Identifying Potential Regions of Copy Number Variation for Bipolar Disorder

Abstract
:1. Introduction
2. Methods
2.1. Subjects, DNA Pooling Construction and Genotyping
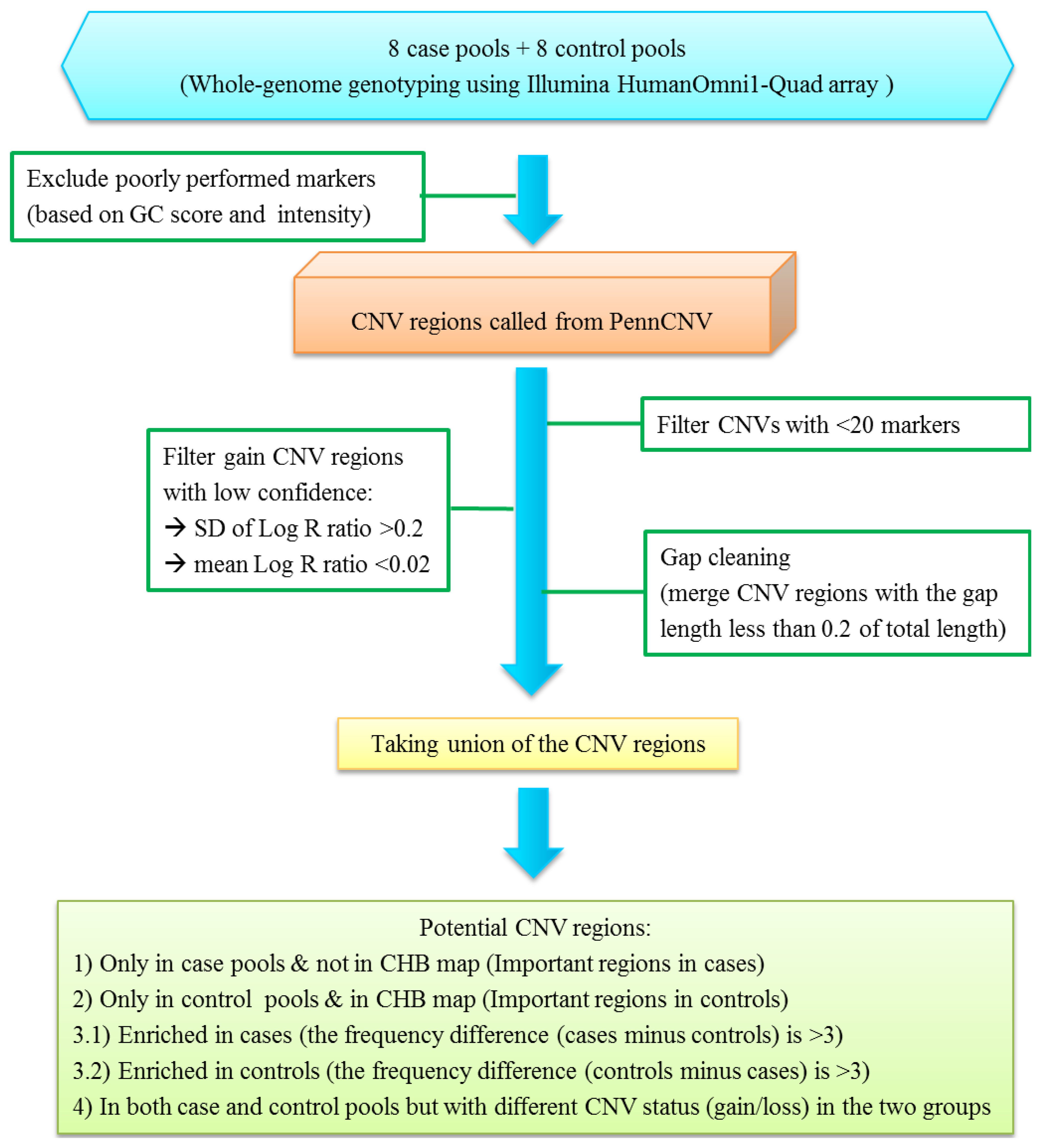
2.2. Quality Control and Filtering Procedures for CNV Analysis

- (1)
- the CNV regions were only found in case pools but not in the Han Chinese CNV map; these CNVs were defined as important regions in cases;
- (2)
- the CNV regions were only found in control pools and also reported in the Han Chinese CNV map; these CNVs were defined as important regions in controls;
- (3)
- the CNV regions were shown in both case and control pools, but the frequency difference in the two groups is large (>3); these CNVs were defined as enriched in cases or controls;
- (4)
- the CNV regions were found in both case and control pools, however the CNV status (gain/loss) was different in the two groups.
