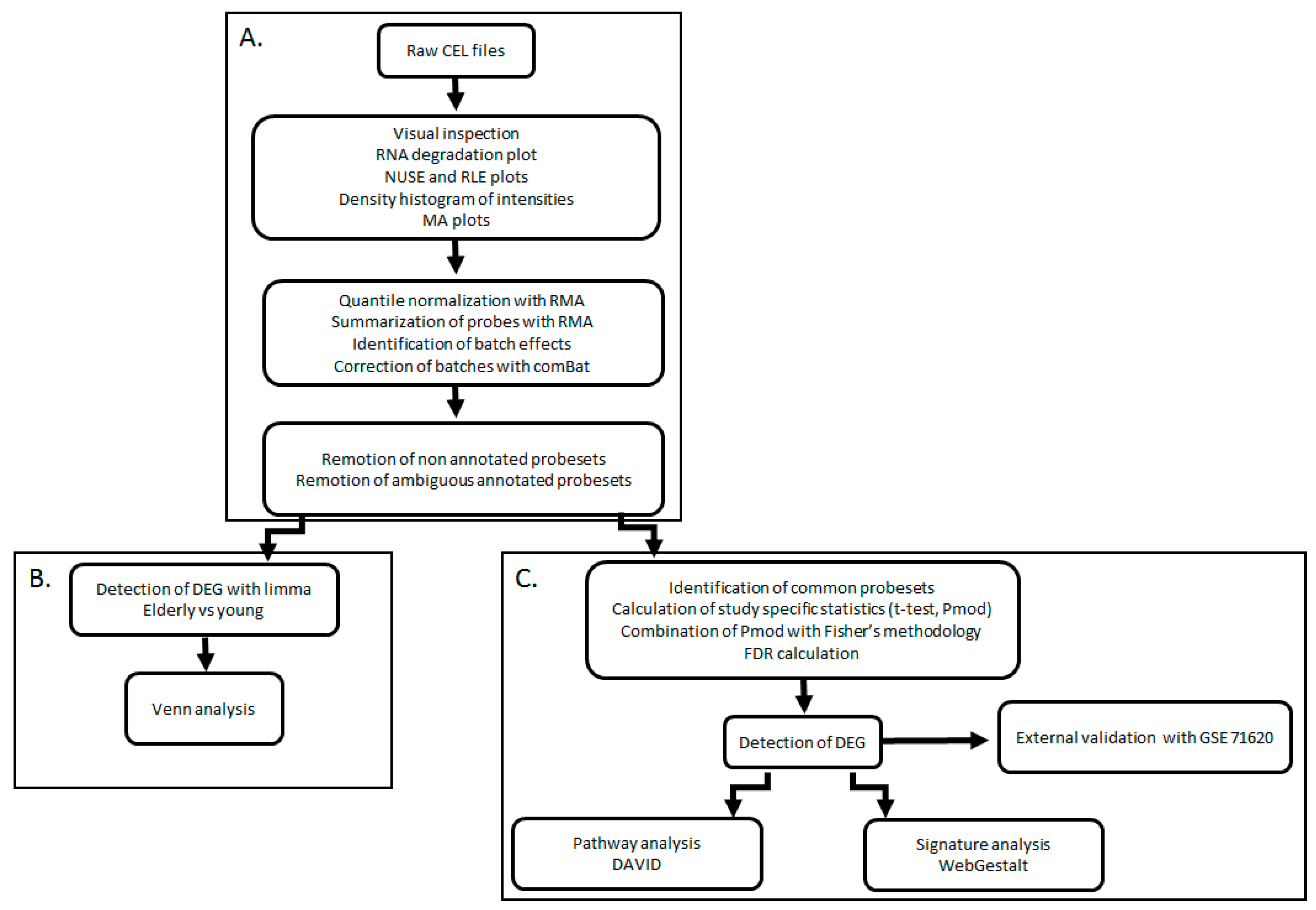
4.1. General Transcriptomic Landscape of Aging in PFC
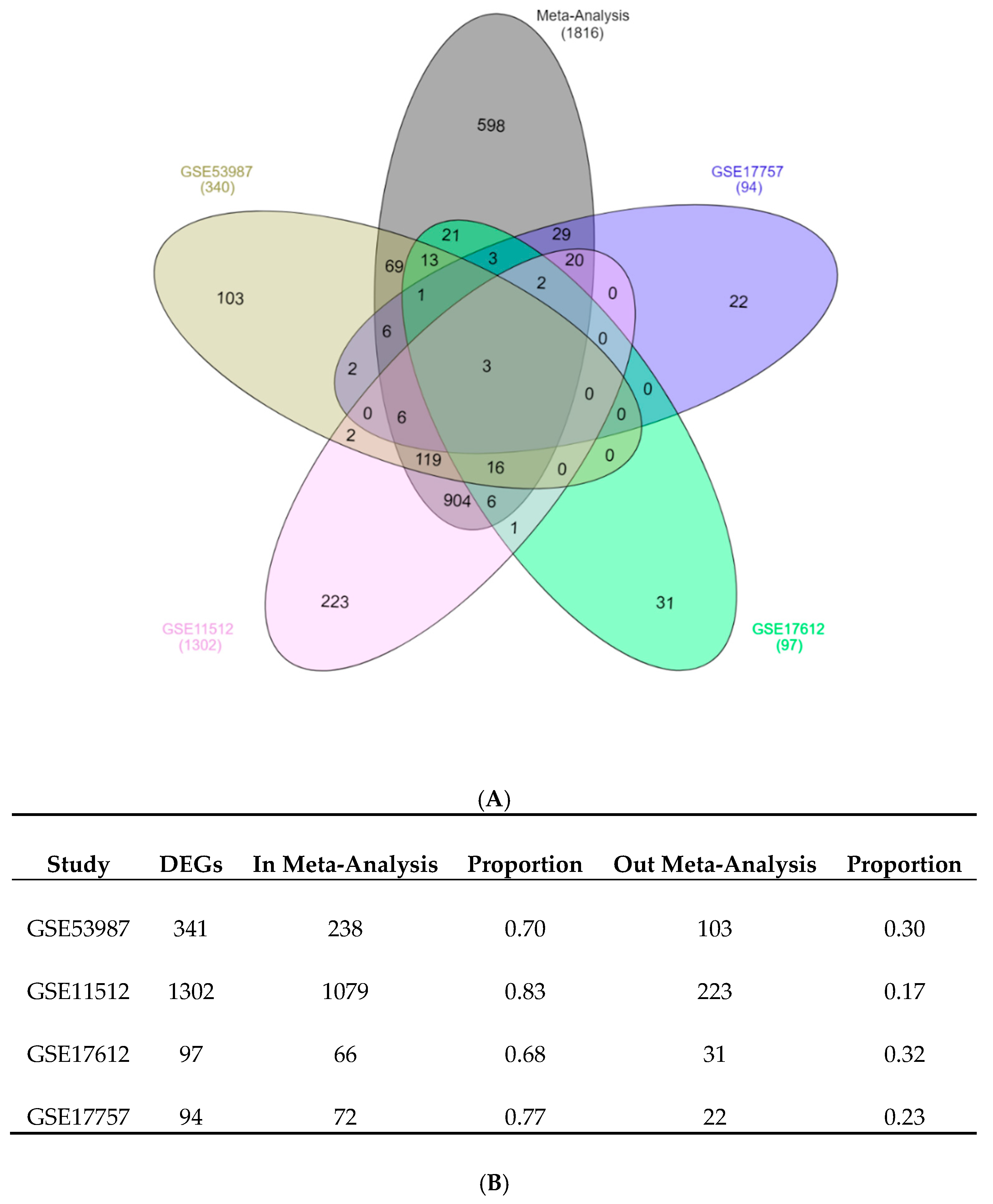
The combination of elderly PFC vs. young PFC samples from several independent studies by meta-analysis identified a list of DEGs that had high overlapping with the validation dataset constituted by many biological replicates. The approach used in our study could detect genes with a consistent and coherent deregulation across several independent studies. The proportion of down-regulated genes vs. up-regulated genes was 2.2:1. In previous analyses of the aging transcriptomic profile on different tissues and organisms, the number and proportion of down-regulated and up-regulated genes were variable. Meta-analysis of the aged liver in mice found a 1:3 ratio of down-regulated to up-regulated genes [
30]. A similar result was obtained in other analyses using several aging organs (kidney, lung, brain cortex, and liver) from humans, mice, and rats [
7]. In human lymphoblastoid cells, the proportion was 1:1 [
31]. Two studies using whole blood cells found a proportion close to 1.4:1 [
32,
33]. In a meta-analysis of human muscles, the proportion was 1.2:1 [
34]. This variation in the proportion of the direction of de-regulated genes could be explained as follows: since post-mitotic cells (such as muscle cells and neurons) accumulate DNA damage over their lifespan, it is more likely that mutations in transcriptionally active genes will induce a down-regulation, while, mitotically, active cells with an accumulation of mutations are negatively selected and removed from the tissues. Our findings support this hypothesis because the down-regulated genes had an enrichment in specific markers for neurons (post-mitotic cells) and the up-regulated genes had an enrichment in specific markers for astrocytes (cells with proliferative ability in the nervous tissue).
