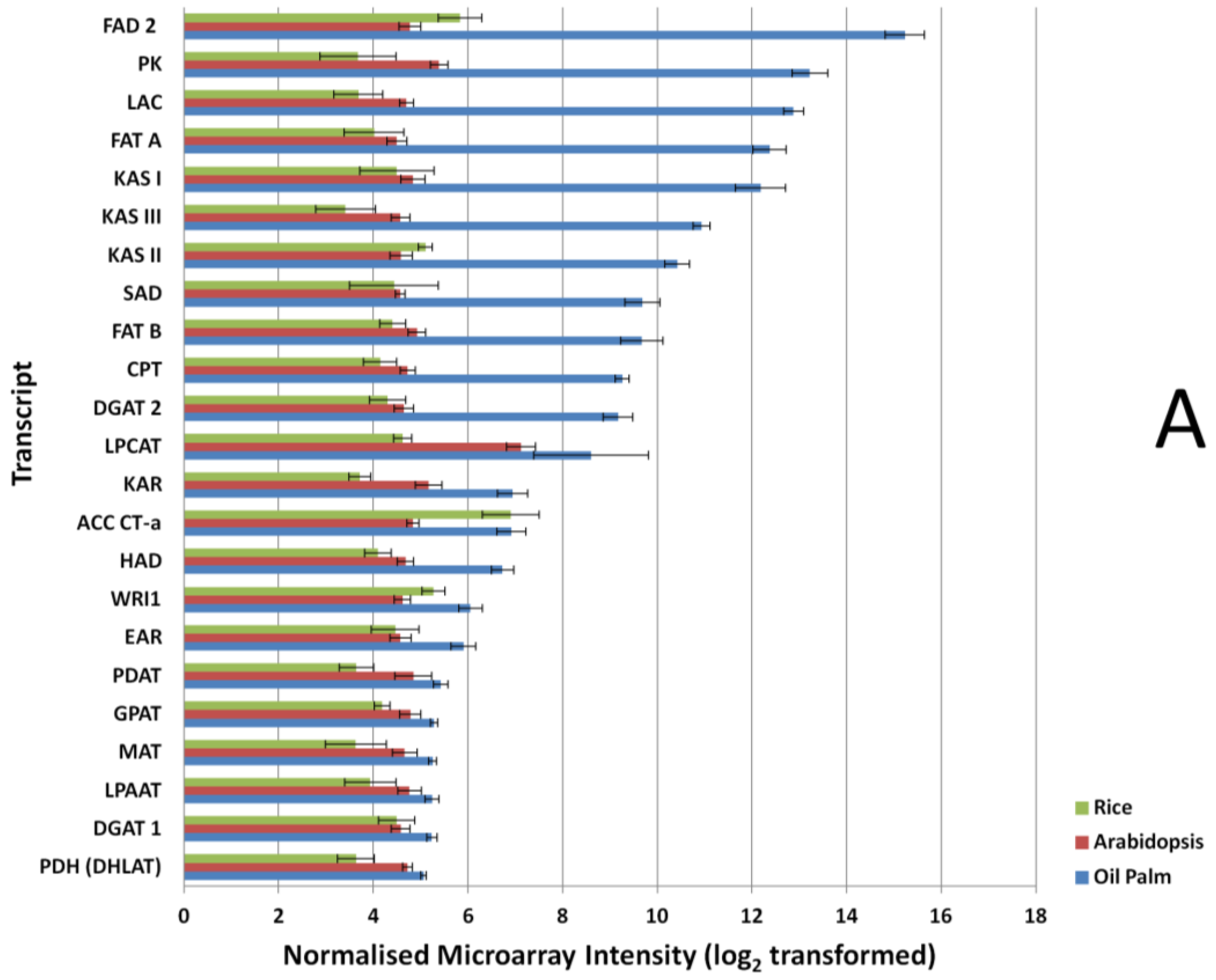
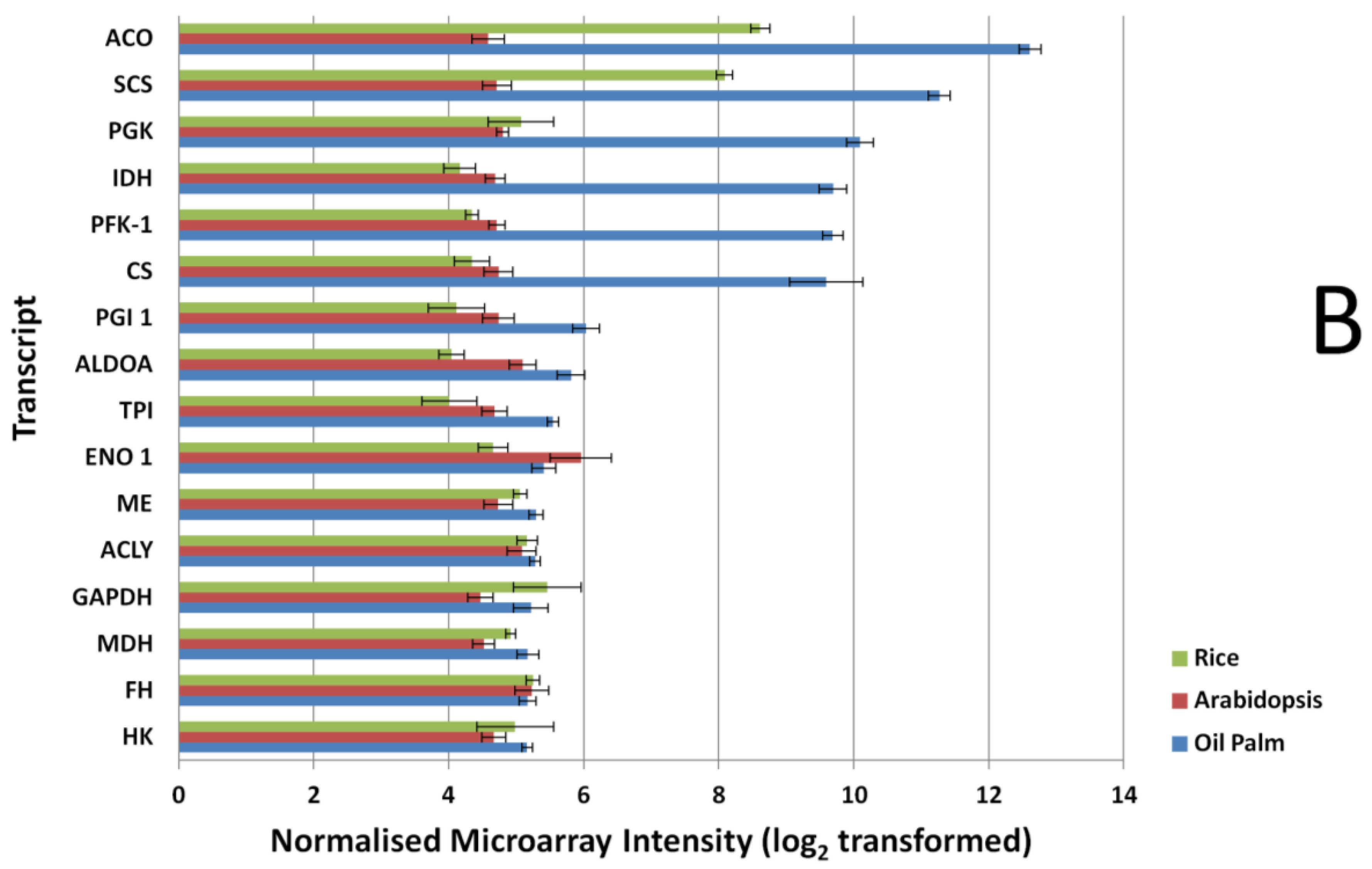
3.2. Expression Comparisons of Fatty Acid (FA), Triacyl Glyceride (TAG) Biosynthesis, Citric Acid Cycle (TCA) and Glycolysis Genes between Custom Oil Palm Mesocarp, Arabidopsis and Rice Microarrays at 16 Week After Anthesis (WAA)
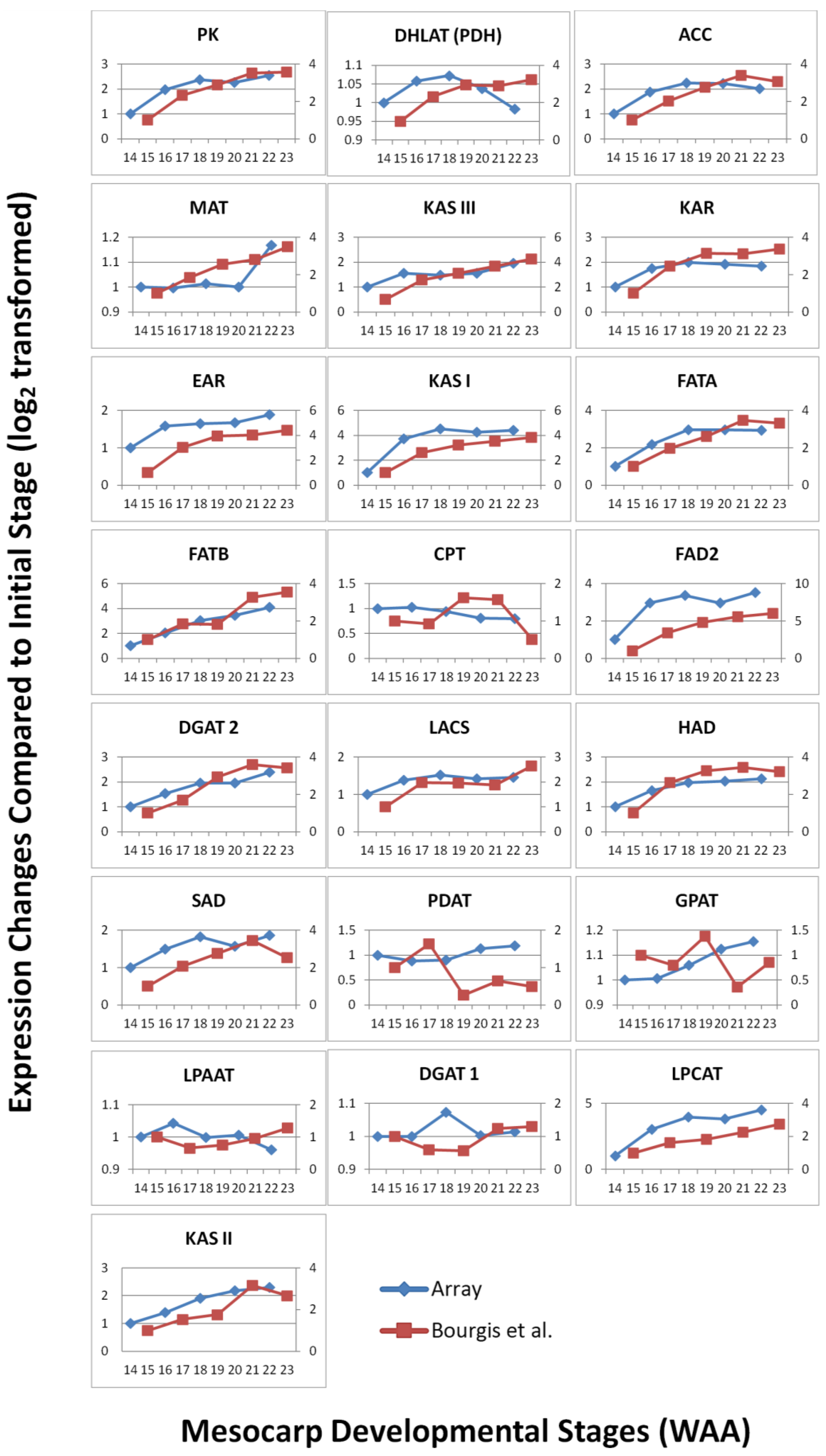
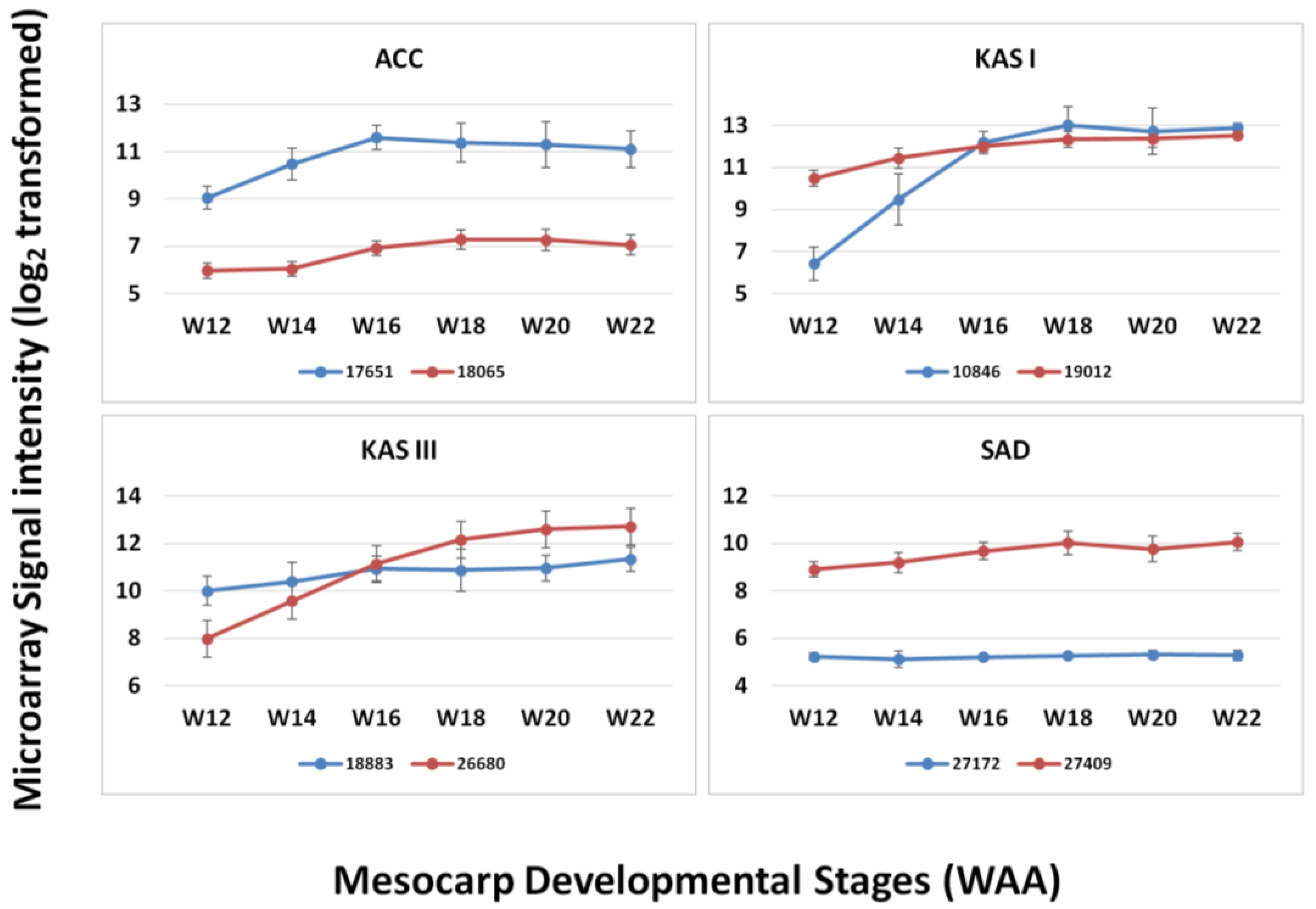
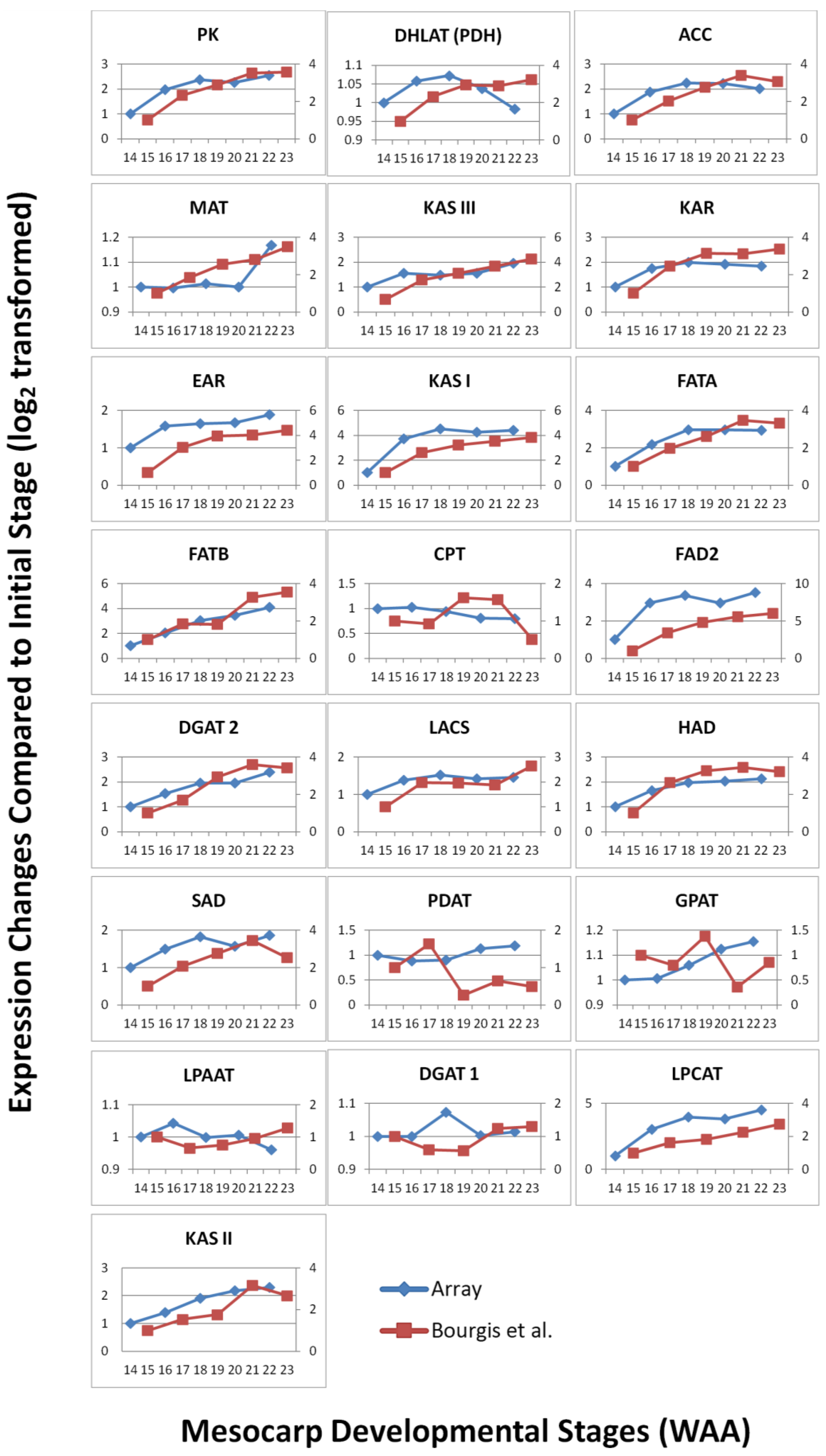
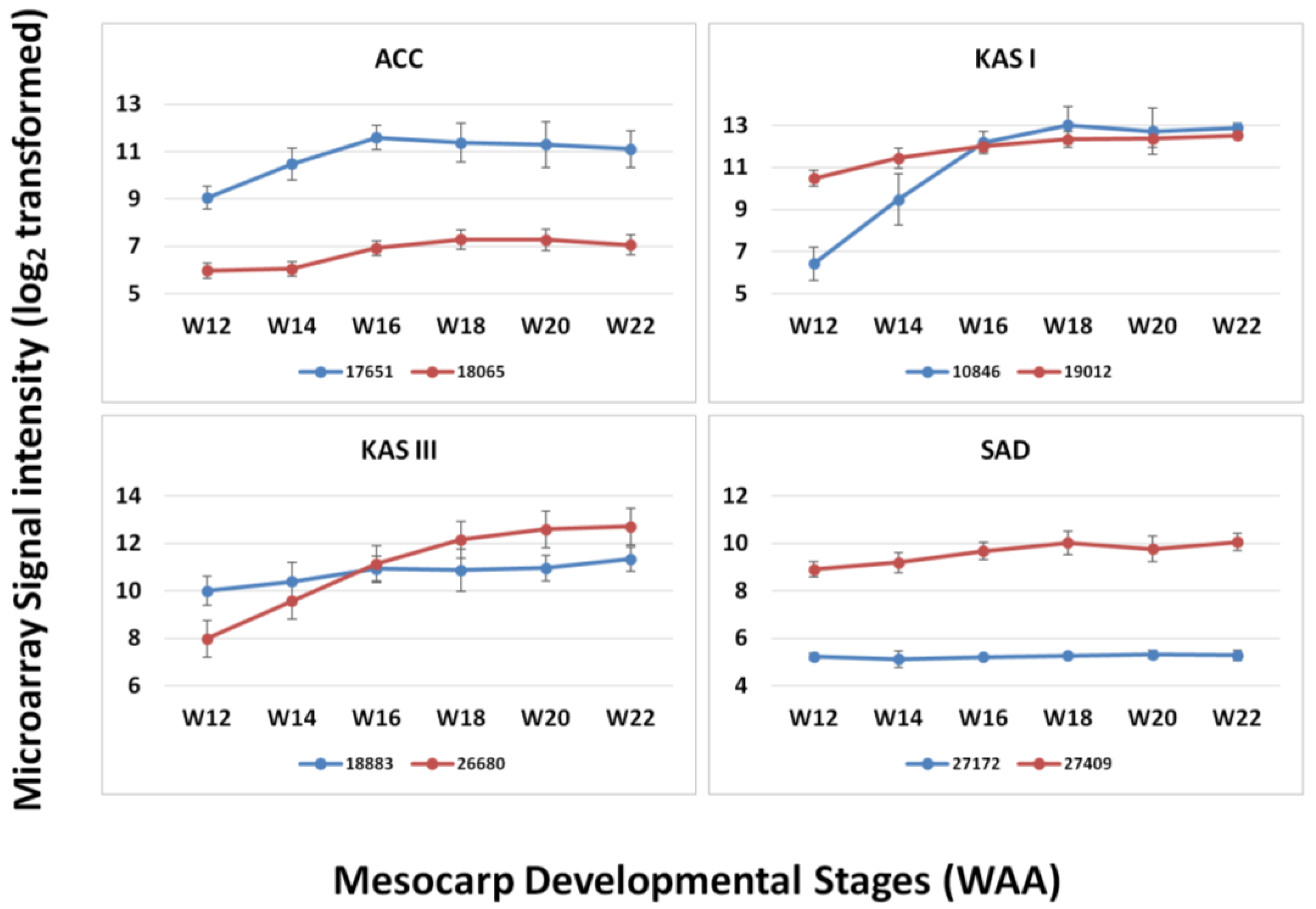
3.3. FA and TAG Biosynthesis Profiling in Oil Palm Mesocarp Development (12 WAA–22 WAA) Using the Oil Palm Mesocarp Microarray
| Genes | R2 | p-value |
|---|---|---|
| HAD | 0.984 | 0.003* |
| EAR | 0.975 | 0.005* |
| KAR | 0.963 | 0.008* |
| KAS I | 0.961 | 0.009* |
| PK | 0.953 | 0.012* |
| KAS III | 0.941 | 0.017* |
| FATA | 0.937 | 0.019* |
| DGAT2 | 0.927 | 0.023* |
| FATB | 0.919 | 0.027* |
| ACC | 0.919 | 0.027* |
| FAD2 | 0.913 | 0.03* |
| LPCAT | 0.911 | 0.031* |
| KAS II | 0.907 | 0.034* |
| LACS | 0.836 | 0.078 |
