Contributions of Molecular and Optical Techniques to the Clinical Diagnosis of Alzheimer’s Disease


1
Fondazione IRCCS Istituto Neurologico Carlo Besta, Division of Neurology 5 and Neuropathology, 20133 Milan, Italy
2
Fondazione IRCCS Istituto Neurologico Carlo Besta, Scientific Directorate, 20133 Milan, Italy
3
IFAC-CNR, Institute of Applied Physics “Nello Carrara”, National Research Council, 50019 Sesto Fiorentino, Italy
*
Authors to whom correspondence should be addressed.
Brain Sci. 2020, 10(11), 815; https://doi.org/10.3390/brainsci10110815
Submission received: 16 October 2020
/
Revised: 29 October 2020
/
Accepted: 31 October 2020
/
Published: 3 November 2020
(This article belongs to the Special Issue Advances in Alzheimer's Disease: Where Do We Stand in 2020?)
Abstract
:Alzheimer’s disease (AD) is the most common neurodegenerative disorder worldwide. The distinctive neuropathological feature of AD is the intracerebral accumulation of two abnormally folded proteins: β-amyloid (Aβ) in the form of extracellular plaques, and tau in the form of intracellular neurofibrillary tangles. These proteins are considered disease-specific biomarkers, and the definite diagnosis of AD relies on their post-mortem identification in the brain. The clinical diagnosis of AD is challenging, especially in the early stages. The disease is highly heterogeneous in terms of clinical presentation and neuropathological features. This phenotypic variability seems to be partially due to the presence of distinct Aβ conformers, referred to as strains. With the development of an innovative technique named Real-Time Quaking-Induced Conversion (RT-QuIC), traces of Aβ strains were found in the cerebrospinal fluid of AD patients. Emerging evidence suggests that different conformers may transmit their strain signature to the RT-QuIC reaction products. In this review, we describe the current challenges for the clinical diagnosis of AD and describe how the RT-QuIC products could be analyzed by a surface-enhanced Raman spectroscopy (SERS)-based systems to reveal the presence of strain signatures, eventually leading to early diagnosis of AD with the recognition of individual disease phenotype.

{kind=link}
1. Alzheimer’s Disease is a Double-Prion Disorder
Alzheimer’s disease (AD) is the most frequent neurodegenerative disorder in elderly people affecting nowadays, about 44 million patients worldwide, and this estimation is expected to double every 20 years [1]. For this reason, AD represents one of the great health-care challenges of the 21st century, but from the discovery of the key component of AD pathogenic mechanism, little is known about the causes of the disease, and no effective treatments have been developed so far. Most cases have a sporadic origin and occur in subjects over the age of 65, while less than 5% of all the cases occur earlier [2]. Mutations in the amyloid precursor protein (APP), presenilin-1 (PSEN1), and presenilin-2 (PSEN2) are known to cause familial forms of AD that are characterized by early onset and rapid progression [3,4,5,6,7,8]. The apolipoprotein E gene (ApoE) represents a genetic risk factor for AD. It has three common alleles: ɛ2, ɛ3, and ɛ4 and the allelic variant ɛ4/ɛ4 is associated with high susceptibility to AD in the general population [9]. AD leads inevitably to dementia with progressive impairment of physiological functions, and the death generally occurs within 5–12 years from symptoms onset [10]. The main neuropathological hallmark of AD is the aggregation in the brain of two different proteins: (1) β-amyloid (Aβ), which deposits extracellularly to form amyloid plaques, and (2) tau, which accumulates intraneuronally to form neurofibrillary tangles (NFTs) made up of hyperphosphorylated-tau. Before aggregating, both Aβ and tau undergo a process of structural rearrangement (misfolding), resulting in a higher content of β-sheet in their secondary structures [11]. Aβ plaques and NFTs are considered specific biomarkers for AD, and their deposition often parallel the cognitive decline [12]. Emerging evidence suggests that both Aβ and tau proteins possess prion-like features [13,14,15]. What does it mean? In 1982 the Nobel prize winner, prof. Stanley Prusiner [16], discovered that an infectious protein was responsible for a group of neurodegenerative disorders, named prion diseases. This discovery has vexed the scientific community because it was against the central dogma of molecular biology [17]. Indeed, in this case, a single protein with aberrant conformations (misfolded) was able to cause disease. Prof. Prusiner coined the term ‘prion’ (proteinaceous infectious only particle) to refer to this misfolded protein that was characterized by infectious and pathological properties, and that was able to ‘replicate’ even without the presence of genetic material. In particular, prion (or PrPSc) arises from the conformational conversion of the normal cellular prion protein (PrPC), which is expressed in many tissues, including the central nervous system (CNS) [18]. Several studies have confirmed that PrPSc alone can cause disease and sustain its progression. Once generated, PrPSc coerces other PrPC to misfold, thus, generating new PrPSc molecules by overcoming the thermodynamic barriers which normally prevent this phenomenon from occurring [19]. In this way, PrPSc spreads throughout the brain and aggregates to form oligomers, which then assemble into insoluble amyloid fibrils. One of the most fascinating aspects of these diseases is that PrPSc can acquire distinct abnormal conformations, referred to as strains, which are responsible for at least six different phenotypes of prion disorders [20,21]. Each prion strain possesses peculiar properties (in terms of pathological and biochemical features), which confer a distinctive signature useful for their identification. Since prions replicate without the presence of genetic instructions, the abnormal structure is believed to encipher and propagate individual prion strain signature [22]. This represents an innovative and unusual biological paradigm of information storage and transmission. The prion paradigm is now expanding to AD and some other neurodegenerative disorders characterized by the presence of misfolded proteins, including Parkinson’s disease (PD), Huntington’s disease (HD), and Amyotrophic Lateral Sclerosis (ALS) [23,24,25,26]. In AD, Aβ and tau undergo misfolding and acquire distinct aberrant structures that significantly influence their interactome, their tropism for defined neuronal cells, and their capacity to modulate specific cellular pathways. Once misfolded, both proteins can template the conformational conversion of their normal counterparts and propagate in the brain following defined spatiotemporal patterns [27,28]. For this reason, they are considered prion-like proteins capable of sustaining disease progression [29,30,31], and AD can be thought of as a double prion disorder. AD is phenotypically heterogeneous, and this variability might be sustained, at least in part, by a spectrum of conformationally diverse Aβ aggregates, referred to as Aβ strains. Thus, similarly to prion diseases, Aβ seems to have important roles in disease pathogenesis and is included among the debated causal factors of AD, as well as the loss of cholinergic neurons, the deregulation of neuronal cell cycle, and the presence of neuroinflammatory molecules and toxic products [32,33,34,35,36,37,38]. Nevertheless, a growing body of evidence sustains that different strains of Aβ are believed to be responsible for different phenotypes of AD. Several factors, including chaperones, inflammatory cytokines, ApoE, microbiota, lifestyle, and other elements still unknown, could contribute to disease variability, especially by influencing the interactions between misfolded Aβ and other cellular components (as well as tau) [39,40,41,42,43,44,45,46,47,48,49].
Recent studies, including electron cryo-microscopy (cryo-EM) and immuno-gold electron microscopy (immuno-EM) analyses, showed that the abnormal conformations of tau between patients with sporadic and genetic forms of AD are not that different. Probably, the intracellular environment limits the range of structural rearrangements adoptable by the protein [50]. In contrast, Aβ aggregates extracellularly, in a different environment, which is likely more permissive to the generation of multiple Aβ strains that can be key determinants of disease heterogeneity. Therefore, at present, there is a strong correlation between the phenotypic heterogeneity of AD and Aβ strains.
2. Phenotypic Heterogeneity of Alzheimer’s Disease and the Role of Aβ Strains
The exact mechanisms through which Aβ misfolds are still not well understood. Nevertheless, it is known that Aβ can adopt a variety of abnormal conformations (Aβ strains), which are believed to account for the phenotypic heterogeneity of AD (in terms of clinical presentation, neuropathological features, and neuroanatomical involvement) [51,52,53]. Several studies have shown that among AD patients, it is possible to identify different types of Aβ deposits characterized by distinct morphology and biochemical features [52,54,55,56,57,58]. These findings strengthen the causal link between the Aβ structures and disease phenotype, but also highlight the possibility that more than one Aβ strain can be present in the same brain, while contributing to disease heterogeneity [59]. A variety of in vivo studies performed using transgenic mice reinforced this concept [51,53,60,61,62,63]. For instance, inoculations of susceptible animals with different Aβ aggregates, either synthetically produced or present in brain homogenates of patients with distinct AD phenotypes, led to cerebral Aβ deposition characterized by different morphology and distribution [45,52,53,61,64,65,66]. In some cases, the types of Aβ aggregates found in animals faithfully reproduced the neuropathological changes found in the brain of the AD donor [62]. All these findings suggest that every Aβ strain tries to transmit its own signature to normal Aβ, either during its pathological spreading in the brain of the patient or when inoculated in susceptible mice. Indeed, as for prions, Aβ strains can interact with the normal Aβ and coerce it to acquire similar abnormal structures, thus, transmitting individual strain signature [52,67]. In 2014, Stohr and colleagues [63] showed that two distinct misfolded Aβ conformers prepared using synthetic peptides composed of either 40 (Aβ40) or 42 (Aβ42) residues were able to generate distinct amyloid pathology in susceptible mice, thus, confirming that the pathological properties of each conformer were enciphered in the structure. A better comprehension of the molecular events which lead to the generation of distinct phenotypes of AD is becoming extremely important, especially in consideration of the fact that they might differentially respond to pharmacological treatments. But, above all, the recognition of the individual disease phenotype in patients in the early stages of AD will consent to develop tailored treatments.
