Chromosomal Microarrays in Prenatal Diagnosis: Time for a Change of Policy?

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Procedures and Methods
2.1. The Current Approach to Prenatal Diagnosis of Chromosome Anomalies: A Brief Introduction
2.2. Technical Aspects
2.3. Limitations of Clinical Relevance
3. Results and Discussion
3.1. Clinical Utility of Microarrays
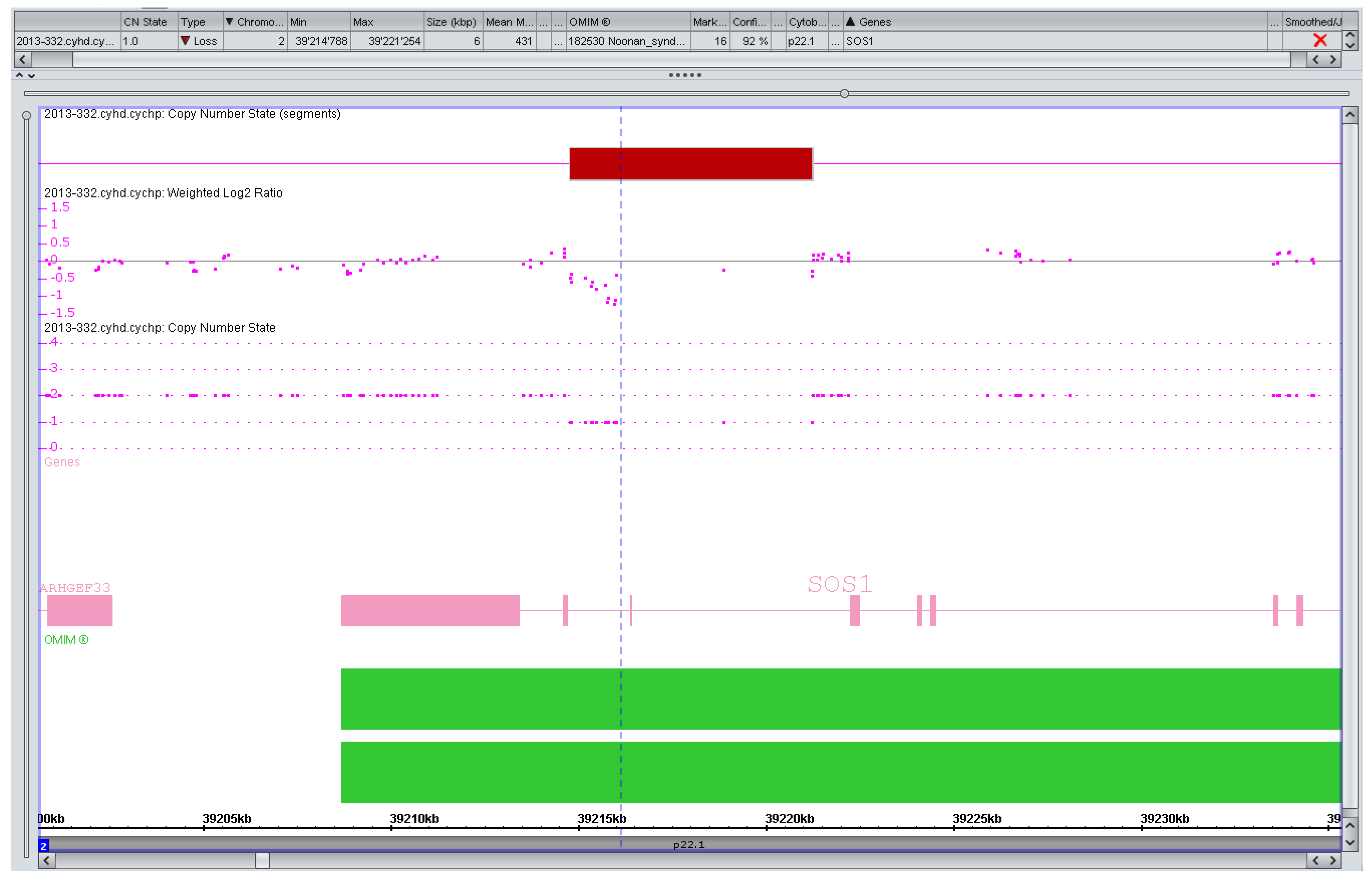
