First-Principles Investigation of Atomic Hydrogen Adsorption and Diffusion on/into Mo-doped Nb (100) Surface

 ,
,
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
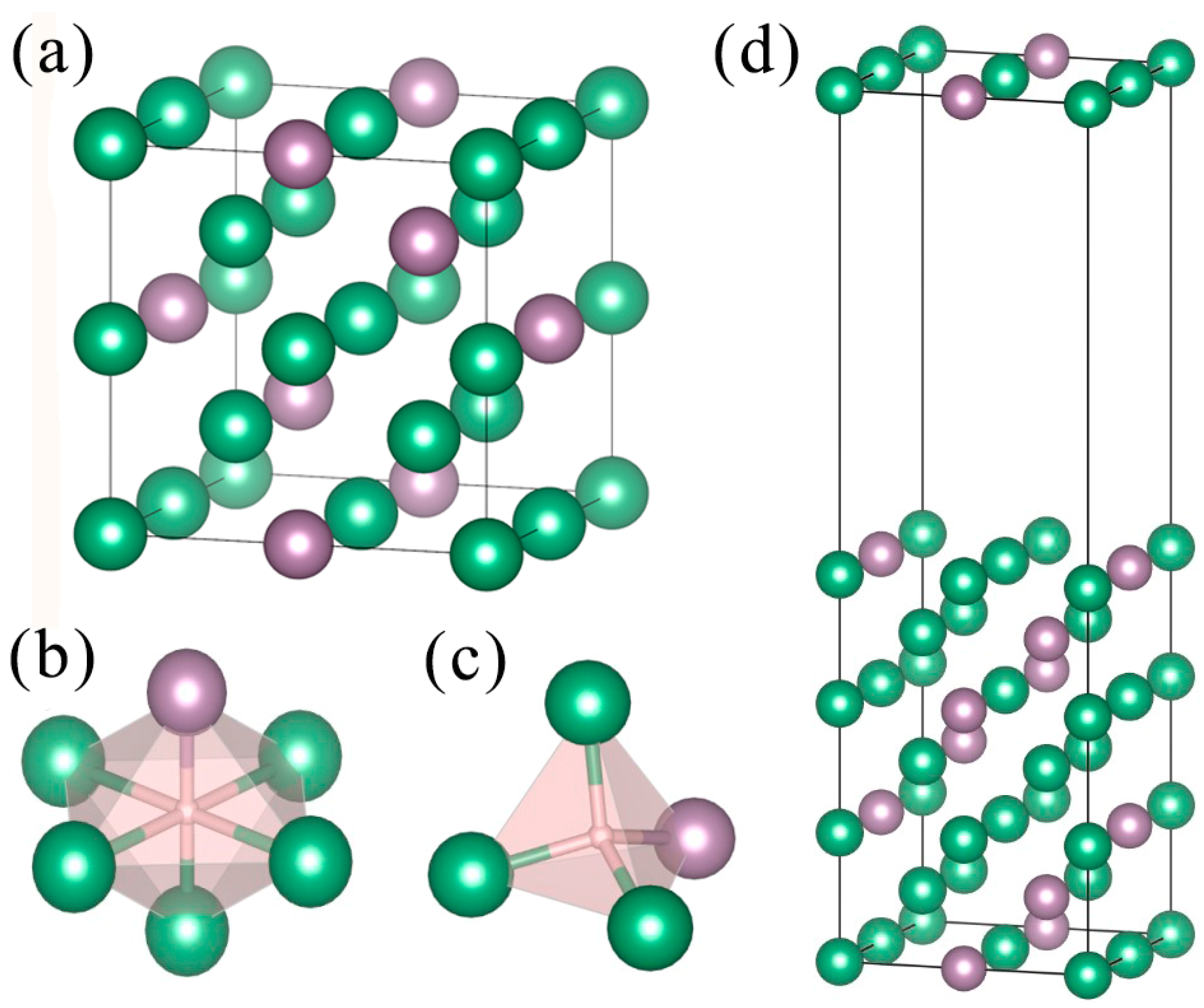
3.1. Surface Model of Mo-doped Nb
3.2. H Atom Adsorption Sites of Mo-doped Nb (100) Surface
3.3. Electronic Properties of Mo-doped Nb (100) Surface
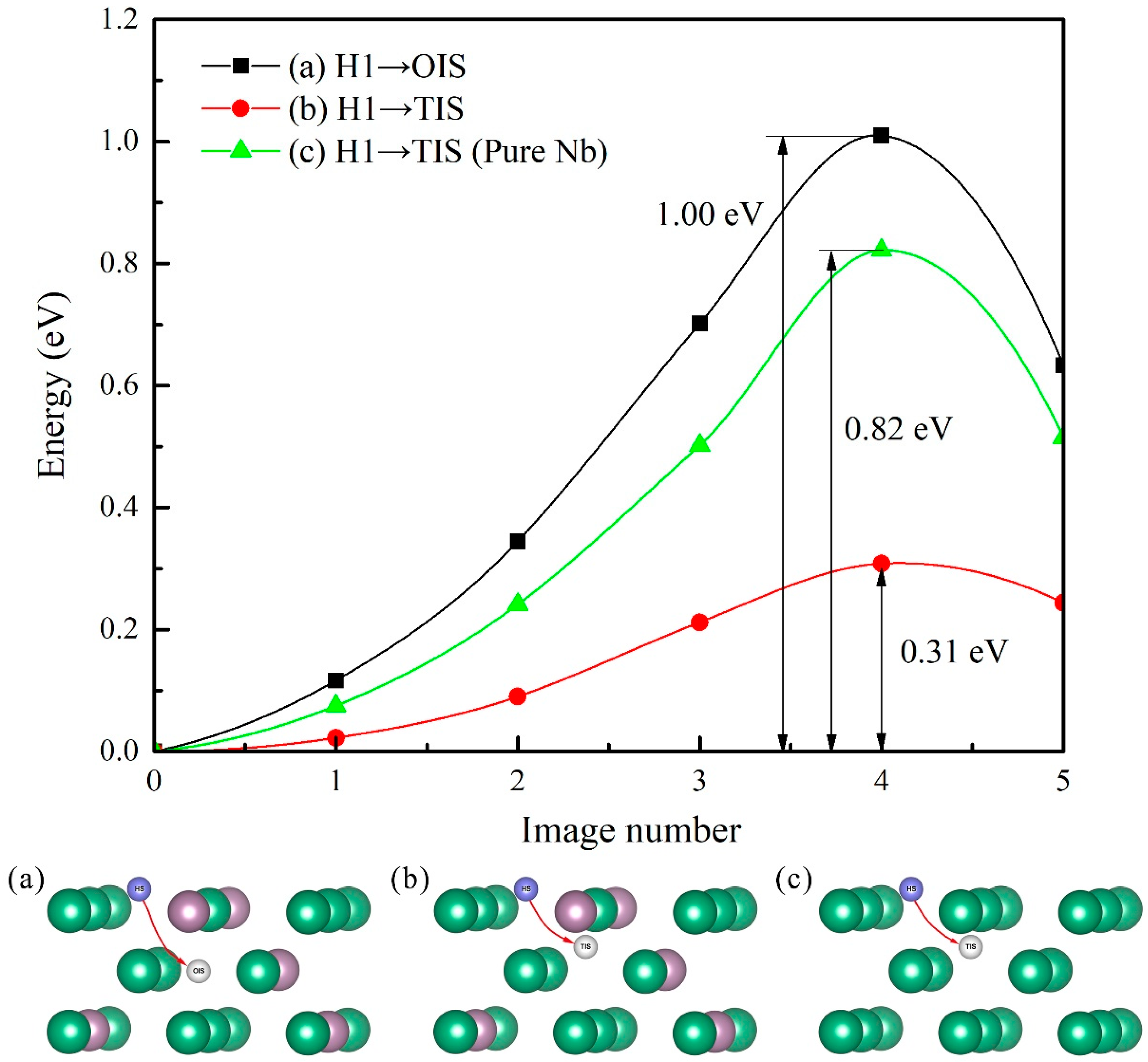
3.4. Diffusion of Hydrogen Atom into Mo-doped Nb (100) Subsurface
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ding, L.; Wei, Y.; Li, L.; Zhang, T.; Wang, H.; Xue, J.; Wang, S.; Caro, J.; Gogotsi, Y. MXene molecular sieving membranes for highly efficient gas separation. Nat. Commun. 2018, 9, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.L.; Hao, X.F.; Jiang, Z.; Sun, X.P.; Xu, D.; Wang, J.; Zhong, H.; Meng, F.; Zhang, X. C and N hybrid coordination derived Co-C-N complex as a highly efficient electrocatalyst for hydrogen evolution reaction. J. Am. Chem. Soc. 2015, 48, 15070–15073. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhong, B.; Xiao, H.; Ye, X.; Lu, L.; Guan, W.; Zhang, Y.; Wang, X.; Chen, C. Effect of degassing treatment on the deuterium permeability of Pd-Nb-Pd composite membranes during deuterium permeation. Sep. Purif. Technol. 2018, 190, 136–142. [Google Scholar] [CrossRef]
- Ferrin, P.; Kandoia, S.; Nilekara, A.U.; Mavrikakisa, M. Hydrogen adsorption, absorption and diffusion on and intransition metal surfaces: A DFT study. Surf. Sci. 2012, 606, 679–689. [Google Scholar] [CrossRef]
- Wang, J.W.; He, Y.H.; Gong, H.R. Various properties of Pd3Ag/TiAl membranes from density functional theory. J. Membr. Sci. 2015, 475, 406–413. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, H.; Wang, Z.; Meng, F.L.; Zhang, X.B. Integrated three-dimensional carbon paper/carbon tubes/cobalt-sulfide sheets as an efficient electrode for overall water splitting. ACS Nano 2016, 2, 2342–2348. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Wang, J.; Meng, F.; Zhang, X. In Situ Activating Ubiquitous Rust towards Low-Cost, Efficient, Free-Standing, and Recoverable Oxygen Evolution Electrodes. Angew. Chem. Int. Ed. 2016, 34, 9937–9941. [Google Scholar] [CrossRef]