Whether Aβ strains are bona fide prions is still a debated issue. In any case, thanks to the prion-like behavior of Aβ, it is now possible to study the process of Aβ protein misfolding in vitro by exploiting an innovative technology developed in the field of prion diseases named Real-Time Quaking-Induced Conversion (RT-QuIC) [68]. This technique is useful to study Aβ misfolding, but also the factors that could drive and influence its structural rearrangement. Finally, it represents a new ultrasensitive platform potentially exploitable for the clinical diagnosis of AD (see below) [69].
3. Challenges in the Clinical Diagnosis of Alzheimer’s Disease
AD progresses toward three stages: An early preclinical stage where no symptoms are observed (although the process of Aβ and tau misfolding, spreading, and aggregation are already in progress); a middle stage characterized by the presence of mild cognitive impairment (MCI) where memory and thinking functions are impaired (without significant interferences with the normal life of the patient); and a late-stage marked by the presence of many symptoms of dementia (memory loss, visual/spatial problems, the patient is no longer independent). MCI not always leads to AD, but can evolve to other forms of dementia, including frontotemporal dementia (FTD) or dementia with Lewy bodies (DLB), or can even revert back to a normal condition [70,71,72]. The main challenge in the field of AD is represented by the impossibility to formulate the correct diagnosis of the disease in its early stages when potential treatments can be provided before extensive brain damages have occurred. At present, the diagnosis of AD is made following criteria that rely on clinical, neuropsychological, instrumental (e.g., magnetic resonance imaging, fludeoxyglucose F 18 positron emission tomography—PET, amyloid PET, tau PET, and computed tomography) and laboratory tests [40]. The first criteria for the diagnosis of AD were defined in 1984 by the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the Alzheimer’s Disease and Related Disorders Association (ADRDA) [73]. In 2011, these criteria were revised and updated by the National Institute on Aging–Alzheimer’s Association (NIA-AA) and classified AD as ‘possible’ (the patient shows predominant amnestic dementia with an atypical course that can be due to other causes) or ‘probable’ (the patient shows predominant amnestic dementia with an insidious onset and history of progressive worsening, without other causes identified) [40,74]. At present, the in vivo diagnostic accuracy for AD is about 77%, even among expert neurologists [75]. At least in part, this uncertainty is because the diagnostic criteria indicate to analyze biomarkers that can be altered in AD, but also in some other conditions. For this reason, ‘proven’ (definitive) diagnosis of AD can be achieved only at post-mortem, after extensive neuropathological analyses aimed at identifying the presence of Aβ and NFTs aggregates in the brain. As previously mentioned, details about the localization and the biochemical/physicochemical features of Aβ (its signature) is important to identify the phenotypes of AD. Unfortunately, the recognition of these pathological variants is not possible when patients are alive. Therefore, there is an urgent need to uncover more sensitive biomarkers for early diagnosis and recognition of AD phenotypes. Recent evidence suggests that misfolded Aβ and tau do not remain confined to the brain, but can spread, in traces, to other tissues, including the cerebrospinal fluid (CSF). It is important to take advantage of these new findings to develop innovative diagnostic approaches that can identify both peripheral AD biomarkers for improving the clinical diagnostic accuracy of AD.
4. Alzheimer’s Disease Biomarkers: The Role of CSF and Other Peripheral Tissues
Nowadays, CSF biomarkers have been shown to have a high diagnostic performance to identify AD patients [76]. CSF can be collected through a lumbar puncture, and its alterations reflect brain pathophysiology [77]. In particular, several CSF biomarkers are investigated for evaluating the main neuropathological changes of AD: (1) Aβ deposition, (2) tau deposition (NFTs), and (3) neuronal loss. Among them, the peptide composed of 42 amino acids (Aβ1-42) is decreased in the CSF of AD patients. This reduction is associated with an increased concentration of the peptide in the brain, in the form of extracellular Aβ1-42 amyloid plaques [78,79].
PET imaging studies with an amyloid-binding agent (Pittsburgh Compound-B, PIB) strengthen the inverse correlation between Aβ1-42 levels in the brain and CSF. In particular, high concentrations of Aβ1-42 in the brain of AD patients were paralleled by low levels in the CSF [80,81]. There are subjects known to hyper produce Aβ proteins, and this might hamper the possibility to observe a real reduction of Aβ1-42 in their CSF samples. For this reason, the Aβ peptide composed of 40 amino acids (Aβ1-40) has been introduced among the CSF biomarkers to limit this potential issue. The concentration of Aβ1-40 in the CSF does not change in patients with AD [82,83]. Therefore, the ratio between Aβ1-42/Aβ1-40 consents to identify a real reduction of Aβ1-42 in the CSF of AD patients and can identify AD from other non-AD dementias with 85% sensitivity and 82% specificity [84,85,86]. Moreover, this indicator well correlates with the amyloid PET results [87,88].
Another important CSF biomarker for AD is the tau protein (T-tau) and its phosphorylated form at threonine 181 (P-tau). During disease progression, as a result of neuronal death, T-tau and P-tau are released in the CSF of AD patients, and their concentrations significantly increase. However, T-tau proteins increase also in the CSF of patients with other neurodegenerative diseases characterized by severe neuronal loss, including among the others, prion disorders (e.g., Creutzfeldt-Jakob disease). Therefore, this marker is not specific for AD even though it well correlates with the cognitive decline in AD patients [89,90,91,92]. In contrast, increased P-tau levels are more specific for AD and generally do not increase in other degenerative conditions [93,94,95]. A lot of work has been done, and is currently in progress, to update, standardize and harmonize the pre-analytical, analytical, and post-analytical procedures related to CSF biomarkers collection and analysis. Several consortia, including the Biomarkers for AD and Parkinson’s Disease (BIOMARKAPD), the Global Biomarkers Standardization Consortium (GBSC), the AD Neuroimaging Initiative (ADNI), the Alzheimer Biomarker Standardization Initiative (ABSI), and the JPND BIOMARKPD run huge efforts to ensure reproducible and consistent CSF biomarkers measurements for facilitating worldwide comparison of the results and maximizing their diagnostic utility [96,97,98]. The analysis of Aβ1–42 (or the ratio Aβ1–42/Aβ1–40) T-tau, and P-tau is currently highly valuable to discriminate between AD and non-AD patients, as well as cognitively healthy controls [99]. However, new potential biomarkers for AD present in the CSF, but also blood and urine are currently under investigation. For instance, the S100B protein was found to be significantly higher in the CSF of AD and FTD patients compared to controls [100]. A very recent publication indicates that tau phosphorylated at residue 217 (P-tau217) seems to perform better than the classical P-tau to identify patients with AD and might represent a better biomarker for the clinical diagnosis of the disease [101]. Remarkably, several studies showed that most of tau in CSF of AD patients is present as fragments. In this regard, the novel N-terminal tau fragments ending at amino acid 224 (N-224) were found to be significantly higher in AD patients compared to controls and correlated to cognitive impairment over time [102].