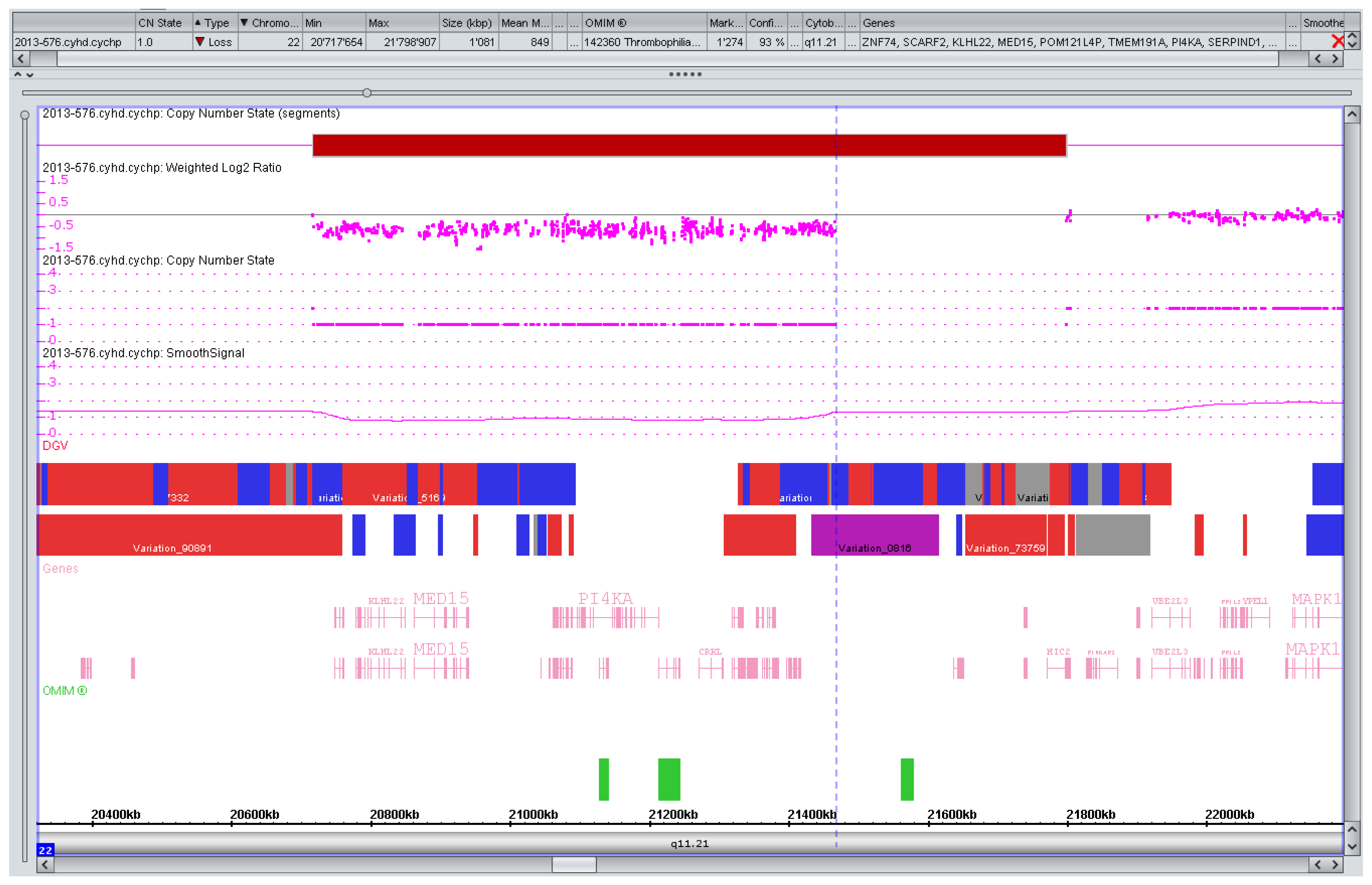
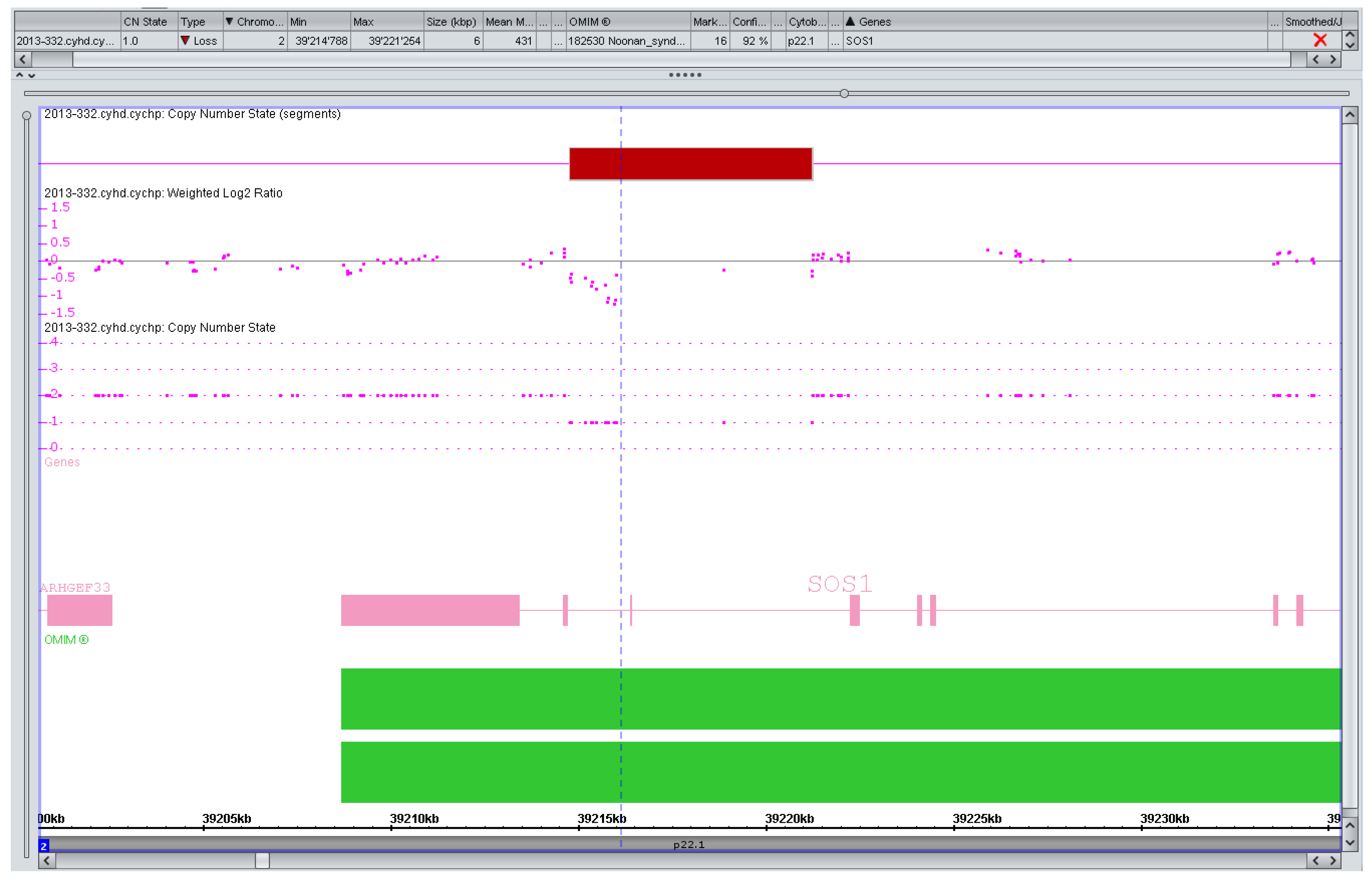
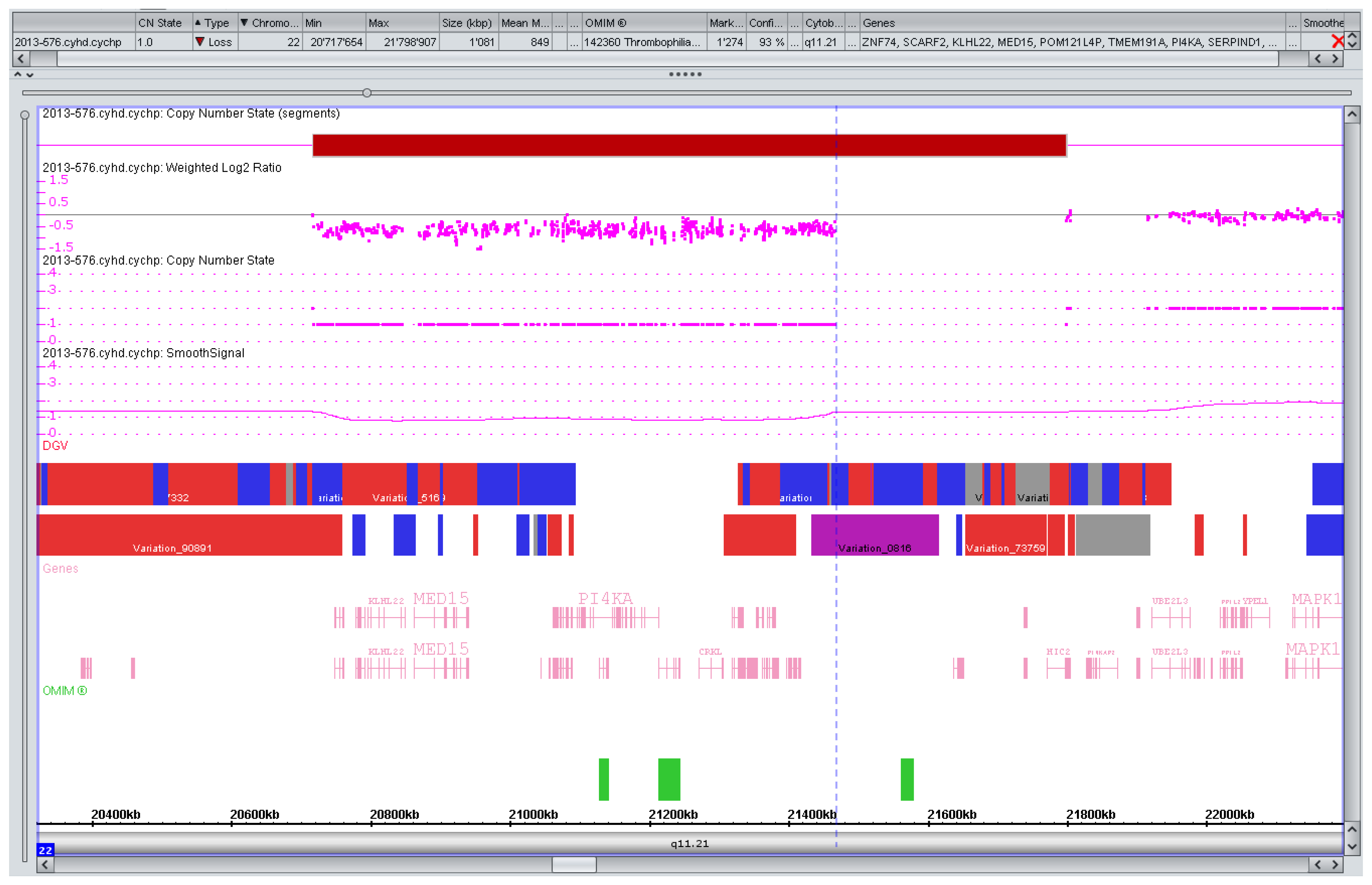
3.2. Counseling Issues
3.3. The Local Approach to Prenatal Microarray Testing
- Highly recommend microarray testing for further characterization of abnormal results obtained by conventional chromosome analysis which are of unclear clinical significance such as de novo translocations, inversions, marker chromosomes and others.