| SAD | 0.753 | 0.142 |
| MAT | 0.702 | 0.186 |
| DHLAT | 0.209 | 0.736 |
| CPT | 0.018 | 0.978 |
| GPAT | −0.373 | 0.536 |
| PDAT | −0.438 | 0.461 |
| DGAT1 | −0.497 | 0.394 |
| WRI1 | −0.665 | 0.221 |
| LPAAT | −0.884 | 0.047 |
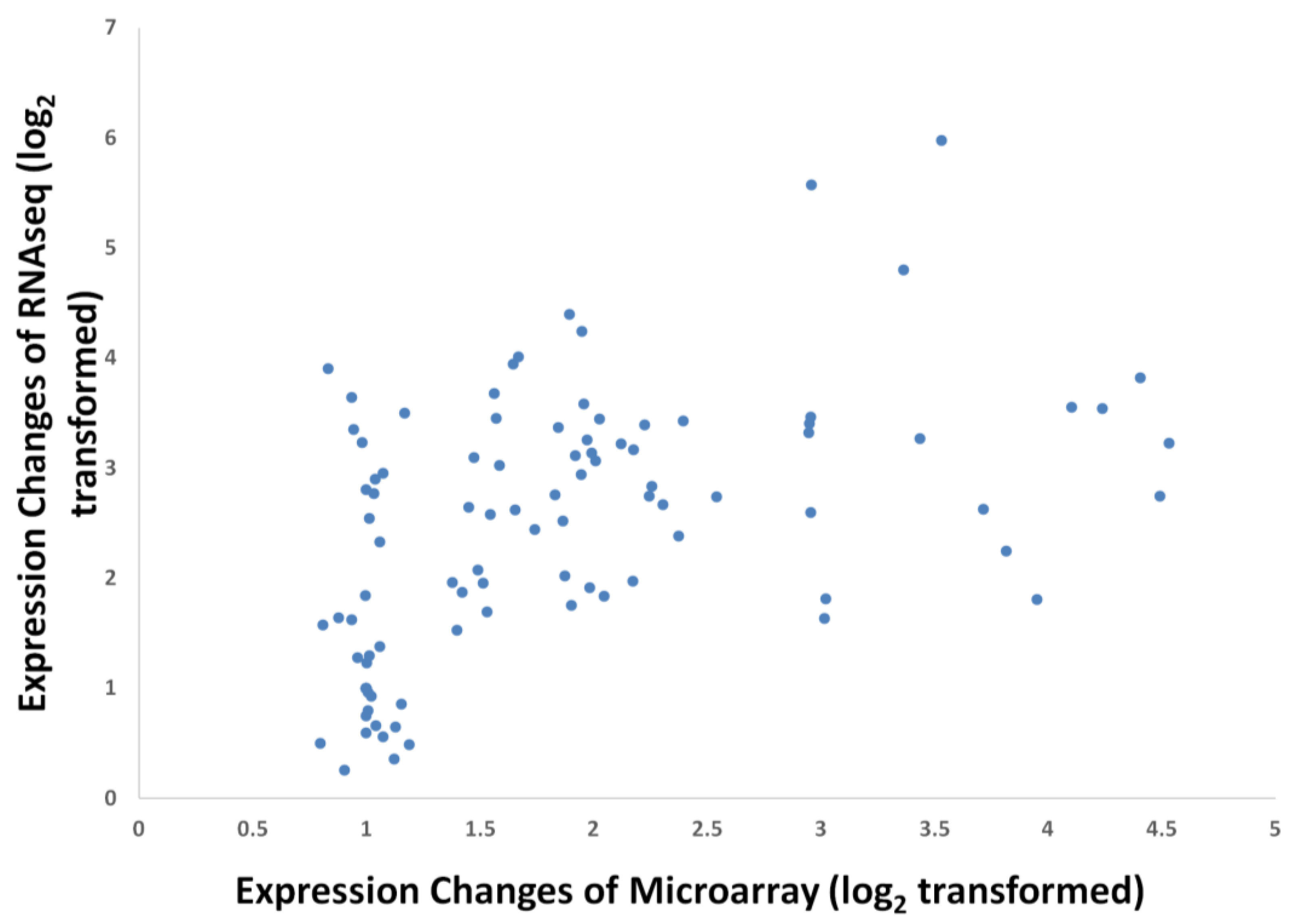
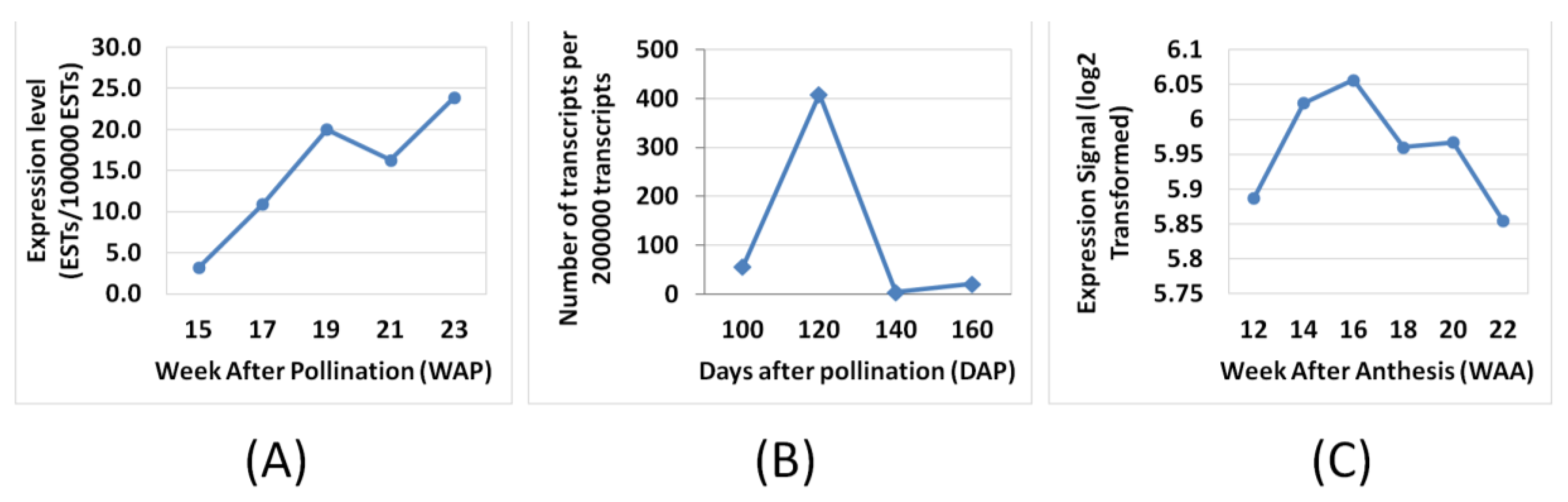
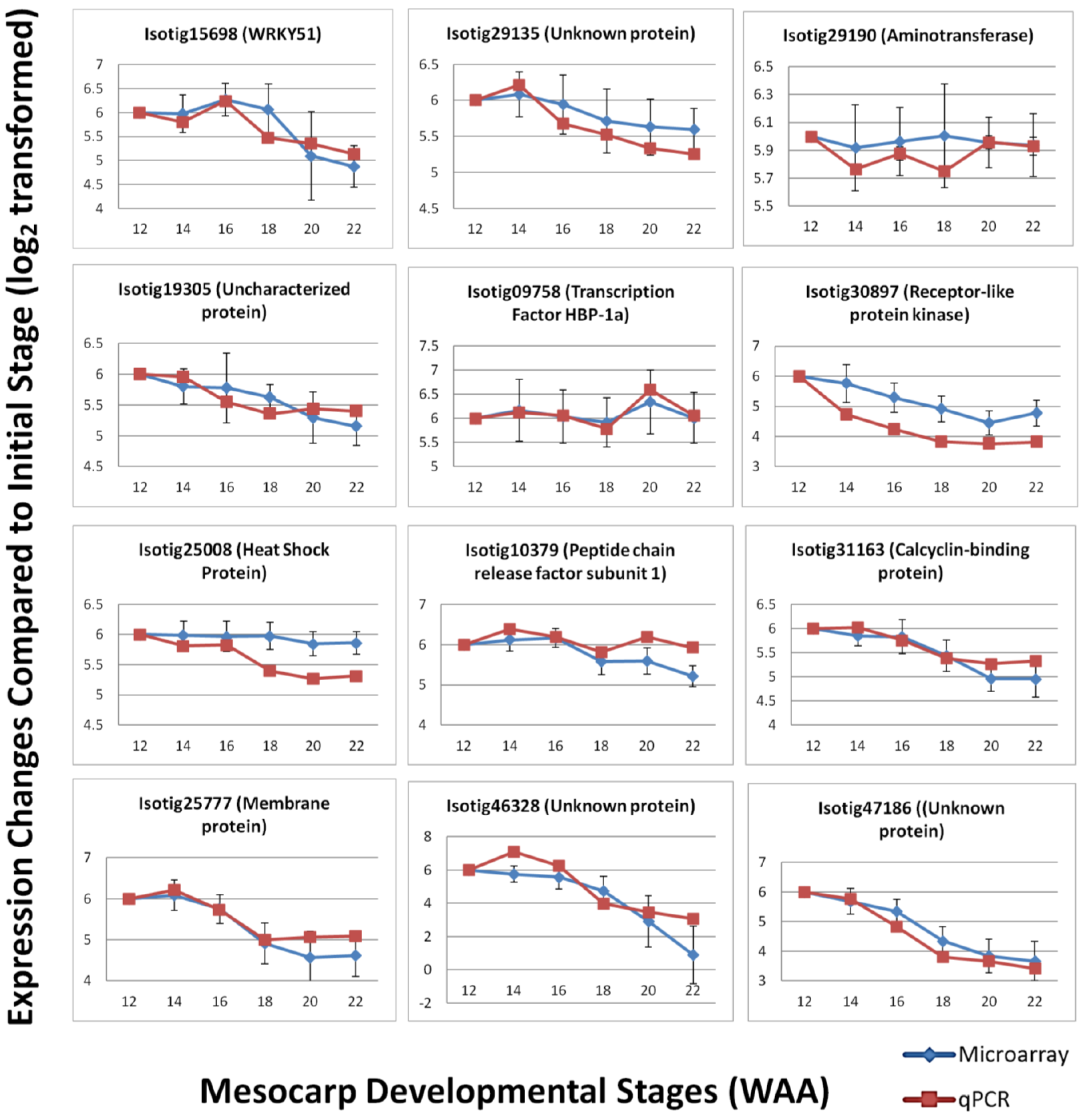
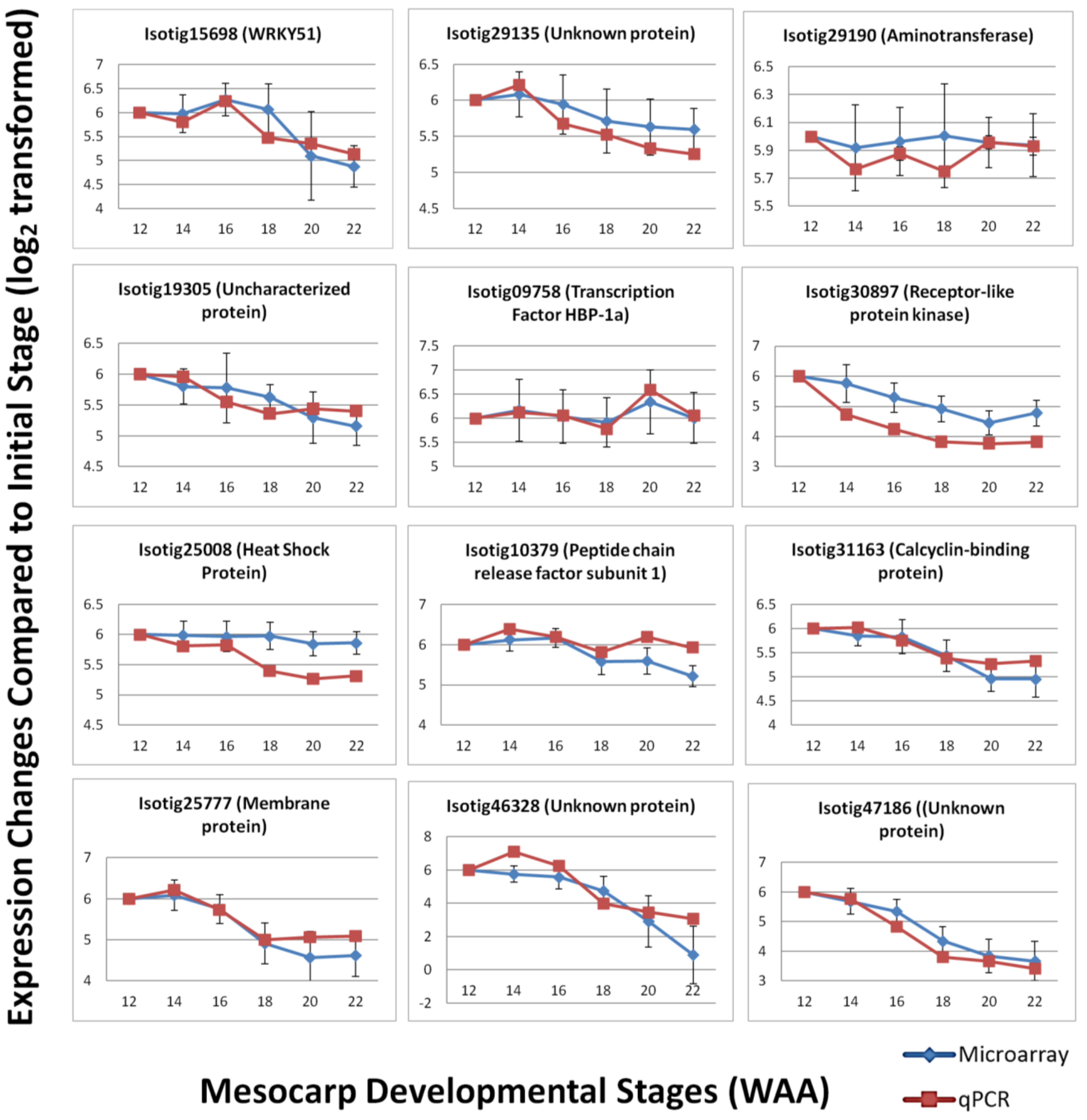
3.4. Validation by Quantitative Real-time PCR (qPCR)
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tan, K.T.; Lee, K.T.; Mohamed, A.R.; Bhatia, S. Palm oil: Addressing issues and towards sustainable development. Renew. Sust. Energ. Rev. 2009, 13, 420–427. [Google Scholar] [CrossRef]
- Montoya, C.; Lopes, R.; Flori, A.; Cros, D.; Cuellar, T.; Summo, M.; Espeout, S.; Rivallan, R.; Risterucci, A.-M.; Bittencourt, D.; et al. Quantitative trait loci (QTLs) analysis of palm oil fatty acid composition in an interspecific pseudo-backcross from Elaeis oleifera (H.B.K.) Cortés and oil palm (Elaeis guineensis Jacq.). Tree Genet. Genomes 2013, 9, 1207–1225. [Google Scholar] [CrossRef] [Green Version]