To identify which types of PFC cells were altered in the de-regulation of the transcriptome of elderly samples compared with young samples, we performed on our list of DEGs over-representation and under-representation analyses with specific markers for neurons, oligodendrocytes, and astrocytes [
23]. As in previous studies on the cerebral cortex [
6], we found an over-representation of neuronal markers in the down-regulated genes and an under-representation of those markers in the up-regulated genes. Astrocyte markers had the opposite over-representation results, with an enrichment of up-regulated genes and fewer down-regulated genes on the list of DEGs. Oligodendrocyte markers had the number of DEGs that would be expected by chance. Taken together, those results indicate that, in aging PFC, there is a down-regulation of neuronal genes without compensatory up-regulation of other neural genes, as well as an increased expression of astrocyte genes. Neuropathological studies show contradictory evidence regarding the change in the number of neurons and neuroglial cells in different regions of the brain with aging. Some results have pointed to a loss of neurons in the rat’s prefrontal cortex [
35], basal forebrain [
36], thalamus [
37], cortex, hypothalamus, cerebellum and olfactory bulb [
38]. However, other studies found no modification in the number of neurons in aging. For instance, in the human substantia nigra, there was no correlation between the number of neurons and age [
39], and, in the Rhesus macaque, the number of white matter neurons did not show a correlation with age [
40]. Therefore, the down-regulation of neuron-specific markers could be explained as a result of a decreased number of neurons or a down-regulation in the expression of genes related to a specific aging phenotype. Likewise, the over-representation of astrocyte genes in the up-regulated genes is due to an increase in the number of cells or their activation.
Our results, as discussed below, support that in aging, there is a down-regulation of gene expression in specific neuronal zones, especially synapses, and opposite patterns of astrocyte activation.
4.3. Astrocyte Transcriptome in Aged PFC: The Opposite Activation
Astrocytes, the most abundant glial cells, are important for adequate central nervous system (CNS) function. They are involved in the formation and elimination of neuronal synapses [
42,
43], and also mediate the uptake and recycling of neurotransmitters [
44]. We found that, in aging, there is an up-regulation of specific astrocyte markers. These results coincide with a previous report using a different source of information [
6]. Current knowledge suggests that astrocyte number is preserved in aging [
45,
46]. Therefore, the up-regulation of astrocyte markers could be explained by an increase in the activation state of those cells. There are distinctive phenotypes of activated astrocytes, which depend on the stimuli that induce the activation. The best-characterized phenotypes of activated astrocytes are A1 and A2. Reactive astrocytes induced by LPS (A1 astrocytes) exhibit a phenotype that suggests they are detrimental, whereas reactive astrocytes induced by ischemia (A2 astrocytes) exhibit a cellular phenotype that suggests that they are beneficial or protective [
24]. The A1 and A2 phenotypes share common genes that are useful for identifying reactive astrocytes in general (activated A1A2 astrocytes). A study in rat brains found an increase of A1-like reactive astrocytes in the hippocampus and striatum with aging [
4] suggesting that, in this animal model, astrocyte activation is mainly toxic and it is associated with the loss of brain function. We found that, in elderly PFC, there is a strong up-regulation of A1A2 signature genes. When we analyzed what kind of activated astrocytes were present in elderly PFC, we found a similar over-representation of A1 and A2 signature genes. Additionally, since there are reports linking methamphetamine abuse with the neurochemical profile of aging [
47] and premature cellular senescence [
26], we compared the molecular profile of the astrocytes activated by methamphetamine abuse with our signature of elderly PFC. However, this profile was not over-represented, indicating that astrocyte activation by methamphetamine is not associated to normal aging astrocyte activation. These joint results indicate that aged human PFC seems to have patterns of gene-expression compatible with heterogeneous astrocyte activation, mixing of protective and toxic astrocyte phenotypes.