2.3. Association Testing for CNV Regions with BPD
2.4. CNV Validation by RT-qPCR
3. Results

{kind=link}
| CNV size | CNV | Sample | No. of | Mean CNVs | Wilcoxon |
|---|---|---|---|---|---|
| type | Group | unique CNVs | per pool | p-value | |
| ≥100 kb | Both | Controls | 1441 | 346.25 | 0.645 |
| Patients | 1446 | 307.25 | |||
| ≥100 kb | Gain | Controls | 1438 | 345.5 | 0.645 |
| Patients | 1434 | 304.375 | |||
| ≥100 kb | Loss | Controls | 3 | 0.75 | 0.105 |
| Patients | 12 | 2.875 | |||
| ≥500 kb | Both | Controls | 43 | 16 | 0.798 |
| Patients | 44 | 15.5 | |||
| ≥500 kb | Gain | Controls | 43 | 16 | 0.798 |
| Patients | 44 | 15.5 | |||
| ≥500 kb | Loss | Controls | 0 | 0 | NA |
| Patients | 0 | 0 |
| Potential CNV regions | No. of CNV (Gain/Loss) | Mean CNV length (kb) | No. of mapped Genes in CNV regions a | Genes overlapped with the list in BPDgenes b |
|---|---|---|---|---|
| Important regions in cases | 882 (859/23) | 120.52 | 982 | ANK3, ARNTL, ASTN2, CHST11, CSMD2, DACH, DLG2, DPP10, DSCAM, GRIK1, HTR6, KALRN, MCTP1, MYO3B, NALCN, NOS1, OPCML, OR6S1, PARK2, PDLIM5, PLCB1, PTPRG, SLC39A3, SYN3, TGFB2, UGT1A10, VAV3 |
| Important regions in controls | 94 (94/0) | 91.36 | 164 | DMGDH |
| Regions enriched in cases | 2 (2/0) | 447.74 | 0 | None |
| Regions enriched in controls | 26 (25/1) | 244.49 | 86 | CSMD2, OPCML |
| Different CNV status in cases and controls | (1 Gain CNV in 1 control /1 Loss CNV in 1 case) | 253.96 | 15 | None |
| Chr | Position a | CNV type b | Length (kb) | Affected Genes c | p-value (t test) | p-value (Wilcoxon test) |
|---|---|---|---|---|---|---|
| 1 | 34,268,681–34,936,979 | Gain in 6 controls and 2 cases | 668.30 | CSMD2, C1orf94 | 0.192 | 0.169 |
| 3 | 61,681,785–61,928,141 | Gain in 1 case | 246.36 | PTPRG | 0.171 | 0.234 |
| 4 | 95,487,295–95,868,284 | Gain in 3 case | 380.99 | PDLIM5, ENH, ENH1, LIM | 0.646 | 0.161 |
| 9 | 118,292,450–118,450,577 | Gain in 1 case | 158.13 | ASTN2, KIAA0634, bA67K19.1 | 0.157 | 0.169 |
| 11 | 13,224,130–13,256,233 | Gain in 1 case | 32.10 | ARNTL, BMAL1, BMAL1c, JAP3, MGC47515, MOP3, PASD3, TIC, bHLHe5 | 0.007 * | 0.010 * |
| 12 | 103,611,282–103,669,104 | Gain in 1 case | 57.82 | CHST11, C4ST, C4ST-1, C4ST1, DKFZp667A035, FLJ41682, HSA269537 | 0.113 | 0.065 |
| 22 | 31,480,536–31,564,931 | Gain in 1 case | 84.40 | SYN3, TIMP3 | 0.029 * | 0.021 * |
| Enriched GO category | Database ID | p-value a | Adjusted p-value b | O c | N d |
|---|---|---|---|---|---|
| Biological process | |||||
| biological adhesion | GO:0022610 | 1.16 × 10−8 | 1.5 × 10−5 | 93 | 905 |
| cell adhesion | GO:0007155 | 2.13 × 10−8 | 1.5 × 10−5 | 92 | 903 |
| cell-cell adhesion | GO:0016337 | 4.03 × 10−5 | 0.0190 | 41 | 374 |
| Cellular component | |||||
| neuron projection | GO:0043005 | 1.03 × 10−7 | 2.2 × 10−5 | 69 | 628 |
| synapse | GO:0045202 | 1.13 × 10−5 | 0.0008 | 51 | 478 |
| cell projection | GO:0042995 | 9.19 × 10−6 | 0.0008 | 102 | 1173 |
| axon | GO:0030424 | 1.85 × 10−5 | 0.0010 | 34 | 276 |
| dendrite | GO:0030425 | 2.61 × 10−5 | 0.0011 | 39 | 341 |
| cell projection part | GO:0044463 | 4.52 × 10−5 | 0.0016 | 59 | 610 |
| synaptic membrane | GO:0097060 | 0.0001 | 0.0031 | 26 | 208 |
| synapse part | GO:0044456 | 0.0002 | 0.0053 | 38 | 361 |
| neuron spine | GO:0044309 | 0.0004 | 0.0086 | 20 | 153 |
| dendritic spine | GO:0043197 | 0.0004 | 0.0086 | 20 | 153 |