- Voelker, T. Secrets of palm oil biosynthesis revealed. Proc. Natl. Acad. Sci. USA 2011, 108, 12193–12194. [Google Scholar] [CrossRef] [PubMed]
- Bourgis, F.; Kilaru, A.; Cao, X.; Ngando-Ebongue, G.F.; Drira, N.; Ohlrogge, J.B.; Arondel, V. Comparative transcriptome and metabolite analysis of oil palm and date palm mesocarp that differ dramatically in carbon partitioning. Proc. Natl. Acad. Sci. USA 2011, 108, 12527–12532. [Google Scholar] [CrossRef] [PubMed]
- Dussert, S.; Guerin, C.; Andersson, M.; Joët, T.; Tranbarger, T.J.; Pizot, M.; Sarah, G.; Omore, A.; Durand-Gasselin, T.; Morcillo, F. Comparative transcriptome analysis of three oil palm fruit and seed tissues that differ in oil content and fatty acid composition. Plant Physiol. 2013, 162, 1337–1358. [Google Scholar] [CrossRef] [PubMed]
- Tranbarger, T.J.; Dussert, S.; Joet, T.; Argout, X.; Summo, M.; Champion, A.; Cros, D.; Omore, A.; Nouy, B.; Morcillo, F. Regulatory mechanisms underlying oil palm fruit mesocarp maturation, ripening, and functional specialization in lipid and carotenoid metabolism. Plant Physiol. 2011, 156, 564–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Low, E.-T.L.; Ooi, L.C.-L.; Ong-Abdullah, M.; Ting, N.-C.; Nagappan, J.; Nookiah, R.; Amiruddin, M.D.; Rosli, R.; Manaf, M.A.A.; et al. The oil palm shell gene controls oil yield and encodes a homologue of seedstick. Nature 2013, 500, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Ong-Abdullah, M.; Low, E.-T.L.; Manaf, M.A.A.; Rosli, R.; Nookiah, R.; Ooi, L.C.-L.; Ooi, S.-E.; Chan, K.-L.; Halim, M.A.; et al. Oil palm genome sequence reveals divergence of interfertile species in old and new worlds. Nature 2013, 500, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, W.W.; Alba, R.; Yu, Y.-S.; Bordeaux, J.; Simoes, M.; Dean, J. Microarray analysis and scale-free gene networks identify candidate regulators in drought-stressed roots of loblolly pine (P. taeda L.). BMC Genomics 2011, 12, e264. