1. Introduction
Proteins are fundamental to human life and seldom function as a single unit. They always interact with each other in a specific way to perform cellular processes [
1]. As a consequence, the analysis of protein–protein interactions (PPIs) can help researchers reveal tissue functions and structures and identify the pathogenesis of human diseases and drug targets of gene therapy. Recently, various high-throughput experimental techniques have been discovered for PPI detection, including a yeast two-hybrid system [
2], immunoprecipitation [
3], and protein chips [
4]. However, the biological experiments are generally costly and time-consuming. Moreover, both false negative and false positive rates of these methods are very high [
2,
5]. Therefore, the development of reliable calculating models for the prediction of PPIs has great practical significance.
To construct a computational method for PPI prediction, the most important factor is to extract highly discriminative features that can effectively describe proteins. So far, protein feature extraction methods are based on many data types, such as genomic information [
6,
7], structure information [
8,
9], evolutionary information [
10,
11], and amino acid sequence information. Of these approaches, sequence-based methods are more readily available, and it has demonstrated that protein amino acid sequence information is important for detecting PPIs [
12,
13,
14,
15,
16]. Martin et al. used a descriptor called a “signature product” to discover PPIs [
12]. The signature product is a product of sub-sequences from a protein sequence and extends the signature descriptor from chemical information. Shen et al. presented a conjoint triad (CT) approach to take the characters of amino acids and their adjacent amino acids into account [
13]. Guo et al. proposed an auto covariance (AC) descriptor approach to represent an amino acid sequence with a foundation of seven different physicochemical scales [
14]. When they detected Yeast PPIs, prospective prediction accuracy was achieved. Wong et al. employed the physicochemical property response matrix combined with the local phase quantization descriptor (PR-LPQ) to extract the eigen value of the proteins [
15]. Considering the evolutionary information of protein, Huang et al. adopted substitution matrix representation (SMR) based on BLOSUM62 to construct a feature vector and achieved promising prediction accuracy [
16]. Ding et al. proposed a novel protein sequence representation method based on a matrix to predict PPIs via an ensemble classification method [
17]. Wang et al. proposed a computational method based on a probabilistic classification vector machine (PCVM) model and a Zernike moment (ZM) descriptor to identify PPIs from amino acids sequences [
18]. Lei et al. employed the NABCAM (the neighbor affinity-based core-attachment method) to identify protein complexes from dynamic PPI networks [
19]. Nanni et al. summarized and evaluated a couple of feature extraction methods for describing protein amino acids sequences by verifying them on multiple datasets, and they constructed an ensemble of classifiers for sequence-based protein classification, which not only performed well on many datasets but was also, under certain conditions, superior to the state of the art [
20,
21,
22,
23].
Next, the computational methods for PPI prediction can be formulated as a binary classification problem. A number of machine learning-based computational models for PPI prediction have emerged. Ronald et al. proposed a technique of applying Bayesian networks to detect PPIs on the Yeast dataset [
24]. Qi et al. employed several classifiers, including support vector machine (SVM), decision tree (DT), random forest (RF), and logistic regression (LR), to compare their performances in predicting PPIs [
25].
Of machine-learning-based computational models for PPI prediction, one of the most important challenges is that the high-dimensional features may include unimportant information and noise, leading to the over-fitting of classification systems [
26,
27]. Previous works have shown that random projection (RP) is a high-efficiency and sufficient precision approach that can reduce the dimensions of many high-dimensional datasets [
28,
29,
30]. However, the performance using a single RP method is poor [
31] because of the instability of RP. Therefore, in our study, an RP ensemble method was designed to predict PPIs.
The capacity of the integration model was better and more effective than that of separate runs of the RP approach. Moreover, the ensemble algorithm achieved results that are also superior to those of similar schemes that use principal component analysis (PCA) to reduce the dimensionality of the dataset [
32]. The RP-ensemble-based classifier has useful features [
33]. Firstly, RP maintains the geometrical structure of the dataset with a certain distortion rate when its dimensionality is reduced. This feature can reduce the complicacy of the classifier and the difficulty of new sample classification. In addition, dimensionality reduction can eliminate redundant information and reduce generalization error. Particularly, instead of relying on a single classifier, the RP ensemble method incorporates several classifiers that are superior to each single classifier. This feature can lead to better and more stable classification results.