| cell periphery | GO:0071944 | 0.0010 | 0.0195 | 267 | 3989 |
| keratin filament | GO:0045095 | 0.0013 | 0.0227 | 10 | 57 |
| plasma membrane | GO:0005886 | 0.0017 | 0.0227 | 260 | 3905 |
| postsynaptic density | GO:0014069 | 0.0016 | 0.0227 | 15 | 111 |
| cytoskeleton | GO:0005856 | 0.0017 | 0.0227 | 119 | 1613 |
| dendritic spine head | GO:0044327 | 0.0016 | 0.0227 | 15 | 111 |
| postsynaptic membrane | GO:0045211 | 0.0028 | 0.0333 | 20 | 178 |
| presynaptic membrane | GO:0042734 | 0.0028 | 0.0333 | 9 | 53 |
| Present Study | Previous Studies | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Location | Position a | CNV type | Length (kb) | Affected Genes | No. of cases/controls | Position a | CNV type | Length (kb) | References |
| 6q16.3 | 101966969:102040222 | Gain | 73.250 | GRIK2 | 1/0 | 101953625:102624651 | Unknown | 671.027 | [32] |
| 6q27 | 168320777:168376820 | Gain | 56.044 | KIF25, FERM, | 1/0 | 168090000:168330000 | Gain | 240.000 | [28,29] |
| MILT4, DACT2 | |||||||||
| 9q34.3 | 138149942:138217164 | Gain | 67.233 | None | 2/0 | 136600001:140273252 | Gain | 3673.252 | [31] |
| 19p12 | 20091264:2029165 | Gain | 200.402 | ZNF682, ZNF90, | 1/0 | 20001614:20177979 | Gain/Loss | 176.365 | [30] |
| ZNF486 | |||||||||
| 19p12 | 24193894:24282139 | Loss | 88.246 | ZNF254 | 2/0 | 24013968-24295825 | Gain | 281.857 | [30] |
4. Discussion
5. Conclusions
Acknowledgments
Authors Contributions
Conflicts of Interest
References
- Fagiolini, A.; Forgione, R.; Maccari, M.; Cuomo, A.; Morana, B.; Dell’Osso, M.C.; Pellegrini, F.; Rossi, A. Prevalence, chronicity, burden and borders of bipolar disorder. J. Affect. Disord. 2013, 148, 161–169. [Google Scholar] [CrossRef]
- Burmeister, M.; McInnis, M.G.; Zollner, S. Psychiatric genetics: Progress amid controversy. Nat. Rev. Genet. 2008, 9, 527–540. [Google Scholar] [CrossRef]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef]
- Tam, G.W.; Redon, R.; Carter, N.P.; Grant, S.G. The role of DNA copy number variation in schizophrenia. Biol. Psychiatry 2009, 66, 1005–1012. [Google Scholar] [CrossRef]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Daly, M.J.; O’Donovan, M. Genetic architectures of psychiatric disorders: The emerging picture and its implications. Nat. Rev. Genet. 2012, 13, 537–551. [Google Scholar] [CrossRef]
- Lachman, H.M.; Pedrosa, E.; Petruolo, O.A.; Cockerham, M.; Papolos, A.; Novak, T.; Papolos, D.F.; Stopkova, P. Increase in gsk3beta gene copy number variation in bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr Genet. 2007, 144B, 259–265. [Google Scholar] [CrossRef]
- Zöllner, S.; Teslovich, T.M. Using gwas data to identify copy number variants contributing to common complex diseases. Stat. Sci. 2009, 24, 530–546. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. Penncnv: An integrated hidden markov model designed for high-resolution copy number variation detection in whole-genome snp genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar]
- Colella, S.; Yau, C.; Taylor, J.M.; Mirza, G.; Butler, H.; Clouston, P.; Bassett, A.S.; Seller, A.; Holmes, C.C.; Ragoussis, J. Quantisnp: An objective bayes hidden-markov model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35, 2013–2025. [Google Scholar] [CrossRef]
- Wineinger, N.E.; Tiwari, H.K. The impact of errors in copy number variation detection algorithms on association results. PLoS One 2012, 7, e32396. [Google Scholar] [CrossRef]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef]