The neurofilament light chain protein (NfL) was also proposed as an innovative CSF biomarker for predicting brain atrophy, cognition, and Aβ accumulation in AD. In particular, the elevated levels of NfL in CSF correlate with white matter changes and axonal degeneration [103]. Moreover, the increase of NfL in CSF predicts hippocampal atrophy in cognitively healthy subjects at risk for AD [104] and correlate with disease progression [105]. Another protein that is currently being investigated as a new potential biomarker for AD is the neurogranin [106,107]. This protein is increased in the CSF of AD patients and correlates with the level of T-tau [108]. Moreover, an inverse correlation between neurogranin and Aβ1–42/Aβ1–40 ratio has been observed in the CSF of AD patients [108]. Finally, it seems that the ratio between neurogranin and the protein BACE1 is a potential correlate of cognitive decline in patients with AD [109]. The European Union-North American Clinical Trials in Alzheimer’s Disease Task Force (EU/US CTAD Task Force) has recently evaluated the status of blood-based AD biomarker and confirmed that (1) plasma Aβ levels accurately estimates the brain amyloid deposition, (2) plasma NfL levels correlates with neurodegeneration (although not specific for AD), and (3) plasma T-tau levels do not correlate with those of the CSF [110]. Thanks to the use of an innovative method based on immunoprecipitation coupled with mass spectrometry (IP-MS), it was demonstrated a reduction of Aβ1-42/Aβ1-40 ratio in plasma of AD patients versus control with a diagnostic accuracy of about 90% [111,112]. Similarly, the concentration of plasma tau was found to be higher in AD patients compared to healthy individuals [82,113,114,115]. Another study showed the levels of plasmatic P-tau181 were higher in AD patients than controls, and they correlated with disease severity [116]. Many other studies (including metabolomics approaches, miRNA and exosomes analyses) are currently ongoing to evaluate the diagnostic significance of blood-based or even urine-based AD biomarkers, including the last described proteins SPP1, GSN, and IGFBP7 that were found to be differentially expressed in the urine of AD patients compared to healthy subjects [117,118,119,120,121,122]. However, compared to CSF, these tissues have completely different matrix components, in terms of proteins (especially those binding to Aβ and tau) and factors, that might affect the efficient identification of AD biomarkers. Moreover, their alterations can be identified during the MCI stage, thus, limiting the possibility to identify AD patients in the very early stages of the disease [76].
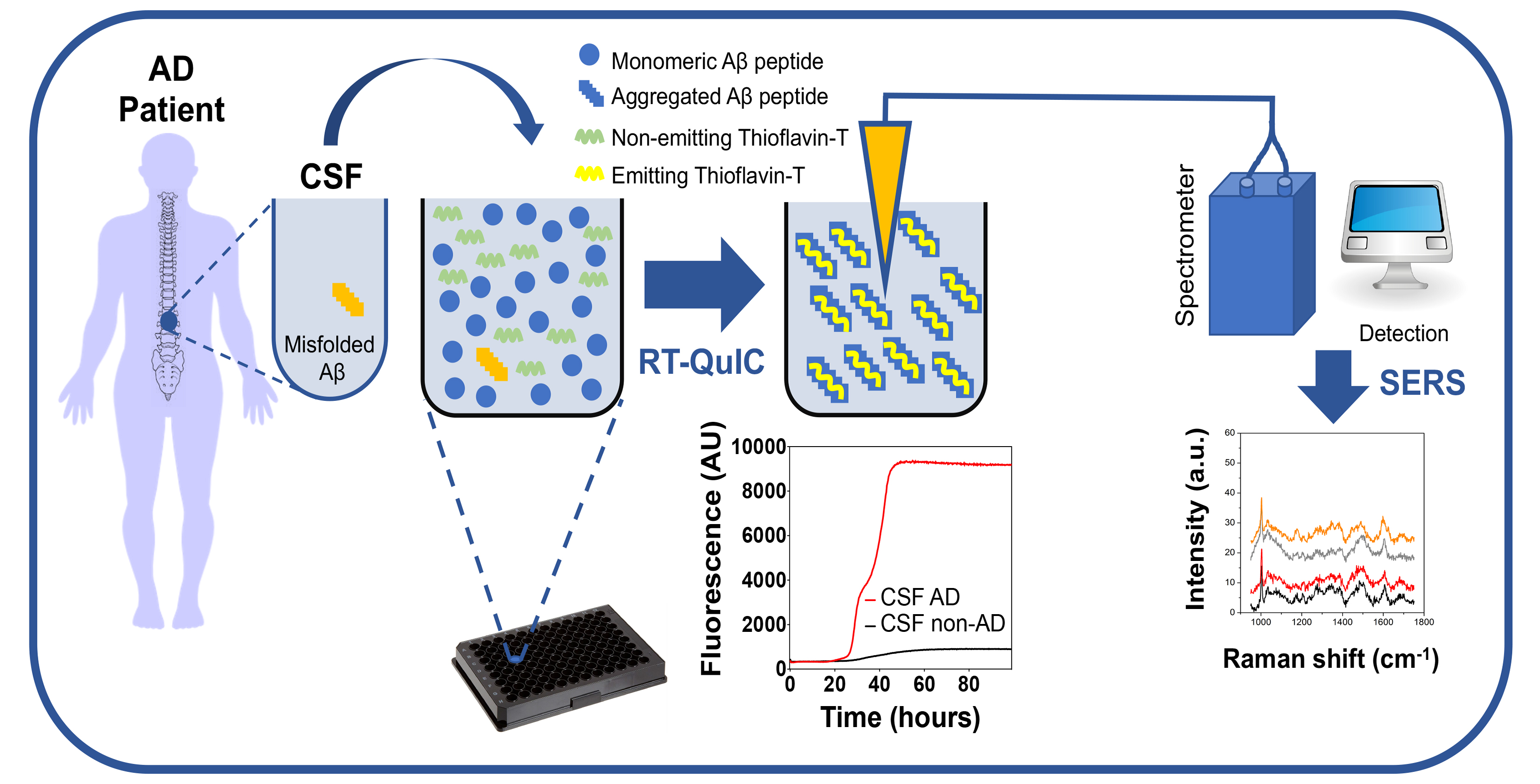
With the development of highly sensitive techniques, traces of Aβ strains were found in the CSF of patients with AD [123]. Certainly, the identification of misfolded Aβ in this tissue will consent to formulate a considerably more accurate diagnosis of AD. Based on this premise, we decided to set-up an ultrasensitive and nanotechnologies-inspired method for early detection of misfolded Aβ in the CSF of AD patients, which meets the needs of reliability, ultrasensitivity, effectiveness, and low cost. Our approach is not only aimed at detecting Aβ strains, but also at identifying their structural features eventually exploitable for the recognition of individual AD phenotype when patients are alive (see the paragraph Our contribution to the field and future perspectives).
5. Technological Innovations and the Future of Alzheimer’s Disease Biomarker Discovery
In 2014, the group of Claudio Soto described a sensitive method for detecting misfolded Aβ in the CSF of patients with AD, known as Real-Time Quaking-Induced Conversion (RT-QuIC) assay. They have found that the concentration of misfolded Aβ in this fluid was in the femtomolar range; therefore, undetectable with the current diagnostic techniques. To enable its detection, they exploited the ability of the misfolded Aβ to seed the polymerization of normal Aβ in vitro by reproducing the phenomenon of protein misfolding, which occurs in vivo. This work included the analysis of 50 CSF collected from AD patients, 39 CSF from patients affected by non-degenerative neurological conditions, and 37 CSF from patients affected by other neurodegenerative diseases (non-AD). These samples were incubated with the synthetically produced Aβ1–42 peptide (used as a reaction substrate) and subjected to alternated cycles of incubation and shaking. Only CSF-AD samples were able to promote (seed) the aggregation of Aβ1–42, while the others did not. The aggregation of the substrate was monitored with the thioflavin T (ThT) fluorescent dye [123]. The kinetics of Aβ1–42 aggregation was significantly accelerated in the presence of CSF-AD, and the fluorescent signal increased earlier (seeding effect) than that of the other samples. Immunodepletion of misfolded Aβ from the CSF of AD patients led to a total inhibition of the Aβ1–42 aggregation, thus, confirming that the seeding effect was specifically triggered by misfolded Aβ present in the CSF. This was the first and only demonstration that traces of Aβ strains were present in the CSF of AD patients. Similar RT-QuIC studies have been performed in the context of other neurodegenerative diseases caused by misfolded proteins, including Parkinson’s disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA), which are caused by misfolded α-synuclein [124,125,126,127]; frontotemporal dementia (FTLD), corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP) that are caused by misfolded tau [128]; and frontotemporal dementia (FTLD) and amyotrophic lateral sclerosis (ALS) that are caused by misfolded TDP-43 [129]. These studies showed that RT-QuIC could detect misfolded α-synuclein in the CSF or olfactory mucosa of patients with PD, MSA and DLB [124,125,126,130,131], misfolded tau in the CSF of patients with PiD [132], CBD and PSP [128], and misfolded TDP-43 in the CSF of patients with FTD and ALS [129].
We mentioned these studies because they led to an important discovery that can be extended to AD diagnosis. Indeed, also α-synuclein, tau, or TDP-43 can misfold and adopt a variety of aberrant structures (again referred to as strains) that account for the phenotypic heterogeneity of these diseases [50,133,134,135,136,137,138]. What has been observed is that each strain seemed to transfer its peculiar signature (in terms of biochemical and structural features) to the RT-QuIC substrate. Hence, the RT-QuIC reaction products acquired different biochemical, morphological, and structural properties extremely important for strain’s identification. In the light of these observations, we, along with few other research groups, have decided to couple the RT-QuIC with highly specialized biophysical and optical technologies (e.g., transmission electron microscopy, protein-NMR, cryogenic electron microscopy, Raman spectroscopy, etc.) to dissect the morphological, and structural properties of the RT-QuIC reaction products obtained from the analysis of CSF or olfactory mucosa samples of patients with PD, MSA or DLB. The results of these preliminary experiments indicated that this approach could successfully identify different α-synuclein strains allowing disease discrimination [131,139,140]. Similarly, it is possible that different misfolded Aβ can transmit their strain signature to the RT-QuIC substrate. For this reason, we decided to set-up an innovative approach for the analysis of CSF collected from patients with AD. Undoubtedly, one of the critical aspects related to the ultrasensitive analyses of these samples is represented by the limited accuracy of AD diagnosis that can lead to misinterpretation of the results.