- Recommend microarray testing in pregnancies at high risk for chromosomal abnormalities due to abnormal ultrasound findings or first trimester test results in the high positive range.
- Not encourage but accept the occasional parental request for a comprehensive exclusion of fetal chromosomal conditions even without a significant risk increase.
- Address microarray testing routinely with all patients seeking general advice on prenatal risk screening and testing options.
3.4. Should Microarrays Replace Conventional Karyotyping as a First-Tier Prenatal Diagnostic Test?
4. Conclusions
Conflicts of Interest
References
- Schaaf, C.P.; Wiszniewska, J.; Beaudet, A.L. Copy number and SNP arrays in clinical diagnostics. Annu. Rev. Genom. Hum. Genet. 2011, 12, 25–51. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Manning, M.; Hudgins, L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet. Med. 2010, 12, 742–745. [Google Scholar] [CrossRef]
- Cooper, G.M.; Coe, B.P.; Girirajan, S.; Rosenfeld, J.A.; Vu, T.H.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011, 43, 838–846. [Google Scholar] [CrossRef]
- Vissers, L.E.; Stankiewicz, P. Microdeletion and microduplication syndromes. Methods Mol. Biol. 2012, 838, 29–75. [Google Scholar]
- Carvill, G.L.; Mefford, H.C. Microdeletion syndromes. Curr. Opin. Genet. Dev. 2013, 23, 232–239. [Google Scholar] [CrossRef]
- ACOG Committee Opinion No. 446: Array comparative genomic hybridization in prenatal diagnosis (Replaced by Committee Opinion No. 581). Obstet. Gynecol. 2009, 114, 1161–1163. [CrossRef]
- Novelli, A.; Grati, F.R.; Ballarati, L.; Bernardini, L.; Bizzoco, D.; Camurri, L.; Casalone, R.; Cardarelli, L.; Cavalli, P.; Ciccone, R.; et al. Microarray application in prenatal diagnosis: A position statement from the cytogenetics working group of the italian society of human genetics (sigu), November 2011. Ultrasound Obstet. Gynecol. 2012, 39, 384–388. [Google Scholar]
- Vetro, A.; Bouman, K.; Hastings, R.; McMullan, D.J.; Vermeesch, J.R.; Miller, K.; Sikkema-Raddatz, B.; Ledbetter, D.H.; Zuffardi, O.; van Ravenswaaij-Arts, C.M. The introduction of arrays in prenatal diagnosis: A special challenge. Hum. Mutat. 2012, 33, 923–929. [Google Scholar] [CrossRef]
- Stark, Z.; Gillam, L.; Walker, S.P.; McGillivray, G. Ethical controversies in prenatal microarray. Curr. Opin. Obstet. Gynecol. 2013, 25, 133–137. [Google Scholar] [CrossRef]
- Benn, P.; Cuckle, H.; Pergament, E. Non-invasive prenatal testing for aneuploidy: Current status and future prospects. Ultrasound Obstet. Gynecol. 2013, 42, 15–33. [Google Scholar] [CrossRef]
- Simpson, J.L. Invasive procedures for prenatal diagnosis: Any future left? Best Pract. Res. Clin. Obstet. Gynaecol. 2012, 26, 625–638. [Google Scholar] [CrossRef]
- Stumm, M.; Tonnies, H. Fluorescence in situ hybridization techniques in medical diagnostics. Expert Opin. Med. Diagn. 2008, 2, 1381–1390. [Google Scholar] [CrossRef]
- Mann, K.; Ogilvie, C.M. QF-PCR: Application, overview and review of the literature. Prenat. Diagn. 2012, 32, 309–314. [Google Scholar] [CrossRef]