- McCarroll, S.A.; Altshuler, D.M. Copy-number variation and association studies of human disease. Nat. Genet. 2007, 39, S37–S42. [Google Scholar] [CrossRef]
- Sham, P.; Bader, J.S.; Craig, I.; O’Donovan, M.; Owen, M. DNA pooling: A tool for large-scale association studies. Nat. Rev. Genet. 2002, 3, 862–871. [Google Scholar] [CrossRef]
- Butcher, L.; Meaburn, E.; Liu, L.; Fernandes, C.; Hill, L.; Al-Chalabi, A.; Plomin, R.; Schalkwyk, L.; Craig, I. Genotyping pooled DNA on microarrays: A systematic genome screen of thousands of snps in large samples to detect qtls for complex traits. Behav. Genet. 2004, 34, 549–555. [Google Scholar] [CrossRef]
- Zuo, Y.; Zou, G.; Wang, J.; Zhao, H.; Liang, H. Optimal two-stage design for case-control association analysis incorporating genotyping errors. Ann. Hum. Genet. 2008, 72, 375–387. [Google Scholar] [CrossRef]
- Wang, H.; Thomas, D.C.; Pe’er, I.; Stram, D.O. Optimal two-stage genotyping designs for genome-wide association scans. Genet. Epidemiol. 2006, 30, 356–368. [Google Scholar] [CrossRef]
- Lin, C.H.; Huang, M.C.; Li, L.H.; Wu, J.Y.; Chen, Y.T.; Fann, C.S. Genome-wide copy number analysis using copy number inferring tool (CNIT) and DNA pooling. Hum. Mutat. 2008, 29, 1055–1062. [Google Scholar] [CrossRef]
- Tsai, H.C.; Lu, M.K.; Yang, Y.K.; Huang, M.C.; Yeh, T.L.; Chen, W.J.; Lu, R.B.; Kuo, P.H. Empirically derived subgroups of bipolar i patients with different comorbidity patterns of anxiety and substance use disorders in han chinese population. J. Affect. Disord. 2012, 136, 81–89. [Google Scholar] [CrossRef]
- Wu, P.J.; Chang, S.M.; Lu, M.K.; Chen, W.J.; Yang, Y.K.; Yeh, T.L.; Liao, S.C.; Lu, R.B.; Kuo, P.H. The profile and familiality of personality traits in mood disorder families. J. Affect. Disord. 2012, 138, 367–374. [Google Scholar] [CrossRef]
- Kuo, P.H.; Chuang, L.C.; Liu, J.R.; Liu, C.M.; Huang, M.C.; Lin, S.K.; Sun, H.F.S.; Hsieh, M.H.; Hung, H.; Lu, R.B. Identification of novel loci for bipolar I disorder in a multi-stage genome-wide association study. Progr. Neuro. Psychopharmacol. Biologic. Psychiatr. 2014, in press. [Google Scholar]
- Lin, C.H.; Lin, Y.C.; Wu, J.Y.; Pan, W.H.; Chen, Y.T.; Fann, C.S. A genome-wide survey of copy number variations in han chinese residing in taiwan. Genomics 2009, 94, 241–246. [Google Scholar] [CrossRef]
- Lou, H.; Li, S.; Yang, Y.; Kang, L.; Zhang, X.; Jin, W.; Wu, B.; Jin, L.; Xu, S. A map of copy number variations in chinese populations. PLoS One 2011, 6, e27341. [Google Scholar] [CrossRef]
- Malhotra, D.; McCarthy, S.; Michaelson, J.J.; Vacic, V.; Burdick, K.E.; Yoon, S.; Cichon, S.; Corvin, A.; Gary, S.; Gershon, E.S.; et al. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron 2011, 72, 951–963. [Google Scholar] [CrossRef]
- Kao, C.F.; Kuo, P.H. Risk and information evaluation of prioritized genes for complex traits: Application to bipolar disorder. Am. J. Med. Genet. B 2014. submitted. [Google Scholar]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. Web-based gene set analysis toolkit (webgestalt): Update 2013. Nucleic Acids Res. 2013, 41, W77–W83. [Google Scholar] [CrossRef]
- CopyCaller® Software. v2.0. Available online: http://www.appliedbiosystems.com/absite/us/en/home/support/software/real-time-pcr/copycaller.html (accessed on 15 January 2014).