Since we used whole tissue with a mixture of cells in our study, we could not delineate more precisely the proportions and specific pathways activated in each type of activated astrocyte. Single cell transcriptomic analysis of astrocytes in aging samples, along with phenotypic analysis of these cells, must be performed to answer this question.
Nonetheless, with the pathway and signature analysis of up-regulated genes, it is possible to suggest the molecular phenotype of astrogliosis in elderly PFC. The fact that mineral absorption was the main up-regulated pathway in the top cluster of activated pathways was an unexpected result of the transcriptomic analysis of the CNS. However, the pathway was statistically significant, because it contained several metallothionein (MT) genes. In the meta-analysis, seven MT genes were analyzed, all of which were from the MT I family, and were up-regulated in elderly PFC. There is an increasing interest in the role of MT in normal and pathological CNS function. The MT superfamily has four isoforms (I–IV); isoforms I and II are expressed in the brain, mainly in astrocytes, while isoform III is expressed in neurons [
48]. Metallothioneins I/II are up-regulated in astrocytes in response to neuronal injury [
49], and their expression is induced by several stimuli such as metals, hormones, cytokines, oxidative stress and inflammation [
50]. The over-expression of MTs is in general protective, for example, when MTs are overexpressed, the mouse lifespan is increased [
51]. Metallothioneins I/II play a neuroprotective role in several forms of brain injury and are able to augment the regenerative capacity of astrocytes [
52]. These molecules also induce a form of astrogliosis that is permissive with the neurite outgrowth and associated with decreased chondroitin sulfate proteoglycan (CSPG) accumulation. CSPGs are involved in maintaining the structure and function of adult neurons, and in the regulation of proliferation, migration, and neurite outgrowth of neural stem cells in the brain. Aged rats show a significant increase in aggrecan expression throughout the PFC and in the hippocampus [
53]. We found up-regulation of the expression of two CSPG genes (BCAN and CD44), and thus the up-regulation of MT I could be related to an astrocyte effort to degrade the increased deposition of CSPGs as a response to synapse malfunction.
Organelle assembly, the second cluster of up-regulated genes, includes the all three ERM family proteins (ezrin, radixin, and moesin). These proteins play a crucial role in organizing membrane domains and regulating signal transduction pathways such as SHH [
54]. In the brain, this family is important in the regulation of plasticity and neuroprotection: ezrin (EZR) is required for the structural plasticity of peripheral astrocyte processes associated with synapses [
55], moesin (MSN) regulates dendrite arborization and spine-like protrusion growth [
56], and radixin (RDX) stimulates adult neural progenitor cell migration and proliferation [
57]. Activation of the three members of the family promotes the migration of subventricular zone-derived neuroblasts in response to traumatic brain damage [
58]. In elderly PFC, neuronal synapse dysfunction could be sensed by the astrocytes as local damage, and part of the protective response could be the up-regulation of ERM genes. Activation of ERM proteins is mediated by RhoA in HeLa cells [
59] and fibroblasts [
60], but is independent of RhoA in kidney-derived cells [
61]. RhoA was not up-regulated in our analysis nor in previous studies [
24] of elderly PFC, but other Rho proteins as RhoJ and RhoU were up-regulated. If those proteins can interact with ERM proteins, then it is plausible that ERM protein activation is caused by other Rho family proteins in the brain and accessory proteins such as ARHGDIA, which is also up-regulated in aging; however, additional analysis of interaction of those proteins are necessary to probe this hypothesis.
Smoothened (SMO) signaling pathway, the representative pathway in the third cluster of up-regulated genes, is the intracellular effector of the activation of Sonic Hedgehog (SHH) pathway. The SHH plays a key role in the development and patterning of the CNS. In the adult brain, SHH is one of the regulators of astrocyte function and activation. Given the importance of this pathway in the biology of astrocytes, we explored in detail their complete regulation in aged PFC. SHH regulates the activity of the GLI transcription factor family, in which there are three members: GLI1, 2 and 3, each with a different role in SHH responsive gene regulation. GLI1 is a transcriptional activator, GLI2 is mainly a transcriptional activator with slight repressor activity, and GLI3 is a transcriptional repressor of target genes [
62]. We looked if the transcriptional targets of each GLI were over-represented on the list of DEGs. When all the targets were interrogated, none of GLI targets were over-represented on the list of up-regulated genes and only GLI1 targets were under-represented on the list of down-regulated genes. These results indicate that there is no general deregulation of the SHH pathway in elderly PFC. However, when we selected the GLI targets that are expressed specifically in astrocytes, there was an over-representation of GLI1, 2 and 3 astrocyte targets on the list of up-regulated genes and under-representation of those targets on the list of down-regulated genes. GLI1 and 2 are transcriptional activators and GLI3 is a repressor, thus there is an activation of GLI1 and 2 and inactivation of GLI3 in astrocytes in aging. As a result, there is an overall activation of the SHH pathway, specifically in astrocytes.