- Singh, M.E.; Rezac, P.H.; Pfromm, P. Hydrogenation of soybean oil using metal decorated integral-asymmetric polymer membranes: Effects of morphology and membrane properties. J. Membr. Sci. 2010, 348, 99–108. [Google Scholar] [CrossRef]
- Singh, U.K.; Vannice, M.A. Kinetics of liquid-phase hydrogenation reactions over supported metal catalysts—A review. Appl. Catal. A 2001, 213, 1–24. [Google Scholar] [CrossRef]
- Wang, J.; Li, K.; Zhong, H.; Xu, D.; Wang, Z.L.; Jiang, Z.; Wu, Z.; Zhang, X.B. Synergistic effect between metal-nitrogen-carbon sheets and NiO nanoparticles for enhanced electrochemical water-oxidation performance. Angew. Chem. Int. Ed. 2015, 36, 10530–10534. [Google Scholar] [CrossRef]
- Zacher, A.H.; Olarte, M.V.; Santosa, D.M.; Elliott, D.C.; Jones, S.B. A review and perspective of recent bio-oil hydrotreating research. Green Chem. 2014, 16, 491–515. [Google Scholar] [CrossRef]
- Carrette, L.; Friedrich, K.A.; Stimming, U. Fuel cells: Principles, types, fuels, and applications. J. Chem. Phys. 2000, 1162–1193. [Google Scholar] [CrossRef]
- Edwards, P.P.; Kuznetsov, V.L.; David, W.I.F.; Brandon, N.P. Hydrogen and fuel cells: towards a sustainable energy future. Energy Policy 2008, 36, 4356–4362. [Google Scholar] [CrossRef]
- Kikuchi, E. Membrane reactor application to hydrogen production. Catal. Today 2000, 56, 97–101. [Google Scholar] [CrossRef]
- Dong, W.; Ledentu, V.; Sautet, P.; Eichler, A.; Hafner, J. Hydrogen adsorption on palladium: A comparative theoretical study of different surfaces. Surf. Sci. 1998, 411, 123–136. [Google Scholar] [CrossRef]
- Lauhon, L.J.; Ho, W. Direct observation of the quantum tunneling of single hydrogen atoms with a scanning tunneling microscope. Phys. Rev. Lett. 2000, 85, 4566. [Google Scholar] [CrossRef] [PubMed]
- Gómez, E.D.V.; Amayaroncancio, S.; Avalle, L.B.; Linares, D.H.; Gimenez, M.C. DFT study of adsorption and diffusion of atomic hydrogen on metal surfaces. Appl. Surf. Sci. 2017, 420, 1–8. [Google Scholar] [CrossRef]