- Priebe, L.; Degenhardt, F.A.; Herms, S.; Haenisch, B.; Mattheisen, M.; Nieratschker, V.; Weingarten, M.; Witt, S.; Breuer, R.; Paul, T.; et al. Genome-wide survey implicates the influence of copy number variants (CNVs) in the development of early-onset bipolar disorder. Mol. Psychiatry 2012, 17, 421–432. [Google Scholar]
- Yang, S.; Wang, K.; Gregory, B.; Berrettini, W.; Wang, L.S.; Hakonarson, H.; Bucan, M. Genomic landscape of a three-generation pedigree segregating affective disorder. PLoS One 2009, 4, e4474. [Google Scholar]
- Grozeva, D.; Kirov, G.; Ivanov, D.; Jones, I.R.; Jones, L.; Green, E.K.; St Clair, D.M.; Young, A.H.; Ferrier, N.; Farmer, A.E.; et al. Rare copy number variants: A point of rarity in genetic risk for bipolar disorder and schizophrenia. Arch. Gen. Psychiatry 2010, 67, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Bergen, S.E.; O’Dushlaine, C.T.; Ripke, S.; Lee, P.H.; Ruderfer, D.M.; Akterin, S.; Moran, J.L.; Chambert, K.D.; Handsaker, R.E.; Backlund, L.; et al. Genome-wide association study in a swedish population yields support for greater CNV and mhc involvement in schizophrenia compared with bipolar disorder. Mol. Psychiatry 2012, 17, 880–886. [Google Scholar] [CrossRef]
- McQuillin, A.; Bass, N.; Anjorin, A.; Lawrence, J.; Kandaswamy, R.; Lydall, G.; Moran, J.; Sklar, P.; Purcell, S.; Gurling, H. Analysis of genetic deletions and duplications in the university college london bipolar disorder case control sample. Eur. J. Hum. Genet. 2011, 19, 588–592. [Google Scholar] [CrossRef]
- Karlsson, R.; Graae, L.; Lekman, M.; Wang, D.; Favis, R.; Axelsson, T.; Galter, D.; Belin, A.C.; Paddock, S. Magi1 copy number variation in bipolar affective disorder and schizophrenia. Biol. Psychiatry 2012, 71, 922–930. [Google Scholar] [CrossRef]
- Hackstadt, A.J.; Hess, A.M. Filtering for increased power for microarray data analysis. BMC Bioinformatics 2009, 10, 11. [Google Scholar] [CrossRef]
- Donner, Y.; Feng, T.; Benoist, C.; Koller, D. Imputing gene expression from selectively reduced probe sets. Nat. Methods 2012, 9, 1120–1125. [Google Scholar] [CrossRef]
- Chiang, C.W.; Gajdos, Z.K.; Korn, J.M.; Kuruvilla, F.G.; Butler, J.L.; Hackett, R.; Guiducci, C.; Nguyen, T.T.; Wilks, R.; Forrester, T.; et al. Rapid assessment of genetic ancestry in populations of unknown origin by genome-wide genotyping of pooled samples. PLoS Genet. 2010, 6, e1000866. [Google Scholar] [CrossRef]
- Malhotra, D.; Sebat, J. CNVs: Harbingers of a rare variant revolution in psychiatric genetics. Cell 2012, 148, 1223–1241. [Google Scholar] [CrossRef]
- Zhang, D.; Cheng, L.; Qian, Y.; Alliey-Rodriguez, N.; Kelsoe, J.R.; Greenwood, T.; Nievergelt, C.; Barrett, T.B.; McKinney, R.; Schork, N.; et al. Singleton deletions throughout the genome increase risk of bipolar disorder. Mol. Psychiatry 2009, 14, 376–380. [Google Scholar] [CrossRef]
- Mansour, H.A.; Talkowski, M.E.; Wood, J.; Chowdari, K.V.; McClain, L.; Prasad, K.; Montrose, D.; Fagiolini, A.; Friedman, E.S.; Allen, M.H.; et al. Association study of 21 circadian genes with bipolar i disorder, schizoaffective disorder, and schizophrenia. Bipolar Disord. 2009, 11, 701–710. [Google Scholar] [CrossRef]