Neurons in elderly PFC have a wide down-regulation of expression of synaptic genes, including the genes related to biosynthesis, transport, and release of neurotransmitters. A study recently described that neurons use SHH to control different properties of the astrocytes [
28,
63]. SHH stimulation of Bergmann glial cells—a type of cerebellar astrocytes—promotes glutamate detection and recovery and potassium homeostasis by up-regulation of SLC1A3 (GLAST) and KCNJ10 (KIR 4.1) [
63]. Those genes are up-regulated in elderly PFC, suggesting that the activation of SHH in PFC astrocytes could be a protective response induced by down-regulation in the expression of neuronal synaptic genes. Furthermore, SHH is also involved in neural progenitor proliferation, neovascularization, and synaptogenesis [
64]. SHH reduces astrocyte reactivity and the inflammatory response after a brain injury [
64], and astrocytes stimulated by SHH protect neurons from cell death [
28]. This is compatible with the finding of over-representation of the protective astrocyte signature on the list of up-regulated genes.
On the other hand, we found an over-representation of the neurotoxic astrocyte signature, suggesting that there are parallel pathways of astrocyte activation inducing diverse astrocyte phenotypes in brain aging. Our analysis identified up-regulation of related inflammatory pathways (Cluster 4 of up-regulated genes). This cluster consisted of the enrichment of genes annotated in response to interferon alpha, beta, and gamma. In the aging brain, it is well characterized that interferon signaling at the choroid plexus negatively affects brain function [
65] and that the interferon pathways are induced in LPS-reactive astrogliosis [
24]. Inflammation is one of the hallmarks of aging, and the hypothalamus integrates inflammatory responses with systemic control of aging through nuclear factor κB (NF-κB) and microglia-neuron neuroimmune crosstalk [
66,
67]. Inflammation is so important in aging brains that chronic treatment with an IFN-I activator contributes to the development of neurodegenerative disease in wild-type mice [
68]. In the context of astrocytes, neurotoxic phenotype development after exposure to LPS is characterized by the induction of interferon pathways [
24]. The activation of IFN pathways is also compatible with the aging model that describes inflammatory astrocyte (A1) activation. Moreover, the direct analysis of astrocytes in normal aging showed that one of the up-regulated pathways in mouse old brain astrocytes was interferon signaling [
4].
Circadian entrainment pathway was over-represented in the list of down-regulated pathways. Circadian dysfunction is a common symptom of aging, elderly people have a modification in rhythms of behaviors, temperature regulation, and hormone release. Although the samples analyzed in the meta-analysis did not have the time of death available, the meta-analysis identified 28 down-regulated genes annotated in the KEGG pathway of circadian entrainment. The original analysis of the validation dataset GSE71620, which includes this information, found that circadian patterns of gene expression are modified by aging [
5]. These results provide an additional validation of our analysis indicating that integrative analysis can identify biologically relevant pathways in aging. The circadian clock is regulated synergistically by neurons and astrocytes [
69]. The suprachiasmatic nucleus (SCN) of the hypothalamus coordinates daily rhythms, neurons are in phase with a higher metabolic activity during the circadian daytime, while astrocytes are anti-phasic. They are active during the circadian night and they inhibit neuronal activation by releasing glutamate in the extracellular space [
69]. Recently, it is reported that the deletion of Bmal1—one of the master regulators of the molecular clock—induces activation of astrocytes and inflammatory gene expression [
70]. The identification in our study of the alteration in the circadian entrainment pathway in the PFC represents one of the possible triggers of the activation of astrocytes in A1-like phenotype in aging.
These results suggest that the up-regulated pathways we found are mainly due to astrocyte activation and they represent two divergent astrocyte molecular and cellular phenotypes of astrogliosis.







{kind=link}
{kind=link}
{kind=link}
{kind=link}