- Mitsui, T.; Rose, M.K.; Fomin, E.; Ogletree, D.F.; Salmeron, M. Hydrogen adsorption and diffusion on Pd(111). Surf. Sci. 2003, 540, 5–11. [Google Scholar] [CrossRef]
- Liu, Z.H.; Shang, J.X. Elastic properties of Nb-based alloys by using the density functional theory. Chin. Phys. B 2012, 21, 016202. [Google Scholar] [CrossRef]
- Liu, G.; Besedin, S.; Irodova, A.; Liu, H.; Gao, G.; Eremets, M.; Wang, X.; Ma, Y. Nb-H system at high pressures and temperatures. Phys. Rev. B 2017, 95, 104110. [Google Scholar] [CrossRef]
- Peterson, D.T.; Hull, A.B.; Loomis, B.A. Hydrogen embrittlement considerations in niobium-base alloys for application in the ITER divertor. J. Nucl. Mater. 2012, 191–194, 430–432. [Google Scholar]
- Watanabe, N.; Yukawa, H.; Nambu, T.; Matsumoto, Y.; Zhang, G.X.; Morinaga, M. Alloying effects of Ru and W on the resistance to hydrogen embrittlement and hydrogen permeability of niobium. J. Alloys Compd. 2009, 477, 851–854. [Google Scholar] [CrossRef]
- Zhang, G.X.; Yukawa, H.; Nambu, T.; Matsumoto, Y.; Morinaga, M. Alloying effects of Ru and W on hydrogen diffusivity during hydrogen permeation through Nb-based hydrogen permeable membranes. Int. J. Hydrogen Energy 2010, 35, 1245–1249. [Google Scholar] [CrossRef]
- Yukawa, H.; Nambu, T.; Matsumoto, Y.; Watanabe, N.; Zhang, G.; Morinaga, M. Alloy design of Nb-based hydrogen permeable membrane with strong resistance to hydrogen embrittlement. Mater Trans. 2008, 49, 2202–2207. [Google Scholar] [CrossRef]
- Nambu, T.; Shimizu, K.; Matsumoto, Y.; Rong, R.; Watanabe, N.; Yukawa, H.; Morinaga, M.; Yasuda, I. Enhanced hydrogen embrittlement of Pd-coated niobium metal membrane detected by in situ small punch test under hydrogen permeation. J. Alloys Compd. 2007, 446, 588–592. [Google Scholar] [CrossRef]
- Long, J.H.; Gong, H. Phase stability and mechanical properties of niobium dihydride. Int. J. Hydrogen Energy 2014, 39, 18989–18996. [Google Scholar] [CrossRef]
- Slining, J.R.; Koss, D.A. Solid solution strengthening of high purity niobium alloys. Metall. Trans. 1973, 4, 1261–1264. [Google Scholar] [CrossRef]