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Dey, B.; Tyagi, A. Development of the first oligonucleotide microarray for global gene expression profiling in guinea pigs: Defining the transcription signature of infectious diseases. BMC Genomics 2012, 13, e520. [Google Scholar] [CrossRef] [PubMed]
- Gardner, L.; Jayasundara, N.; Castilho, P.; Block, B. Microarray gene expression profiles from mature gonad tissues of Atlantic bluefin tuna, Thunnus thynnus in the gulf of mexico. BMC Genomics 2012, 13, e530. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, H.; Liu, G.; Xu, C.; Zhang, D.; Ban, Q. Analysis of gene expression profile of Limonium bicolor under NaHCO3 stress using cDNA microarray. Plant Mol. Biol. Rep. 2008, 26, 241–254. [Google Scholar] [CrossRef]
- Lee, Y.-P.; Yu, G.-H.; Seo, Y.; Han, S.; Choi, Y.-O.; Kim, D.; Mok, I.-G.; Kim, W.; Sung, S.-K. Microarray analysis of apple gene expression engaged in early fruit development. Plant Cell Rep. 2007, 26, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Kathiresan, A.; Lafitte, H.R.; Chen, J.; Mansueto, L.; Bruskiewich, R.; Bennett, J. Gene expression microarrays and their application in drought stress research. Field Crops Res. 2006, 97, 101–110. [Google Scholar] [CrossRef]
- Payton, P.; Kottapalli, K.; Rowland, D.; Faircloth, W.; Guo, B.; Burow, M.; Puppala, N.; Gallo, M. Gene expression profiling in peanut using high density oligonucleotide microarrays. BMC Genomics 2009, 10, e265. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Narusaka, M.; Ishida, J.; Nanjo, T.; Fujita, M.; Oono, Y.; Kamiya, A.; Nakajima, M.; Enju, A.; Sakurai, T.; et al. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 2002, 31, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Bagnaresi, P.; Moschella, A.; Beretta, O.; Vitulli, F.; Ranalli, P.; Perata, P. Heterologous microarray experiments allow the identification of the early events associated with potato tuber cold sweetening. BMC Genomics 2008, 9, e176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.; Payton, P.; Wright, M.; Tanksley, S.; Giovannoni, J. Utilization of tomato microarrays for comparative gene expression analysis in the Solanaceae. J. Exp. Bot. 2005, 56, 2885–2895. [Google Scholar] [CrossRef] [PubMed]
- Teh, H.F.; Neoh, B.K.; Hong, M.P.L.; Low, J.Y.S.; Ng, T.L.M.; Ithnin, N.; Thang, Y.M.; Mohamed, M.; Chew, F.T.; Yusof, H.M.; et al. Differential metabolite profiles during fruit development in high-yielding oil palm mesocarp. PLoS One 2013, 8, e61344. [Google Scholar]
- Neoh, B.K.; Teh, H.F.; Ng, T.L.M.; Tiong, S.H.; Thang, Y.M.; Ersad, M.A.; Mohamed, M.; Chew, F.T.; Kulaveerasingam, H.; Appleton, D.R. Profiling of metabolites in oil palm mesocarp at different stages of oil biosynthesis. J. Agric. Food Chem. 2013, 61, 1920–1927. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Uniprot Database. Available online: http://www.uniprot.org/ (accessed on 20 September 2013).