- Shi, J.; Wittke-Thompson, J.K.; Badner, J.A.; Hattori, E.; Potash, J.B.; Willour, V.L.; McMahon, F.J.; Gershon, E.S.; Liu, C. Clock genes may influence bipolar disorder susceptibility and dysfunctional circadian rhythm. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147B, 1047–1055. [Google Scholar] [CrossRef]
- Hamshere, M.L.; Green, E.K.; Jones, I.R.; Jones, L.; Moskvina, V.; Kirov, G.; Grozeva, D.; Nikolov, I.; Vukcevic, D.; Caesar, S.; et al. Genetic utility of broadly defined bipolar schizoaffective disorder as a diagnostic concept. Br. J. Psychiatry 2009, 195, 23–29. [Google Scholar] [CrossRef]
- Glessner, J.T.; Reilly, M.P.; Kim, C.E.; Takahashi, N.; Albano, A.; Hou, C.; Bradfield, J.P.; Zhang, H.; Sleiman, P.M.A.; Flory, J.H.; et al. Strong synaptic transmission impact by copy number variations in schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 10584–10589. [Google Scholar] [CrossRef]
- Sampaio, A.S.; Fagerness, J.; Crane, J.; Leboyer, M.; Delorme, R.; Pauls, D.L.; Stewart, S.E. Association between polymorphisms in grik2 gene and obsessive-compulsive disorder: A family-based study. CNS Neurosci. Ther. 2011, 17, 141–147. [Google Scholar] [CrossRef]
- Kim, S.A.; Kim, J.H.; Park, M.; Cho, I.H.; Yoo, H.J. Family-based association study between grik2 polymorphisms and autism spectrum disorders in the korean trios. Neurosci. Res. 2007, 58, 332–335. [Google Scholar] [CrossRef]
- Olsen, L.; Hansen, T.; Djurovic, S.; Haastrup, E.; Albrecthsen, A.; Hoeffding, L.K.; Secher, A.; Gustafsson, O.; Jakobsen, K.D.; Nielsen, F.C.; et al. Copy number variations in affective disorders and meta-analysis. Psychiatr. Genet. 2011, 21, 319–322. [Google Scholar] [CrossRef]
- Ioannidis, J.P.; Ntzani, E.E.; Trikalinos, T.A. “Racial” differences in genetic effects for complex diseases. Nat. Genet. 2004, 36, 1312–1318. [Google Scholar] [CrossRef]
- Greene, C.S.; Penrod, N.M.; Williams, S.M.; Moore, J.H. Failure to replicate a genetic association may provide important clues about genetic architecture. PLoS One 2009, 4, e5639. [Google Scholar] [CrossRef]
- Freeman, J.L.; Perry, G.H.; Feuk, L.; Redon, R.; McCarroll, S.A.; Altshuler, D.M.; Aburatani, H.; Jones, K.W.; Tyler-Smith, C.; Hurles, M.E.; et al. Copy number variation: New insights in genome diversity. Genome Res. 2006, 16, 949–961. [Google Scholar] [CrossRef]
- Armengol, L.; Villatoro, S.; Gonzalez, J.R.; Pantano, L.; Garcia-Aragones, M.; Rabionet, R.; Caceres, M.; Estivill, X. Identification of copy number variants defining genomic differences among major human groups. PLoS One 2009, 4, e7230. [Google Scholar] [CrossRef]
- Chang, S.H.; Gao, L.; Li, Z.; Zhang, W.N.; Du, Y.; Wang, J. Bdgene: A genetic database for bipolar disorder and its overlap with schizophrenia and major depressive disorder. Biol. Psychiatry 2013, 74, 727–733. [Google Scholar] [CrossRef]
- Chuang, L.-C.; Kao, C.-F.; Shih, W.-L.; Kuo, P.-H. Pathway analysis using information from allele-specific gene methylation in genome-wide association studies for bipolar disorder. PLoS One 2013, 8, e53092. [Google Scholar]
- Lin, P.; Hartz, S.M.; Wang, J.C.; Krueger, R.F.; Foroud, T.M.; Edenberg, H.J.; Nurnberger, J.I., Jr.; Brooks, A.I.; Tischfield, J.A.; Almasy, L.; et al. Copy number variation accuracy in genome-wide association studies. Hum. Hered. 2011, 71, 141–147. [Google Scholar] [CrossRef]