The scenario is even more complicated by the fact that the RT-QuIC could detect peripheral Aβ strains even in subjects at the prodromal stage of AD when they are still asymptomatic, and therefore, considered ‘healthy’. Therefore, we decided to perform our analyses on samples collected from AD patients that have been extensively characterized by multiple points of view. In this regard, the Italian Ministry of Health has funded a project (concluded in 2019), led by Fabrizio Tagliavini in collaboration with other well-recognized Italian dementia centers, that thoroughly characterized (1) patients with pre-MCI, (2) patients with MCI, (3) patients with AD, (4) patients with a subjective cognitive complaint, and (5) healthy subjects from a clinical, neuropsychological, laboratory and instrumental point of view. Among the studies performed by all partners of the research group, CSF and urine were also collected at different time points for RT-QuIC assessment. These experiments are still ongoing. The objective is to evaluate the performance of the RT-QuIC and compare the results with those obtained from all the other tests. Final reaction products will be subjected to biochemical and highly specialized biophysical assessments to verify the presence of distinct properties, eventually reflecting the presence of different Aβ strains. This would consent to truly estimate whether and to what extent the RT-QuIC can identify peripheral Aβ strains in AD, pre-MCI or MCI, due to AD patients. Considering that the RT-QuIC technology has already been introduced among the clinical diagnostic criteria for prion diseases, and is currently being exploited to diagnose other neurodegenerative diseases [124,126,127,130,131,139,141,142], it is worth extending and optimizing the technology to diagnose AD.
6. Our Contribution to the Field of Alzheimer’s Disease Diagnosis
The urgency dictated by the need of revealing an exhaustive molecular and morphological description of RT-QuIC reaction products has inspired the ground motivations of our ERANET EuroNanoMed III SPEEDY (surface-enhanced Raman scattering with nanophotonic and biomedical amplifying systems for early diagnosis of Alzheimer’s disease pathology) project, comprising four research units from three different EU countries and supported by EU along with local funding agencies (Italian Ministry of Education, University and Research, Italian Ministry of Health, Israeli Ministry of Health, Polish National Centre For Research And Development).
The project aims to confer chemostructural information to RT-QuIC products by supporting the molecular amplification provided by RT-QuIC procedure with an optical amplification as obtained by surface-enhanced Raman spectroscopy (SERS)-based systems. Specifically, a dedicated SERS platform with advanced sensing tools, including disposable 2D-nanostructured chips and noble nanoparticles-containing optical fibers represent the main outcomes of the project, which are implemented for label-free ultradetection of misfolded Aβ, in CSF and olfactory mucosa of extensively characterized AD patients.
Raman techniques recently proved to match the requirements of a label-free characterization of the secondary structure of isolated misfolded Aβ [143] and the amyloid content from brain homogenates of PD patients [140]. Raman spectroscopy can provide information on the molecular vibrations (fingerprint of molecules) of a sample by exploiting the inelastic scattered light under visible or near-infrared excitation light. As a consequence, the noninvasive and nondestructive molecular identification and analysis of a species can be achieved. Main applications of Raman spectroscopy in biomedical research concern the investigation of different materials (biomolecules, cells, tissues, etc.) aimed at revealing their chemical composition and structure, in turn, aimed at sample discrimination, classification, and preclinical/clinical diagnosis [144,145]. However, the low efficiency of the Raman scattering process and the frequent interference by a high fluorescence background pose a limit to this technique for the detection of biomolecules, especially when examined at low concentration as typically occurs in their own biological environment. Additionally, biomolecules are characterized by the lowest Raman cross-section values, hence requiring highly sensitive systems for their detection. The weak Raman signal of biomolecular species can be compensated by taking advantage of a resonance condition, such as in resonance Raman spectroscopy (RRS) [146], by increasing the interaction between analyte molecules and excitation laser as in fiber-enhanced Raman spectroscopy (FERS) [147], by the electromagnetic amplification generated by plasmonic nanoparticles as in surface-enhanced Raman spectroscopy (SERS) [148] or by the use of single nanoantennas also pushing the spatial resolution down to the nanoscale as in tip-enhanced Raman spectroscopy (TERS) [149]. During the course of the last few years, SERS has been recognized as a technique with great potential for the analysis of biosamples.
Early achievements from some of us demonstrated the advantage of using nonspherical concave gold nanoparticles with sharp tips forming effective “hot-spots” at the junctions between multiple adjacent nanoparticles, which are at the basis of a boosted Raman signal of proteins, such as insulin and cytochrome c [150]. The amplification observed is here motivated by the formation of 2D-regular arrays of nanoparticles featuring a distribution of nanoholes for protein entrapment and detection. Afterward, preliminary results have established that SERS can offer a reliable tool for effective detection of amyloid forms by using Au-nanoparticles-decorated beads [151], as well as it was demonstrated for the first time that SERS can be exploited to identify trace proteins in a physiological environment in a quantitative and reproducible manner [152]. A low-cost SERS assay was recently introduced for sensitive detection of Aβ aggregates [153]. Here, experiments on toxic and non-toxic Aβ forms showed the possibility of using SERS for their discrimination, suggesting a high potential of the technique also for tracking Aβ during the course of its different aggregation states. At the same time, recent insights have concerned coupling noble metal nanoparticles with hollow-core optical fibers, working as effective SERS sensors for remote detection of molecular species [147]. These systems are perceived as optical tools allowing the in situ and cost-effective identification of low sampling volumes of small molecular quantities (~µg/mL) of biomarker, thanks to combined FERS and SERS effects. Furthermore, fiber-based systems can be adapted as flexible interfaces between a commercial Raman analyzer and fluid samples contained, e.g., inside multiwell plates, which is particularly attractive because of setting up a liquid biopsy assay for preclinical or clinical detection of AD.
By taking advantage of the ability of RT-QuIC to amplify different strains of Aβ with high fidelity and of SERS to reveal the presence of different misfolded structures, it would be possible to associate an early diagnosis of AD with the recognition of disease subgroup. Overall, a combined RT-QuIC/SERS technology for detecting pathological Aβ in the CSF or olfactory mucosa of patients with AD can be exploited to inform prognosis, allow monitoring of disease progression and act as an indicator of treatment effects in future clinical trials.