- Kozhakhmetov, S.; Sidorov, N.; Piven, V.; Sipatov, I.; Gabis, I.; Arinov, B. Alloys based on Group 5 metals for hydrogen purification membranes. J. Alloys Comp. 2015, 645, S36–S40. [Google Scholar] [CrossRef]
- Xu, Z.J.; Wang, Z.M.; Tang, J.L.; Deng, J.Q.; Yao, Q.R.; Zhou, H.Y. Effects of Mo alloying on the structure and hydrogen-permeation properties of Nb metal. J. Alloys Compd. 2018, 740, 810–815. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Z.M.; Wang, D.H.; Wan, Z.Z.; Zhong, Y.; Hu, C.H.; Zhou, H. Effects of Ni doping on various properties of NbH phases: A first-principles investigation. Sci. Rep. UK 2017, 7, 6535. [Google Scholar] [CrossRef]
- Hu, Y.T.; Gong, H.; Chen, L. Fundamental effects of W alloying on various properties of NbH phases. Int. J. Hydrogen Energy 2015, 40, 12745–12749. [Google Scholar] [CrossRef]
- Kong, X.S.; Wang, S.; Wu, X.; You, Y.W.; Liu, C.S.; Fang, Q.F.; Chen, J.L.; Luo, G.N. First-principles calculations of hydrogen solution and diffusion in tungsten: Temperature and defect-trapping effects. Acta Mater. 2015, 84, 426–435. [Google Scholar] [CrossRef]
- Nagata, S.; Takahiro, K. Deuterium retention in tungsten and molybdenum. J. Nucl. Mater. 2000, 283, 1038–1042. [Google Scholar] [CrossRef]
- Duan, C.; Liu, Y.L.; Zhou, H.B.; Zhang, Y.; Jin, S.; Lu, G.H.; Luo, G.N. First-principles study on dissolution and diffusion properties of hydrogen in molybdenum. J. Nucl. Mater. 2010, 404, 109–115. [Google Scholar] [CrossRef]
- Wright, G.M.; Whyte, D.G.; Lipschultz, B. Measurement of hydrogenic retention and release in molybdenum with the DIONISOS experiment. J. Nucl. Mater. 2009, 390–391, 544–549. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab-initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector agmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jonsson, H.A. Climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Lässer, R.; Bickmann, K. Phase diagram of the Nb-T system. J. Nucl. Mater. 1985, 132, 244–248. [Google Scholar] [CrossRef]
- Baskes, M.I. Modified embedded-atom potentials for cubic materials and impurities. Phys. Rev. B 1992, 46, 2727. [Google Scholar] [CrossRef]