- Willis, A.S.; van den Veyver, I.; Eng, C.M. Multiplex ligation-dependent probe amplification (MLPA) and prenatal diagnosis. Prenat. Diagn. 2012, 32, 315–320. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Spencer, K.; Avgidou, K.; Faiola, S.; Falcon, O. Multicenter study of first-trimester screening for trisomy 21 in 75,821 pregnancies: Results and estimation of the potential impact of individual risk-orientated two-stage first-trimester screening. Ultrasound Obstet. Gynecol. 2005, 25, 221–226. [Google Scholar] [CrossRef]
- Merz, E.; Thode, C.; Alkier, A.; Eiben, B.; Hackeloer, B.J.; Hansmann, M.; Huesgen, G.; Kozlowski, P.; Pruggmaier, M.; Wellek, S. A new approach to calculating the risk of chromosomal abnormalities with first-trimester screening data. Ultraschall Med. 2008, 29, 639–645. [Google Scholar] [CrossRef]
- Ekelund, C.K.; Jørgensen, F.S.; Petersen, O.B.; Sundberg, K.; Tabor, A.; Danish Fetal Medicine Research Group. Impact of a new national screening policy for Down’s syndrome in Denmark: Population based cohort study. BMJ 2008, 337, a2547. [Google Scholar] [CrossRef]
- Morain, S.; Greene, M.F.; Mello, M.M. A new era in noninvasive prenatal testing. N. Engl. J. Med. 2013, 369, 499–501. [Google Scholar] [CrossRef]
- Gregg, A.R.; Gross, S.J.; Best, R.G.; Monaghan, K.G.; Bajaj, K.; Skotko, B.G.; Thompson, B.H.; Watson, M.S. The Noninvasive Prenatal Screening Work Group of the American College of Medical Genetics and Genomics. ACMG statement on noninvasive prenatal screening for fetal aneuploidy. Genet. Med. 2013, 15, 395–398. [Google Scholar] [CrossRef]
- ACOG Committee Opinion No. 545: Noninvasive prenatal testing for fetal aneuploidy. Obstet. Gynecol. 2012, 120, 1532–1534. [CrossRef]
- Benn, P.; Borell, A.; Chiu, R.; Cuckle, H.; Dugoff, L.; Faas, B.; Gross, S.; Johnson, J.; Maymon, R.; Norton, M.; et al. Position statement from the aneuploidy screening committee on behalf of the board of the international society for prenatal diagnosis. Prenat. Diagn. 2013, 33, 622–629. [Google Scholar] [CrossRef]
- Pinkel, D.; Segraves, R.; Sudar, D.; Clark, S.; Poole, I.; Kowbel, D.; Collins, C.; Kuo, W.L.; Chen, C.; Zhai, Y.; et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998, 20, 207–211. [Google Scholar] [CrossRef]
- Vissers, L.E.; de Vries, B.B.; Osoegawa, K.; Janssen, I.M.; Feuth, T.; Choy, C.O.; Straatman, H.; van der Vliet, W.; Huys, E.H.; van Rijk, A.; et al. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am. J. Hum. Genet. 2003, 73, 1261–1270. [Google Scholar] [CrossRef]
- Brady, P.D.; Vermeesch, J.R. Genomic microarrays: A technology overview. Prenat. Diagn. 2012, 32, 336–343. [Google Scholar] [CrossRef]
- Pinto, D.; Darvishi, K.; Shi, X.; Rajan, D.; Rigler, D.; Fitzgerald, T.; Lionel, A.C.; Thiruvahindrapuram, B.; Macdonald, J.R.; Mills, R.; et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 2011, 29, 512–520. [Google Scholar] [CrossRef]
- Tyreman, M.; Abbott, K.M.; Willatt, L.R.; Nash, R.; Lees, C.; Whittaker, J.; Simonic, I. High resolution array analysis: Diagnosing pregnancies with abnormal ultrasound findings. J. Med. Genet. 2009, 46, 531–541. [Google Scholar] [CrossRef]
- Ballif, B.C.; Kashork, C.D.; Saleki, R.; Rorem, E.; Sundin, K.; Bejjani, B.A.; Shaffer, L.G. Detecting sex chromosome anomalies and common triploidies in products of conception by array-based comparative genomic hybridization. Prenat. Diagn. 2006, 26, 333–339. [Google Scholar] [CrossRef]