Author Contributions
E.B., F.T., P.M. and F.M. wrote and reviewed the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported in part by the Italian Ministry of Health (GR-2013-02355724 and Ricerca Corrente), MJFF, ALZ, Alzheimer’s Research UK and the Weston Brain Institute (BAND2015) to FM; European Community, Italian Ministry of Education, University and Research and the Italian Ministry of Health within the EuroNanoMed3 ERANET cofund SPEEDY project to PM and to FM and Italian Ministry of Health grant (NET-2011-02346784) to FT. Italian Ministry of Foreign Affairs and International Cooperation of Italy through the DESWEAT project (No._KR19GR08) to PM.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef]
- 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [CrossRef]
- Swearer, J.M.; O’Donnell, B.F.; Ingram, S.M.; Drachman, D.A. Rate of progression in familial Alzheimer’s disease. J. Geriatr. Psychiatry Neurol. 1996, 9, 22–25. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Review Article: Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimers. Res. Ther. 2010, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLOS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Rosa-Neto, P.; Hsiung, G.-Y.R.; Sadovnick, A.D.; Masellis, M.; Black, S.E.; Jia, J.; Gauthier, S. Early-Onset Familial Alzheimer’s Disease (EOFAD). Can. J. Neurol. Sci. J. Can. des Sci. Neurol. 2012, 39, 436–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.S.; Tangalos, E.G.; Petersen, R.C.; Smith, G.E.; Schaid, D.J.; Kokmen, E.; Ivnik, R.J.; Thibodeau, S.N. Apolipoprotein E: Risk factor for Alzheimer disease. Am. J. Hum. Genet. 1994, 54, 643–649. [Google Scholar]
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.-J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef]
- Kelényi, G. Thioflavin S fluorescent and congo red anisotropic stainings in the histologic demonstration of amyloid. Acta Neuropathol. 1967, 7, 336–348. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 608–655. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C. Prion-like mechanisms in Alzheimer disease. In Handbook of Clinical Neurology; Elsevier BV: Amsterdam, The Netherlands, 2018; Volume 153, pp. 303–319. [Google Scholar]
- Goedert, M.; Clavaguera, F.; Tolnay, M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010, 33, 317–325. [Google Scholar] [CrossRef]
- Morales, R.; Callegari, K.; Soto, C. Prion-like features of misfolded Aβ and tau aggregates. Virus Res. 2015, 207, 106–112. [Google Scholar] [CrossRef]
- Prusiner, S. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [Green Version]
- CRICK, F. Central Dogma of Molecular Biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef]
- Bendheim, P.E.; Brown, H.R.; Rudelli, R.D.; Scala, L.J.; Goller, N.L.; Wen, G.Y.; Kascsak, R.J.; Cashman, N.R.; Bolton, D.C. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 1992, 42, 149. [Google Scholar] [CrossRef]
- Dee, D.R.; Woodside, M.T. Comparing the energy landscapes for native folding and aggregation of PrP. Prion 2016, 10, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Gambetti, P.; Kong, Q.; Zou, W.; Parchi, P.; Chen, S.G. Sporadic and familial CJD: Classification and characterisation. Br. Med. Bull. 2003, 66, 213–239. [Google Scholar] [CrossRef] [Green Version]
- Morales, R. Prion strains in mammals: Different conformations leading to disease. PLoS Pathog. 2017, 13, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, L.C.; Jucker, M. Neurodegenerative Diseases: Expanding the Prion Concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef] [Green Version]
- Brundin, P.; Ma, J.; Kordower, J.H. How strong is the evidence that Parkinsonʼs disease is a prion disorder? Curr. Opin. Neurol. 2016, 29, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Vasili, E.; Dominguez-Meijide, A.; Outeiro, T.F. Spreading of α-Synuclein and Tau: A Systematic Comparison of the Mechanisms Involved. Front. Mol. Neurosci. 2019, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Pecho-Vrieseling, E.; Rieker, C.; Fuchs, S.; Bleckmann, D.; Esposito, M.S.; Botta, P.; Goldstein, C.; Bernhard, M.; Galimberti, I.; Müller, M.; et al. Transneuronal propagation of mutant huntingtin contributes to non–cell autonomous pathology in neurons. Nat. Neurosci. 2014, 17, 1064–1072. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. A Unifying Role for Prions in Neurodegenerative Diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 2017, 140, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Moh, C.; Kubiak, J.Z.; Bajic, V.P.; Zhu, X.; Smith, M.A.; Lee, H. Cell cycle deregulation in the neurons of Alzheimer’s disease. Results Probl. Cell Differ. 2011, 53, 565–576. [Google Scholar]
- Raina, A.K.; Monteiro, M.J.; McShea, A.; Smith, M.A. The role of cell cycle-mediated events in Alzheimer’s disease. Int. J. Exp. Pathol. 1999, 80, 71–76. [Google Scholar] [CrossRef]
- Terry, A.V.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef] [Green Version]
- Passamonti, L.; Tsvetanov, K.A.; Jones, P.S.; Bevan-Jones, W.R.; Arnold, R.; Borchert, R.J.; Mak, E.; Su, L.; O’Brien, J.T.; Rowe, J.B. Neuroinflammation and Functional Connectivity in Alzheimer’s Disease: Interactive Influences on Cognitive Performance. J. Neurosci. 2019, 39, 7218–7226. [Google Scholar] [CrossRef] [Green Version]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers. Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Lau, H.H.C.; Ingelsson, M.; Watts, J.C. The existence of Aβ strains and their potential for driving phenotypic heterogeneity in Alzheimer’s disease. Acta Neuropathol. 2020, 1–23. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Suttkus, A.; Holzer, M.; Morawski, M.; Arendt, T. The neuronal extracellular matrix restricts distribution and internalization of aggregated Tau-protein. Neuroscience 2016, 313, 225–235. [Google Scholar] [CrossRef]
- Warren, J.D.; Fletcher, P.D.; Golden, H.L. The paradox of syndromic diversity in Alzheimer disease. Nat. Rev. Neurol. 2012, 8, 451–464. [Google Scholar] [CrossRef]
- Suárez-González, A.; Henley, S.M.; Walton, J.; Crutch, S.J. Posterior Cortical Atrophy. Psychiatr. Clin. North Am. 2015, 38, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villain, N.; Dubois, B. Alzheimer’s Disease Including Focal Presentations. Semin. Neurol. 2019, 39, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Di Fede, G.; Catania, M.; Maderna, E.; Ghidoni, R.; Benussi, L.; Tonoli, E.; Giaccone, G.; Moda, F.; Paterlini, A.; Campagnani, I.; et al. Molecular subtypes of Alzheimer’s disease. Sci. Rep. 2018, 8, 3269. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D. Attempt to Untangle the Prion-Like Misfolding Mechanism for Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3081. [Google Scholar] [CrossRef] [Green Version]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, T.; Zhang, J.; Liu, Q.; Liu, C.-C.; Zhang, L.; Bu, G. Heparan Sulphate Proteoglycan and the Low-Density Lipoprotein Receptor-Related Protein 1 Constitute Major Pathways for Neuronal Amyloid- Uptake. J. Neurosci. 2011, 31, 1644–1651. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.; Zhang, W.; Schweighauser, M.; Murzin, A.G.; Vidal, R.; Garringer, H.J.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 2018, 136, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, J.; Mahler, J.; Beschorner, N.; Kaeser, S.A.; Häsler, L.M.; Baumann, F.; Nyström, S.; Portelius, E.; Blennow, K.; Lashley, T.; et al. Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 13018–13023. [Google Scholar] [CrossRef] [Green Version]
- Condello, C.; Lemmin, T.; Stöhr, J.; Nick, M.; Wu, Y.; Maxwell, A.M.; Watts, J.C.; Caro, C.D.; Oehler, A.; Keene, C.D.; et al. Structural heterogeneity and intersubject variability of Aβ in familial and sporadic Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E782–E791. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.; Appleby, B.; Safar, J.G. Distinct prion-like strains of amyloid beta implicated in phenotypic diversity of Alzheimer’s disease. Prion 2016, 10, 9–17. [Google Scholar] [CrossRef] [Green Version]