- eArray. Available online: https://earray.chem.agilent.com/earray/ (accessed on 23 January 2014).
- Model Organism Gene Expression Microarrays—Details & Specifications. Available online: http://www.genomics.agilent.com/CollectionSubpage.aspx?PageType=Product&SubPageType=ProductData&PageID=1508 (accessed on 13 September 2013).
- Feature Extraction Software. Available online: http://www.genomics.agilent.com/en/product.jsp? cid=AG-PT-144&tabId=AG-PR-1050&_requestid=336047 (accessed on 15 March 2014).
- Ritchie, M.E.; Diyagama, D.; Neilson, J.; van Laar, R.; Dobrovic, A.; Holloway, A.; Smyth, G. Empirical array quality weights in the analysis of microarray data. BMC Bioinformatics 2006, 7, e261. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Silver, J.; Oshlack, A.; Holmes, M.; Diyagama, D.; Holloway, A.; Smyth, G.K. A comparison of background correction methods for two-colour microarrays. Bioinformatics 2007, 23, 2700–2707. [Google Scholar] [CrossRef]
- Smyth, G. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor, 2005 ed.; Gentleman, R., Carey, V.J., Huber, W., Irizarry, R.A., Dudoit, S., Eds.; Springer: New York, NY, USA, 2005; Volume V, pp. 397–420. [Google Scholar]
- Primer Premier. Available online: http://www.premierbiosoft.com/primerdesign/index.html (accessed on 13 May 2014).
- Hellemans, J.; Mortier, G.; de Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, eR19. [Google Scholar] [CrossRef]
- Yeap, W.-C.; Loo, J.; Wong, Y.; Kulaveerasingam, H. Evaluation of suitable reference genes for qRT-PCR gene expression normalization in reproductive, vegetative tissues and during fruit development in oil palm. Plant Cell Tiss. Organ Cult. 2013, 116, 1–12. [Google Scholar]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1–research0034.11. [Google Scholar] [CrossRef] [Green Version]
- Davey, M.; Graham, N.; Vanholme, B.; Swennen, R.; May, S.; Keulemans, J. Heterologous oligonucleotide microarrays for transcriptomics in a non-model species; a proof-of-concept study of drought stress in Musa. BMC Genomics 2009, 10, e436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, W.; Zhou, W.; Gregg, K.; Yu, N.; Davis, S. A method for cross-species gene expression analysis with high-density oligonucleotide arrays. Nucleic Acids Res. 2004, 32, e93. [Google Scholar] [CrossRef]
- Grigoryev, D.; Ma, S.-F.; Simon, B.; Irizarry, R.; Ye, S.; Garcia, J. In vitro identification and in silico utilization of interspecies sequence similarities using genechip® technology. BMC Genomics 2005, 6, e62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chismar, J.D.; Mondala, T.; Fox, H.S.; Roberts, E.; Langford, D.; Masliah, E.; Salomon, D.R.; Head, S.R. Analysis of result variability from high-density oligonucleotide arrays comparing same-species and cross-species hybridizations. BioTechniques 2002, 33, 516–518, 520, 522 passim. [Google Scholar] [PubMed]
- Rismani-Yazdi, H.; Haznedaroglu, B.; Bibby, K.; Peccia, J. Transcriptome sequencing and annotation of the microalgae dunaliella tertiolecta: Pathway description and gene discovery for production of next-generation biofuels. BMC Genomics 2011, 12, e148. [Google Scholar] [CrossRef] [PubMed]
- Nair, K.P.P. Oil palm (Elaeis guineensis Jacquin). In The Agronomy and Economy of Important Tree Crops of the Developing World, 1st ed.; Nair, K.P.P., Ed.; Elsevier: Burlington, MA, USA, 2010; Volume 7, pp. 210–236. [Google Scholar]
- Sîrbu, A.; Kerr, G.; Crane, M.; Ruskin, H.J. RNA-Seq vs. dual- and single-channel microarray data: Sensitivity analysis for differential expression and clustering. PLoS One 2012, 7, e50986. [Google Scholar] [CrossRef]
- Jakubek, Y.; Cutler, D. A model of binding on DNA microarrays: Understanding the combined effect of probe synthesis failure, cross-hybridization, DNA fragmentation and other experimental details of affymetrix arrays. BMC Genomics 2012, 13, e737. [Google Scholar] [CrossRef] [PubMed]
- Allanach, K.; Mengel, M.; Einecke, G.; Sis, B.; Hidalgo, L.G.; Mueller, T.; Halloran, P.F. Comparing microarray versus RT-PCR assessment of renal allograft biopsies: Similar performance despite different dynamic ranges. Am. J. Transplant. 2008, 8, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Draghici, S.; Khatri, P.; Eklund, A.C.; Szallasi, Z. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet. 2006, 22, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Yuen, T.; Wurmbach, E.; Pfeffer, R.L.; Ebersole, B.J.; Sealfon, S.C. Accuracy and calibration of commercial oligonucleotide and custom cDNA microarrays. Nucleic Acids Res. 2002, 30, e48. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, Y.C.; Kwong, Q.B.; Lee, H.L.; Ong, C.K.; Mayes, S.; Chew, F.T.; Appleton, D.R.; Kulaveerasingam, H. Expression Comparison of Oil Biosynthesis Genes in Oil Palm Mesocarp Tissue Using Custom Array. Microarrays 2014, 3, 263-281. https://doi.org/10.3390/microarrays3040263
Wong YC, Kwong QB, Lee HL, Ong CK, Mayes S, Chew FT, Appleton DR, Kulaveerasingam H. Expression Comparison of Oil Biosynthesis Genes in Oil Palm Mesocarp Tissue Using Custom Array. Microarrays. 2014; 3(4):263-281. https://doi.org/10.3390/microarrays3040263
Chicago/Turabian StyleWong, Yick Ching, Qi Bin Kwong, Heng Leng Lee, Chuang Kee Ong, Sean Mayes, Fook Tim Chew, David R. Appleton, and Harikrishna Kulaveerasingam. 2014. "Expression Comparison of Oil Biosynthesis Genes in Oil Palm Mesocarp Tissue Using Custom Array" Microarrays 3, no. 4: 263-281. https://doi.org/10.3390/microarrays3040263