- Rosner, B.A. Fundamentals of Biostatistics; Cengage Learning: Singapore, 2011. [Google Scholar]
Appendices
| No. | Chromosome | Start Position | End Position | CNV type | Length (kb) | Gene | References |
|---|---|---|---|---|---|---|---|
| 1 * | 6q27 | 168,090,000 | 168,330,000 | duplication | 240 | KIF25, FERM, MILT4, DACT2 | [28,29] |
| 2 * | 6q16.3 | 101,953,625 | 102,624,651 | unknown | 671.027 | GRIK2 | [32] |
| 3 * | 9q34.3 | 136,600,001 | 14,0273,252 | duplication | 3,673.252 | None | [31] |
| 4 * | 19p12 | 20,001,614 | 20,177,979 | both | 176.366 | ZNF682, ZNF90, ZNF486 | [30] |
| 5 * | 19p12 | 24,013,968 | 24,295,825 | duplication | 281.858 | ZNF254 | [30] |
| 6 | 1 | 28,399,376 | 28,842,172 | unknown | 442.797 | DNAJC8, ATPIF1, SESN2, MED18, SNHG3-RCC1, RCC1, TRSPAP1, RAB42, TAF12PHACTR4 | [32] |
| 7 | 1 | 47,415,160 | 47,600,013 | duplication | 184.854 | PDZK1IP1; TAL1; STIL; CMPK1 | [24] |
| 8 | 1 | 144,439,082 | 144,791,590 | unknown | 352.509 | PDZK1, GPR89A, GPR89C, NBPF11, LOC728912, FAM108A3 | [32] |
| 9 | 1q21.1 | 142,400,001 | 148,000,000 | both | 5,600 | None | [31] |
| 10 | 1q25.1 | 173,769,777 | 173,978,862 | duplication | 209.086 | TNR | [30] |
| 11 | 1 | 232,723,219 | 232,828,069 | unknown | 104.851 | IRF2BP2 | [31] |
| 12 | 2 | 196,772,221 | 197,165,580 | unknown | 393.36 | HECW2 | [32] |
| 13 | 3 | 8,896,559 | 8,980,146 | unknown | 83.588 | RAD18 | [32] |
| 14 | 3p14 | 65,649,762 | 65,848,146 | deletion | 198.385 | MAGI1 | [33] |
| 15 | 3p26 | 2,124,587 | 2,955,648 | duplication | 831.062 | CNTN4 | [24] |
| 16 | 3q | 120,920,000 | 121,100,000 | deletion | 180.001 | GSK3beta | [7] |
| 17 | 4q34.3 | 180,892,619 | 180,921,485 | unknown | 28.867 | None | [31] |
| 18 | 5 | 180,098,728 | 180,099,664 | unknown | 0.937 | OR2Y1 | [32] |
| 19 | 6 | 56,430,743 | 56,816,422 | unknown | 385.68 | DST | [32] |
| 20 | 6 | 57,290,380 | 57,621,335 | unknown | 330.956 | PRIM2 | [32] |
| 21 | 6 | 157,140,777 | 157,572,094 | unknown | 431.318 | ARID1B | [32] |
| 22 | 7 | 34,935,017 | 35,044,178 | unknown | 109.162 | DPY19L1 | [32] |
| 23 | 7 | 75,975,221 | 76,052,734 | unknown | 77.514 | UPK3B | [32] |
| 24 | 7 | 88,226,688 | 89,777,622 | unknown | 1,550.94 | ZNF804B, MGC26647, STEAP1, STEAP2, FLJ21062 | [32] |
| 25 | 7 | 132,588,362 | 133,401,053 | unknown | 812.692 | EXOC4 | [32] |
| 26 | 8 | 13,236,908 | 13,304,907 | unknown | 68 | DLC1 | [31] |
| 27 | 9 | 111,037 | 169,075 | unknown | 58.039 | CBWD1 | [32] |
| 28 | 9 | 71,289,871 | 71,308,782 | duplication | 18.912 | None | [29] |
| 29 | 9 | 134,871,014 | 134,890,520 | unknown | 19.507 | GTF3C5, GFI1B | [31] |
| 30 | 9q31.1 | 104,826,097 | 104,885,068 | both | 58.972 | None | [30] |
| 31 | 10 | 8,108,359 | 8,192,845 | duplication | 84.487 | GATA3 | [24] |
| 32 | 10q11 | 47,010,000 | 47,170,000 | duplication | 160 | ANTXRL | [28] |
| 33 | 10 | 50,334,496 | 50,490,772 | unknown | 156.277 | ERCC6, PGBD3, CHAT, SLC18A3 | [32] |
| 34 | 10 | 51,497,689 | 52,053,743 | unknown | 556.055 | FAM21A, FAM21B, ASAH2, SGMS1 | [32] |
| 35 | 12 | 7,884,583 | 8,017,012 | duplication | 132.43 | SCL2A3M, SLC2A14 | [29] |
| 36 | 12p11.21 | 31,202,250 | 31,301,551 | duplication | 99.302 | OVOS2 | [30] |