- LeVine, H.; Walker, L.C. Molecular polymorphism of Aβ in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Piccini, A.; Russo, C.; Gliozzi, A.; Relini, A.; Vitali, A.; Borghi, R.; Giliberto, L.; Armirotti, A.; D’Arrigo, C.; Bachi, A.; et al. β-Amyloid Is Different in Normal Aging and in Alzheimer Disease. J. Biol. Chem. 2005, 280, 34186–34192. [Google Scholar] [CrossRef] [Green Version]
- Maarouf, C.L.; Daugs, I.D.; Spina, S.; Vidal, R.; Kokjohn, T.A.; Patton, R.L.; Kalback, W.M.; Luehrs, D.C.; Walker, D.G.; Castaño, E.M.; et al. Histopathological and molecular heterogeneity among individuals with dementia associated with Presenilin mutations. Mol. Neurodegener. 2008, 3, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, K.P.R.; Åslund, A.; Berg, I.; Nyström, S.; Konradsson, P.; Herland, A.; Inganäs, O.; Stabo-Eeg, F.; Lindgren, M.; Westermark, G.T.; et al. Imaging Distinct Conformational States of Amyloid-β Fibrils in Alzheimer’s Disease Using Novel Luminescent Probes. ACS Chem. Biol. 2007, 2, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.; Ghetti, B. Characterization of Amyloid Deposits in Neurodegenerative Diseases. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; pp. 241–258. ISBN 9781617793271. [Google Scholar]
- Steinerman, J.R.; Irizarry, M.; Scarmeas, N.; Raju, S.; Brandt, J.; Albert, M.; Blacker, D.; Hyman, B.; Stern, Y. Distinct Pools of β-Amyloid in Alzheimer Disease–Affected Brain. Arch. Neurol. 2008, 65, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Condello, C.; Stohr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of A prions from Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef] [Green Version]
- Stohr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Purified and synthetic Alzheimer’s amyloid beta (A) prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030. [Google Scholar] [CrossRef] [Green Version]
- Heilbronner, G.; Eisele, Y.S.; Langer, F.; Kaeser, S.A.; Novotny, R.; Nagarathinam, A.; Åslund, A.; Hammarström, P.; Nilsson, K.P.R.; Jucker, M. Seeded strain-like transmission of β-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013, 14, 1017–1022. [Google Scholar] [CrossRef] [Green Version]
- Stohr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic A prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10329–10334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Riquelme, A.; Lau, H.H.C.; Stuart, E.; Goczi, A.N.; Wang, Z.; Schmitt-Ulms, G.; Watts, J.C. Prion-like propagation of β-amyloid aggregates in the absence of APP overexpression. Acta Neuropathol. Commun. 2018, 6, 26. [Google Scholar] [CrossRef]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for Seeding of β-Amyloid by Intracerebral Infusion of Alzheimer Brain Extracts in β-Amyloid Precursor Protein-Transgenic Mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Luehmann, M. Exogenous Induction of Cerebral -Amyloidogenesis Is Governed by Agent and Host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef]
- Masters, C.L.; Selkoe, D.J. Biochemistry of Amyloid -Protein and Amyloid Deposits in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef]
- Atarashi, R.; Sano, K.; Satoh, K.; Nishida, N. Real-time quaking-induced conversion. Prion 2011, 5, 150–153. [Google Scholar] [CrossRef] [Green Version]
- Paravastu, A.K.; Qahwash, I.; Leapman, R.D.; Meredith, S.C.; Tycko, R. Seeded growth of -amyloid fibrils from Alzheimer’s brain-derived fibrils produces a distinct fibril structure. Proc. Natl. Acad. Sci. USA 2009, 106, 7443–7448. [Google Scholar] [CrossRef] [Green Version]
- Petersen, R.C.; Caracciolo, B.; Brayne, C.; Gauthier, S.; Jelic, V.; Fratiglioni, L. Mild cognitive impairment: A concept in evolution. J. Intern. Med. 2014, 275, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Ferman, T.J.; Smith, G.E.; Kantarci, K.; Boeve, B.F.; Pankratz, V.S.; Dickson, D.W.; Graff-Radford, N.R.; Wszolek, Z.; Van Gerpen, J.; Uitti, R.; et al. Nonamnestic mild cognitive impairment progresses to dementia with Lewy bodies. Neurology 2013, 81, 2032–2038. [Google Scholar] [CrossRef] [Green Version]
- de Mendonça, A.; Ribeiro, F.; Guerreiro, M.; Garcia, C. Frontotemporal mild cognitive impairment. J. Alzheimers. Dis. 2004, 6, 1–9. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939. [Google Scholar] [CrossRef] [Green Version]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, M.N.; Lue, L.-F.; Fayard, D.; Shi, J. Increasing Precision of Clinical Diagnosis of Alzheimer’s Disease Using a Combined Algorithm Incorporating Clinical and Novel Biomarker Data. Neurol. Ther. 2017, 6, 83–95. [Google Scholar] [CrossRef]
- Niemantsverdriet, E.; Valckx, S.; Bjerke, M.; Engelborghs, S. Alzheimer’s disease CSF biomarkers: Clinical indications and rational use. Acta Neurol. Belg. 2017, 117, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Engelborghs, S.; Niemantsverdriet, E.; Struyfs, H.; Blennow, K.; Brouns, R.; Comabella, M.; Dujmovic, I.; Flier, W.; Frölich, L.; Galimberti, D.; et al. Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimer’s Dement. Diagnosis, Assess. Dis. Monit. 2017, 8, 111–126. [Google Scholar] [CrossRef]
- Catania, M.; Di Fede, G.; Tonoli, E.; Benussi, L.; Pasquali, C.; Giaccone, G.; Maderna, E.; Ghidoni, R.; Tagliavini, F. Mirror Image of the Amyloid-β Species in Cerebrospinal Fluid and Cerebral Amyloid in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 47, 877–881. [Google Scholar] [CrossRef]
- Strozyk, D.; Blennow, K.; White, L.R.; Launer, L.J. CSF Aβ 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology 2003, 60, 652–656. [Google Scholar] [CrossRef]
- Fagan, A.M.; Mintun, M.A.; Mach, R.H.; Lee, S.-Y.; Dence, C.S.; Shah, A.R.; LaRossa, G.N.; Spinner, M.L.; Klunk, W.E.; Mathis, C.A.; et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ 42 in humans. Ann. Neurol. 2006, 59, 512–519. [Google Scholar] [CrossRef]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.-K.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttilä, T. Cerebrospinal Fluid β-Amyloid 42 and Tau Proteins as Biomarkers of Alzheimer-Type Pathologic Changes in the Brain. Arch. Neurol. 2009, 66, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Portelius, E.; Tran, A.J.; Andreasson, U.; Persson, R.; Brinkmalm, G.; Zetterberg, H.; Blennow, K.; Westman-Brinkmalm, A. Characterization of Amyloid β Peptides in Cerebrospinal Fluid by an Automated Immunoprecipitation Procedure Followed by Mass Spectrometry. J. Proteome Res. 2007, 6, 4433–4439. [Google Scholar] [CrossRef]
- Biscetti, L.; Salvadori, N.; Farotti, L.; Cataldi, S.; Eusebi, P.; Paciotti, S.; Parnetti, L. The added value of Aβ42/Aβ40 in the CSF signature for routine diagnostics of Alzheimer’s disease. Clin. Chim. Acta 2019, 494, 71–73. [Google Scholar] [CrossRef]
- Shahpasand-Kroner, H.; Klafki, H.-W.; Bauer, C.; Schuchhardt, J.; Hüttenrauch, M.; Stazi, M.; Bouter, C.; Wirths, O.; Vogelgsang, J.; Wiltfang, J. A two-step immunoassay for the simultaneous assessment of Aβ38, Aβ40 and Aβ42 in human blood plasma supports the Aβ42/Aβ40 ratio as a promising biomarker candidate of Alzheimer’s disease. Alzheimers. Res. Ther. 2018, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Lewczuk, P.; Lelental, N.; Spitzer, P.; Maler, J.M.; Kornhuber, J. Amyloid-β 42/40 Cerebrospinal Fluid Concentration Ratio in the Diagnostics of Alzheimer’s Disease: Validation of Two Novel Assays. J. Alzheimer’s Dis. 2014, 43, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Lewczuk, P.; Matzen, A.; Blennow, K.; Parnetti, L.; Molinuevo, J.L.; Eusebi, P.; Kornhuber, J.; Morris, J.C.; Fagan, A.M. Cerebrospinal Fluid Aβ42/40 Corresponds Better than Aβ42 to Amyloid PET in Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 55, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janelidze, S.; Zetterberg, H.; Mattsson, N.; Palmqvist, S.; Vanderstichele, H.; Lindberg, O.; Westen, D.; Stomrud, E.; Minthon, L.; Blennow, K.; et al. CSF A β 42/A β 40 and A β 42/A β 38 ratios: Better diagnostic markers of Alzheimer disease. Ann. Clin. Transl. Neurol. 2016, 3, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Savva, G.M.; Wharton, S.B.; Ince, P.G.; Forster, G.; Matthews, F.E.; Brayne, C. Age, Neuropathology, and Dementia. N. Engl. J. Med. 2009, 360, 2302–2309. [Google Scholar] [CrossRef]