- Filges, I.; Kang, A.; Klug, V.; Wenzel, F.; Heinimann, K.; Tercanli, S.; Miny, P. aCGH on chorionic villi mirrors the complexity of fetoplacental mosaicism in prenatal diagnosis. Prenat. Diagn. 2011, 31, 473–478. [Google Scholar] [CrossRef]
- Campbell, C.D.; Eichler, E.E. Properties and rates of germline mutations in humans. Trends Genet. 2013, 29, 575–584. [Google Scholar] [CrossRef]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Dabell, M.P.; Fisher, A.J.; Coppinger, J.; Bandholz, A.M.; Ellison, J.W.; Ravnan, J.B.; Torchia, B.S.; Ballif, B.C.; Rosenfeld, J.A. Experience with microarray-based comparative genomic hybridization for prenatal diagnosis in over 5000 pregnancies. Prenat. Diagn. 2012, 32, 976–985. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Rosenfeld, J.A.; Dabell, M.P.; Coppinger, J.; Bandholz, A.M.; Ellison, J.W.; Ravnan, J.B.; Torchia, B.S.; Ballif, B.C.; Fisher, A.J. Detection rates of clinically significant genomic alterations by microarray analysis for specific anomalies detected by ultrasound. Prenat. Diagn. 2012, 32, 986–995. [Google Scholar] [CrossRef]
- Mademont-Soler, I.; Morales, C.; Soler, A.; Martinez-Crespo, J.M.; Shen, Y.; Margarit, E.; Clusellas, N.; Obon, M.; Wu, B.L.; Sanchez, A. Prenatal diagnosis of chromosomal abnormalities in fetuses with abnormal cardiac ultrasound findings: Evaluation of chromosomal microarray-based analysis. Ultrasound Obstet. Gynecol. 2013, 41, 375–382. [Google Scholar] [CrossRef]
- Hillman, S.C.; McMullan, D.J.; Hall, G.; Togneri, F.S.; James, N.; Maher, E.J.; Meller, C.H.; Williams, D.; Wapner, R.J.; Maher, E.R.; et al. Use of prenatal chromosomal microarray: Prospective cohort study and systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2013, 41, 610–620. [Google Scholar] [CrossRef]
- De Wit, M.C.; Srebniak, M.I.; Govaerts, L.C.; van Opstal, D.; Galjaard, R.J.; Go, A.T. The additional value of prenatal genomic array testing in fetuses with (isolated) structural ultrasound abnormalities and a normal karyotype: A systematic review of the literature. Ultrasound Obstet. Gynecol. 2013. [Google Scholar] [CrossRef]
- Breman, A.; Pursley, A.N.; Hixson, P.; Bi, W.; Ward, P.; Bacino, C.A.; Shaw, C.; Lupski, J.R.; Beaudet, A.; Patel, A.; et al. Penatal chromosomal microarray analysis in a diagnostic laboratory; experience with >1000 cases and review of the literature. Prenat. Diagn. 2012, 32, 351–361. [Google Scholar] [CrossRef]
- Callaway, J.L.; Shaffer, L.G.; Chitty, L.S.; Rosenfeld, J.A.; Crolla, J.A. The clinical utility of microarray technologies applied to prenatal cytogenetics in the presence of a normal conventional karyotype: A review of the literature. Prenat. Diagn. 2013. [Google Scholar] [CrossRef]
- Hillman, S.C.; McMullan, D.J.; Silcock, L.; Maher, E.R.; Kilby, M.D. How does altering the resolution of chromosomal microarray analysis in the prenatal setting affect the rates of pathological and uncertain findings? J. Matern. Fetal Neonatal Med. 2013. [Google Scholar] [CrossRef]