- Strayer, R.W.; Mackie, W.; Swanson, L.W. Work function measurements by the field emission retarding potential method. Surf. Sci. 1973, 34, 225–248. [Google Scholar] [CrossRef] [Green Version]
- Michaelson, H.B. The work function of the elements and its periodicity. J. Chem. Phys. 1977, 48, 4729–4733. [Google Scholar] [CrossRef]
- Gong, L.; Su, Q.; Deng, H.; Xiao, S.; Hu, W. The stability and diffusion properties of foreign impurity atoms on the surface and in the bulk of vanadium: A first-principles study. Comput. Mater. Sci. 2014, 81, 191–198. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface Energies (eV) | |||||||
|---|---|---|---|---|---|---|---|
| 4 Layers | 5 Layers | 6 Layers | 7 Layers | 8 Layers | 9 Layers | 10 Layers | |
| Pure Nb (100) | 2.63 | 2.45 | 2.43 | 2.42 | 2.44 | 2.44 | 2.44 |
| Pure Nb (100) a | 2.3–2.7 | ||||||
| Nb12Mo4 (100) | 2.72 | 2.73 | 2.74 | 2.75 | 2.75 | 2.75 | 2.75 |
| Nb12Mo4 (111) | 3.28 | 3.31 | 3.32 | 3.32 | 3.32 | 3.32 | 3.32 |
| Site | Eads (eV) | dH-TM (Å) | dH-Surf (Å) |
|---|---|---|---|
| T2 | −3.41 | 1.77 | 1.85 |
| T1 | −3.26 | 1.85 | 1.93 |
| B2 | −3.77 | 1.97 | 1.30 |
| B1 | −3.75 | 1.97 | 1.21 |
| H2 | −4.24 | 1.98 | 0.46 |
| H1 | −4.27 | 1.99 | 0.44 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Wang, Z.; Wang, D.; Qin, J.; Wan, Z.; Zhong, Y.; Hu, C.; Zhou, H. First-Principles Investigation of Atomic Hydrogen Adsorption and Diffusion on/into Mo-doped Nb (100) Surface. Appl. Sci. 2018, 8, 2466. https://doi.org/10.3390/app8122466
Wu Y, Wang Z, Wang D, Qin J, Wan Z, Zhong Y, Hu C, Zhou H. First-Principles Investigation of Atomic Hydrogen Adsorption and Diffusion on/into Mo-doped Nb (100) Surface. Applied Sciences. 2018; 8(12):2466. https://doi.org/10.3390/app8122466
Chicago/Turabian StyleWu, Yang, Zhongmin Wang, Dianhui Wang, Jiayao Qin, Zhenzhen Wan, Yan Zhong, Chaohao Hu, and Huaiying Zhou. 2018. "First-Principles Investigation of Atomic Hydrogen Adsorption and Diffusion on/into Mo-doped Nb (100) Surface" Applied Sciences 8, no. 12: 2466. https://doi.org/10.3390/app8122466