- Hansson, O.; Seibyl, J.; Stomrud, E.; Zetterberg, H.; Trojanowski, J.Q.; Bittner, T.; Lifke, V.; Corradini, V.; Eichenlaub, U.; Batrla, R.; et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimer’s Dement. 2018, 14, 1470–1481. [Google Scholar] [CrossRef]
- Wallin, A.K.; Blennow, K.; Zetterberg, H.; Londos, E.; Minthon, L.; Hansson, O. CSF biomarkers predict a more malignant outcome in Alzheimer disease. Neurology 2010, 74, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Zetterberg, H.; Mattsson, N.; Johansson, P.; Minthon, L.; Blennow, K.; Olsson, M.; Hansson, O. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 2015, 85, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Wallin, A.; Ågren, H.; Spenger, C.; Siegfried, J.; Vanmechelen, E. tau protein in cerebrospinal fluid. Mol. Chem. Neuropathol. 1995, 26, 231–245. [Google Scholar] [CrossRef]
- Buerger, K.; Zinkowski, R.; Teipel, S.J.; Tapiola, T.; Arai, H.; Blennow, K.; Andreasen, N.; Hofmann-Kiefer, K.; DeBernardis, J.; Kerkman, D.; et al. Differential Diagnosis of Alzheimer Disease With Cerebrospinal Fluid Levels of Tau Protein Phosphorylated at Threonine 231. Arch. Neurol. 2002, 59, 1267. [Google Scholar] [CrossRef]
- Riemenschneider, M.; Wagenpfeil, S.; Vanderstichele, H.; Otto, M.; Wiltfang, J.; Kretzschmar, H.; Vanmechelen, E.; Förstl, H.; Kurz, A. Phospho-tau/total tau ratio in cerebrospinal fluid discriminates Creutzfeldt–Jakob disease from other dementias. Mol. Psychiatry 2003, 8, 343–347. [Google Scholar] [CrossRef]
- Bjerke, M.; Andreasson, U.; Kuhlmann, J.; Portelius, E.; Pannee, J.; Lewczuk, P.; Umek, R.M.; Vanmechelen, E.; Vanderstichele, H.; Stoops, E.; et al. Assessing the commutability of reference material formats for the harmonization of amyloid-β measurements. Clin. Chem. Lab. Med. 2016, 54, 1177–1191. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, N.; Andreasson, U.; Persson, S.; Carrillo, M.C.; Collins, S.; Chalbot, S.; Cutler, N.; Dufour-Rainfray, D.; Fagan, A.M.; Heegaard, N.H.H.; et al. CSF biomarker variability in the Alzheimer’s Association quality control program. Alzheimers. Dement. 2013, 9, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef]
- Engelborghs, S.; De Vreese, K.; Van de Casteele, T.; Vanderstichele, H.; Van Everbroeck, B.; Cras, P.; Martin, J.-J.; Vanmechelen, E.; De Deyn, P.P. Diagnostic performance of a CSF-biomarker panel in autopsy-confirmed dementia. Neurobiol. Aging 2008, 29, 1143–1159. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Jenkins, R.; Watt, H.C.; Green, A.J.E.; Thompson, E.J.; Keir, G.; Fox, N.C.; Rossor, M.N. Cerebrospinal fluid S100B correlates with brain atrophy in Alzheimer’s disease. Neurosci. Lett. 2003, 336, 167–170. [Google Scholar] [CrossRef]
- Janelidze, S.; Stomrud, E.; Smith, R.; Palmqvist, S.; Mattsson, N.; Airey, D.C.; Proctor, N.K.; Chai, X.; Shcherbinin, S.; Sims, J.R.; et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat. Commun. 2020, 11, 1683. [Google Scholar] [CrossRef] [Green Version]
- Cicognola, C.; Brinkmalm, G.; Wahlgren, J.; Portelius, E.; Gobom, J.; Cullen, N.C.; Hansson, O.; Parnetti, L.; Constantinescu, R.; Wildsmith, K.; et al. Novel tau fragments in cerebrospinal fluid: Relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 279–296. [Google Scholar] [CrossRef] [Green Version]
- Sjögren, M.; Blomberg, M.; Jonsson, M.; Wahlund, L.O.; Edman, A.; Lind, K.; Rosengren, L.; Blennow, K.; Wallin, A. Neurofilament protein in cerebrospinal fluid: A marker of white matter changes. J. Neurosci. Res. 2001, 66, 510–516. [Google Scholar] [CrossRef]
- Idland, A.-V.; Sala-Llonch, R.; Borza, T.; Watne, L.O.; Wyller, T.B.; Brækhus, A.; Zetterberg, H.; Blennow, K.; Walhovd, K.B.; Fjell, A.M. CSF neurofilament light levels predict hippocampal atrophy in cognitively healthy older adults. Neurobiol. Aging 2017, 49, 138–144. [Google Scholar] [CrossRef]
- Zetterberg, H.; Skillbäck, T.; Mattsson, N.; Trojanowski, J.Q.; Portelius, E.; Shaw, L.M.; Weiner, M.W.; Blennow, K. Association of Cerebrospinal Fluid Neurofilament Light Concentration With Alzheimer Disease Progression. JAMA Neurol. 2016, 73, 60. [Google Scholar] [CrossRef]
- Thorsell, A.; Bjerke, M.; Gobom, J.; Brunhage, E.; Vanmechelen, E.; Andreasen, N.; Hansson, O.; Minthon, L.; Zetterberg, H.; Blennow, K. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res. 2010, 1362, 13–22. [Google Scholar] [CrossRef]
- Tarawneh, R.; D’Angelo, G.; Crimmins, D.; Herries, E.; Griest, T.; Fagan, A.M.; Zipfel, G.J.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. Diagnostic and Prognostic Utility of the Synaptic Marker Neurogranin in Alzheimer Disease. JAMA Neurol. 2016, 73, 561. [Google Scholar] [CrossRef]
- De Vos, A.; Jacobs, D.; Struyfs, H.; Fransen, E.; Andersson, K.; Portelius, E.; Andreasson, U.; De Surgeloose, D.; Hernalsteen, D.; Sleegers, K.; et al. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1461–1469. [Google Scholar] [CrossRef] [Green Version]
- De Vos, A.; Struyfs, H.; Jacobs, D.; Fransen, E.; Klewansky, T.; De Roeck, E.; Robberecht, C.; Van Broeckhoven, C.; Duyckaerts, C.; Engelborghs, S.; et al. The Cerebrospinal Fluid Neurogranin/BACE1 Ratio is a Potential Correlate of Cognitive Decline in Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 53, 1523–1538. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Blennow, K.; Doody, R.; Hendrix, S.; Lovestone, S.; Salloway, S.; Schindler, R.; Weiner, M.; Zetterberg, H.; Aisen, P.; et al. Plasma Biomarkers of AD Emerging as Essential Tools for Drug Development: An EU/US CTAD Task Force Report. J. Prev. Alzheimer’s Dis. 2019, 6, 169–173. [Google Scholar]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.-X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Ovod, V.; Ramsey, K.N.; Mawuenyega, K.G.; Bollinger, J.G.; Hicks, T.; Schneider, T.; Sullivan, M.; Paumier, K.; Holtzman, D.M.; Morris, J.C.; et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimer’s Dement. 2017, 13, 841–849. [Google Scholar] [CrossRef]
- Zetterberg, H.; Wilson, D.; Andreasson, U.; Minthon, L.; Blennow, K.; Randall, J.; Hansson, O. Plasma tau levels in Alzheimer’s disease. Alzheimers. Res. Ther. 2013, 5, 9. [Google Scholar] [CrossRef]
- Mielke, M.M.; Hagen, C.E.; Wennberg, A.M.V.; Airey, D.C.; Savica, R.; Knopman, D.S.; Machulda, M.M.; Roberts, R.O.; Jack, C.R.; Petersen, R.C.; et al. Association of Plasma Total Tau Level With Cognitive Decline and Risk of Mild Cognitive Impairment or Dementia in the Mayo Clinic Study on Aging. JAMA Neurol. 2017, 74, 1073. [Google Scholar] [CrossRef]
- Pase, M.P.; Beiser, A.S.; Himali, J.J.; Satizabal, C.L.; Aparicio, H.J.; DeCarli, C.; Chêne, G.; Dufouil, C.; Seshadri, S. Assessment of Plasma Total Tau Level as a Predictive Biomarker for Dementia and Related Endophenotypes. JAMA Neurol. 2019, 76, 598. [Google Scholar] [CrossRef]
- Mielke, M.M.; Hagen, C.E.; Xu, J.; Chai, X.; Vemuri, P.; Lowe, V.J.; Airey, D.C.; Knopman, D.S.; Roberts, R.O.; Machulda, M.M.; et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimer’s Dement. 2018, 14, 989–997. [Google Scholar] [CrossRef]
- Lim, C.Z.J.; Zhang, Y.; Chen, Y.; Zhao, H.; Stephenson, M.C.; Ho, N.R.Y.; Chen, Y.; Chung, J.; Reilhac, A.; Loh, T.P.; et al. Subtyping of circulating exosome-bound amyloid β reflects brain plaque deposition. Nat. Commun. 2019, 10, 1144. [Google Scholar] [CrossRef] [Green Version]
- Zetterberg, H.; Burnham, S.C. Blood-based molecular biomarkers for Alzheimer’s disease. Mol. Brain 2019, 12, 26. [Google Scholar] [CrossRef]
- Yao, F.; Hong, X.; Li, S.; Zhang, Y.; Zhao, Q.; Du, W.; Wang, Y.; Ni, J. Urine-Based Biomarkers for Alzheimer’s Disease Identified Through Coupling Computational and Experimental Methods. J. Alzheimer’s Dis. 2018, 65, 421–431. [Google Scholar] [CrossRef]
- Watanabe, Y.; Hirao, Y.; Kasuga, K.; Tokutake, T.; Semizu, Y.; Kitamura, K.; Ikeuchi, T.; Nakamura, K.; Yamamoto, T. Molecular Network Analysis of the Urinary Proteome of Alzheimer’s Disease Patients. Dement. Geriatr. Cogn. Dis. Extra 2019, 9, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Orešič, M.; Hyötyläinen, T.; Herukka, S.-K.; Sysi-Aho, M.; Mattila, I.; Seppänan-Laakso, T.; Julkunen, V.; Gopalacharyulu, P.V.; Hallikainen, M.; Koikkalainen, J.; et al. Metabolome in progression to Alzheimer’s disease. Transl. Psychiatry 2011, 1, e57. [Google Scholar] [CrossRef]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef]