- Ganesamoorthy, D.; Bruno, D.L.; McGillivray, G.; Norris, F.; White, S.M.; Adroub, S.; Amor, D.J.; Yeung, A.; Oertel, R.; Pertile, M.D.; et al. Meeting the challenge of interpreting high-resolution single nucleotide polymorphism array data in prenatal diagnosis: Does increased diagnostic power outweigh the dilemma of rare variants? BJOG 2013, 120, 594–606. [Google Scholar] [CrossRef]
- Srebniak, M.I.; Mout, L.; van Opstal, D.; Galjaard, R.J. 0.5 Mb array as a first-line prenatal cytogenetic test in cases without ultrasound abnormalities and its implementation in clinical practice. Hum. Mutat. 2013, 34, 1298–1303. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Dabell, M.P.; Rosenfeld, J.A.; Neill, N.J.; Ballif, B.C.; Coppinger, J.; Diwan, N.R.; Chong, K.; Shohat, M.; Chitayat, D. Referral patterns for microarray testing in prenatal diagnosis. Prenat. Diagn. 2012, 32, 344–350. [Google Scholar] [CrossRef]
- Fiorentino, F.; Napoletano, S.; Caiazzo, F.; Sessa, M.; Bono, S.; Spizzichino, L.; Gordon, A.; Nuccitelli, A.; Rizzo, G.; Baldi, M. Chromosomal microarray analysis as a first-line test in pregnancies with a priori low risk for the detection of submicroscopic chromosomal abnormalities. Eur. J. Hum. Genet. 2013, 21, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Verhagen, J.M.; Diderich, K.E.; Oudesluijs, G.; Mancini, G.M.; Eggink, A.J.; Verkleij-Hagoort, A.C.; Groenenberg, I.A.; Willems, P.J.; du Plessis, F.A.; de Man, S.A.; et al. Phenotypic variability of atypical 22q11.2 deletions not including tbx1. Am. J. Med. Genet. A 2012, 158a, 2412–2420. [Google Scholar] [CrossRef]
- Dixit, A.; McKee, S.; Mansour, S.; Mehta, S.G.; Tanteles, G.A.; Anastasiadou, V.; Patsalis, P.C.; Martin, K.; McCullough, S.; Suri, M.; et al. 7q11.23 Microduplication: A recognizable phenotype. Clin. Genet. 2013, 83, 155–161. [Google Scholar]
- Pichert, G.; Mohammed, S.N.; Ahn, J.W.; Ogilvie, C.M.; Izatt, L. Unexpected findings in cancer predisposition genes detected by array comparative genomic hybridisation: What are the issues? J. Med. Genet. 2011, 48, 535–539. [Google Scholar] [CrossRef]
- McGillivray, G.; Rosenfeld, J.A.; McKinlay Gardner, R.J.; Gillam, L.H. Genetic counselling and ethical issues with chromosome microarray analysis in prenatal testing. Prenat. Diagn. 2012, 32, 389–395. [Google Scholar] [CrossRef]
- ACOG Practice Bulletin No. 77: Screening for fetal chromosomal abnormalities. Obstet. Gynecol. 2007, 109, 217–227.
- Bui, T.H.; Vetro, A.; Zuffardi, O.; Shaffer, L.G. Current controversies in prenatal diagnosis 3: Is conventional chromosome analysis necessary in the post-array CGH era? Prenat. Diagn. 2011, 31, 235–243. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Miny, P.; Wenzel, F.; Tercanli, S.; Filges, I. Chromosomal Microarrays in Prenatal Diagnosis: Time for a Change of Policy? Microarrays 2013, 2, 304-317. https://doi.org/10.3390/microarrays2040304
Miny P, Wenzel F, Tercanli S, Filges I. Chromosomal Microarrays in Prenatal Diagnosis: Time for a Change of Policy? Microarrays. 2013; 2(4):304-317. https://doi.org/10.3390/microarrays2040304
Chicago/Turabian StyleMiny, Peter, Friedel Wenzel, Sevgi Tercanli, and Isabel Filges. 2013. "Chromosomal Microarrays in Prenatal Diagnosis: Time for a Change of Policy?" Microarrays 2, no. 4: 304-317. https://doi.org/10.3390/microarrays2040304