| 37 | 12 | 107,243,140 | 107,266,950 | unknown | 23.811 | CMKLR1 | [31] |
| 38 | 13 | 49,932,650 | 49,982,221 | deletion | 49.572 | AJ412031; AJ412041 | [24] |
| 39 | 13 | 90,848,887 | 92,317,488 | unknown | 1,468.60 | GPC5 | [32] |
| 40 | 14 | 24,044,551 | 24,047,311 | unknown | 2.761 | CMA1 | [32] |
| 41 | 15q.2 | 21,905,523 | 22,023,095 | deletion | 117.573 | None | [29] |
| 42 | 15q13.2 | 28,000,001 | 29,000,000 | both | 1,000 | CHRFAM7A, MRMR15 | [31] |
| 43 | 16p13.11 | 14,700,001 | 16,700,000 | duplication | 2,000 | None | [31] |
| 44 | 16 | 15,435,825 | 15,889,948 | unknown | 454.124 | C16orf45, KIAA0430, NDE1, MYH11, C16orf63 | [32] |
| 45 | 16 | 15,950,934 | 16,296,168 | unknown | 345.235 | ABCC1, ABCC6, NOMO3, | [32] |
| 46 | 16 | 16,333,234 | 16351940 | unknown | 18.707 | LOC339047 | [32] |
| 47 | 16 | 68,705,029 | 69,071,678 | unknown | 366.65 | PDPR, MGC34761, EXOSC6, AARS, DDX19B, DDX19A, ST3GAL2, FUK | [32] |
| 48 | 17 | 36,465,156 | 36,477,177 | deletion | 12.022 | KRTAP2-4; KRTAP2-4 | [24] |
| 49 | 17q25.1 | 68,400,001 | 72,200,000 | duplication | 3800 | None | [31] |
| 50 | 18p11.21-11.1 | 14,694,694 | 15,092,421 | duplication | 397.728 | ANKRD30B | [30] |
| 51 | 18 | 27,210,737 | 27,312,663 | unknown | 101.927 | DSG4, DSG3 | [32] |
| 52 | 19 | 49,581,647 | 49,644,505 | unknown | 62.859 | ZNF285A, ZNF229 | [32] |
| 53 | 19 | 58,644,961 | 58,689,358 | unknown | 44.398 | ZNF761, ZNF813, ZNF765, ZNF331 | [32] |
| 54 | 21q11.2 | 13,200,001 | 15,300,000 | both | 2,100 | ANKRD21, LOC441956, LIPI, RBM11 | [31] |
| 55 | 21 | 36,429,132 | 36,440,730 | unknown | 11.599 | CBR3 | [32] |
| Functional Category | Genes on CNV | p-value c | Adjusted p-value d | O a | N b |
|---|---|---|---|---|---|
| Bipolar Disorder (DB_ID:PA447199) | PCDH17, ANK3, GABRR3, GABRG2, NOS1, CNTNAP2, ADCYAP1, PTPRG, NRXN1, PCLO, TACR1, TCF4, JMJD8, ADCY3, CSMD1, DPP10, CNTN5, RELN, NALCN, HTR5A, AGAP1, DFNB31, HTR4, GPC6, ATP8A2, GABRA1, CNTN6, ASTN2, FAT1, ADCY8, ARNTL, RPL14, PPP3CC, NRG1, MAGI1, PDLIM5, MMP16, HTR2A, CHRM2 | 2.40 × 10 −5 | 0.0252 | 39 | 286 |
| Mood Disorders (DB_ID:PA447209) | ANK3, GABRR3, GABRG2, NOS1, SST, CNTNAP2, ADCYAP1, PTPRG, PCLO, GRM5, TACR1, TCF4, HTR6, DPP10, CNR2, CNTN5, GPM6A, NALCN, RELN, HTR5A, GRIK1, DFNB31, AGAP1, CNTN6, GABRA1, ASTN2, NXPH1, FAT1, ARNTL, NRG1, PDLIM5, HTR2A, CHRM2 | 5.53 × 10 −5 | 0.0290 | 33 | 235 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, Y.-H.; Lu, R.-B.; Hung, H.; Kuo, P.-H. Identifying Potential Regions of Copy Number Variation for Bipolar Disorder. Microarrays 2014, 3, 52-71. https://doi.org/10.3390/microarrays3010052
Chen Y-H, Lu R-B, Hung H, Kuo P-H. Identifying Potential Regions of Copy Number Variation for Bipolar Disorder. Microarrays. 2014; 3(1):52-71. https://doi.org/10.3390/microarrays3010052
Chicago/Turabian StyleChen, Yi-Hsuan, Ru-Band Lu, Hung Hung, and Po-Hsiu Kuo. 2014. "Identifying Potential Regions of Copy Number Variation for Bipolar Disorder" Microarrays 3, no. 1: 52-71. https://doi.org/10.3390/microarrays3010052
APA StyleChen, Y.-H., Lu, R.-B., Hung, H., & Kuo, P.-H. (2014). Identifying Potential Regions of Copy Number Variation for Bipolar Disorder. Microarrays, 3(1), 52-71. https://doi.org/10.3390/microarrays3010052