- Salvadores, N.; Shahnawaz, M.; Scarpini, E.; Tagliavini, F.; Soto, C. Detection of Misfolded Aβ Oligomers for Sensitive Biochemical Diagnosis of Alzheimer’s Disease. Cell Rep. 2014, 7, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Bongianni, M.; Ladogana, A.; Capaldi, S.; Klotz, S.; Baiardi, S.; Cagnin, A.; Perra, D.; Fiorini, M.; Poleggi, A.; Legname, G.; et al. α-Synuclein RT-QuIC assay in cerebrospinal fluid of patients with dementia with Lewy bodies. Ann. Clin. Transl. Neurol. 2019, 6, 2120–2126. [Google Scholar] [CrossRef]
- Rossi, M.; Candelise, N.; Baiardi, S.; Capellari, S.; Giannini, G.; Orrù, C.D.; Antelmi, E.; Mammana, A.; Hughson, A.G.; Calandra-Buonaura, G.; et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol. 2020, 140, 49–62. [Google Scholar] [CrossRef]
- Fairfoul, G.; McGuire, L.I.; Pal, S.; Ironside, J.W.; Neumann, J.; Christie, S.; Joachim, C.; Esiri, M.; Evetts, S.G.; Rolinski, M.; et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 2016, 3, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Tokuda, T.; Waragai, M.; Mendez, N.; Ishii, R.; Trenkwalder, C.; Mollenhauer, B.; Soto, C. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of α-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol. 2017, 74, 163. [Google Scholar] [CrossRef]
- Saijo, E.; Metrick, M.A.; Koga, S.; Parchi, P.; Litvan, I.; Spina, S.; Boxer, A.; Rojas, J.C.; Galasko, D.; Kraus, A.; et al. 4-Repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol. 2020, 139, 63–77. [Google Scholar] [CrossRef]
- Scialò, C.; Tran, T.H.; Salzano, G.; Novi, G.; Caponnetto, C.; Chiò, A.; Calvo, A.; Canosa, A.; Moda, F.; Caroppo, P.; et al. TDP-43 real time quaking induced conversion reaction optimization and detection of seeding activity in CSF of amyotrophic lateral sclerosis and frontotemporal dementia patients. Brain Commun. 2020, 2, fcaa142. [Google Scholar] [CrossRef]
- Manne, S.; Kondru, N.; Hepker, M.; Jin, H.; Anantharam, V.; Lewis, M.; Huang, X.; Kanthasamy, A.; Kanthasamy, A.G. Ultrasensitive Detection of Aggregated α-Synuclein in Glial Cells, Human Cerebrospinal Fluid, and Brain Tissue Using the RT-QuIC Assay: New High-Throughput Neuroimmune Biomarker Assay for Parkinsonian Disorders. J. Neuroimmune Pharmacol. 2019, 14, 423–435. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.M.G.; Elia, A.E.; Portaleone, S.M.; Cazzaniga, F.A.; Rossi, M.; Bistaffa, E.; De Cecco, E.; Narkiewicz, J.; Salzano, G.; Carletta, O.; et al. Efficient RT-QuIC seeding activity for α-synuclein in olfactory mucosa samples of patients with Parkinson’s disease and multiple system atrophy. Transl. Neurodegener. 2019, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Saijo, E.; Ghetti, B.; Zanusso, G.; Oblak, A.; Furman, J.L.; Diamond, M.I.; Kraus, A.; Caughey, B. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol. 2017, 133, 751–765. [Google Scholar] [CrossRef]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Lawton, M.; Baig, F.; Rolinski, M.; Ruffman, C.; Nithi, K.; May, M.T.; Ben-Shlomo, Y.; Hu, M.T.M. Parkinson’s Disease Subtypes in the Oxford Parkinson Disease Centre (OPDC) Discovery Cohort. J. Parkinsons. Dis. 2015, 5, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Peelaerts, W.; Baekelandt, V. ɑ-Synuclein strains and the variable pathologies of synucleinopathies. J. Neurochem. 2016, 139, 256–274. [Google Scholar] [CrossRef] [Green Version]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Candelise, N.; Schmitz, M.; Llorens, F.; Villar-Piqué, A.; Cramm, M.; Thom, T.; Silva Correia, S.M.; Cunha, J.E.G.; Möbius, W.; Outeiro, T.F.; et al. Seeding variability of different alpha synuclein strains in synucleinopathies. Ann. Neurol. 2019, 85, 691–703. [Google Scholar] [CrossRef]
- Groveman, B.R.; Orrù, C.D.; Hughson, A.G.; Raymond, L.D.; Zanusso, G.; Ghetti, B.; Campbell, K.J.; Safar, J.; Galasko, D.; Caughey, B. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol. Commun. 2018, 6, 1–10. [Google Scholar] [CrossRef]
- Kang, U.J.; Boehme, A.K.; Fairfoul, G.; Shahnawaz, M.; Ma, T.C.; Hutten, S.J.; Green, A.; Soto, C. Comparative study of cerebrospinal fluid α-synuclein seeding aggregation assays for diagnosis of Parkinson’s disease. Mov. Disord. 2019, 34, 536–544. [Google Scholar] [CrossRef]
- Capitini, C.; Patel, J.R.; Natalello, A.; D’Andrea, C.; Relini, A.; Jarvis, J.A.; Birolo, L.; Peduzzo, A.; Vendruscolo, M.; Matteini, P.; et al. Structural differences between toxic and nontoxic HypF-N oligomers. Chem. Commun. 2018, 54, 8637–8640. [Google Scholar] [CrossRef]
- Kong, K.; Kendall, C.; Stone, N.; Notingher, I. Raman spectroscopy for medical diagnostics — From in-vitro biofluid assays to in-vivo cancer detection. Adv. Drug Deliv. Rev. 2015, 89, 121–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krafft, C.; Popp, J. The many facets of Raman spectroscopy for biomedical analysis. Anal. Bioanal. Chem. 2015, 407, 699–717. [Google Scholar] [CrossRef] [PubMed]
- Banchelli, M.; de Angelis, M.; D’Andrea, C.; Pini, R.; Matteini, P. Triggering molecular assembly at the mesoscale for advanced Raman detection of proteins in liquid. Sci. Rep. 2018, 8, 1033. [Google Scholar] [CrossRef] [Green Version]
- Eravuchira, P.; Banchelli, M.; D’Andrea, C.; De Angelis, M.; Matteini, P.; Gannot, I. Hollow core photonic crystal fiber-assisted Raman spectroscopy as a tool for the detection of Alzheimer’s disease biomarkers. J. Biomed. Opt. 2020, 25, 1–10. [Google Scholar] [CrossRef]
- Banchelli, M.; Amicucci, C.; Ruggiero, E.; D’Andrea, C.; Cottat, M.; Ciofini, D.; Osticioli, I.; Ghini, G.; Siano, S.; Pini, R.; et al. Spot-on SERS Detection of Biomolecules with Laser-Patterned Dot Arrays of Assembled Silver Nanowires. ChemNanoMat 2019, 5, 1036–1043. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, C.; Foti, A.; Cottat, M.; Banchelli, M.; Capitini, C.; Barreca, F.; Canale, C.; de Angelis, M.; Relini, A.; Maragò, O.M.; et al. Nanoscale Discrimination between Toxic and Nontoxic Protein Misfolded Oligomers with Tip-Enhanced Raman Spectroscopy. Small 2018, 14, 1800890. [Google Scholar] [CrossRef]
- Matteini, P.; de Angelis, M.; Ulivi, L.; Centi, S.; Pini, R. Concave gold nanocube assemblies as nanotraps for surface-enhanced Raman scattering-based detection of proteins. Nanoscale 2015, 7, 3474–3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrini, L.; Arenal, R.; Mannini, B.; Chiti, F.; Pini, R.; Matteini, P.; Alvarez-Puebla, R.A. SERS Detection of Amyloid Oligomers on Metallorganic-Decorated Plasmonic Beads. ACS Appl. Mater. Interfaces 2015, 7, 9420–9428. [Google Scholar] [CrossRef]
- Matteini, P.; Cottat, M.; Tavanti, F.; Panfilova, E.; Scuderi, M.; Nicotra, G.; Menziani, M.C.; Khlebtsov, N.; de Angelis, M.; Pini, R. Site-Selective Surface-Enhanced Raman Detection of Proteins. ACS Nano 2017, 11, 918–926. [Google Scholar] [CrossRef]
- Banchelli, M.; Cascella, R.; D’Andrea, C.; Cabaj, L.; Osticioli, I.; Ciofini, D.; Li, M.S.; Skupień, K.; de Angelis, M.; Siano, S.; et al. Nanoscopic insights into the surface conformation of neurotoxic amyloid β oligomers. RSC Adv. 2020, 10, 21907–21913. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bistaffa, E.; Tagliavini, F.; Matteini, P.; Moda, F. Contributions of Molecular and Optical Techniques to the Clinical Diagnosis of Alzheimer’s Disease. Brain Sci. 2020, 10, 815. https://doi.org/10.3390/brainsci10110815
AMA Style
Bistaffa E, Tagliavini F, Matteini P, Moda F. Contributions of Molecular and Optical Techniques to the Clinical Diagnosis of Alzheimer’s Disease. Brain Sciences. 2020; 10(11):815. https://doi.org/10.3390/brainsci10110815
Chicago/Turabian StyleBistaffa, Edoardo, Fabrizio Tagliavini, Paolo Matteini, and Fabio Moda. 2020. "Contributions of Molecular and Optical Techniques to the Clinical Diagnosis of Alzheimer’s Disease" Brain Sciences 10, no. 11: 815. https://doi.org/10.3390/brainsci10110815
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.