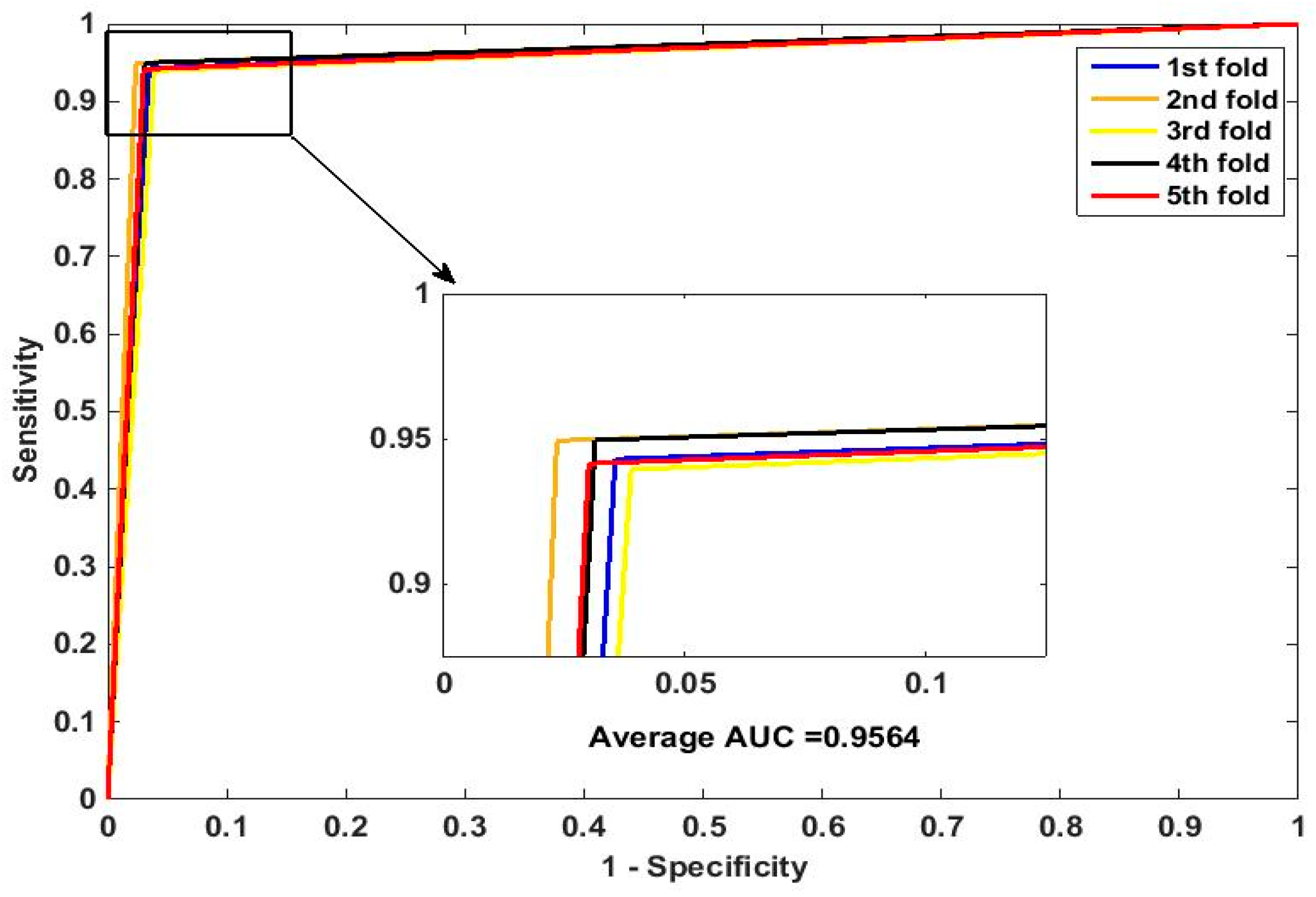
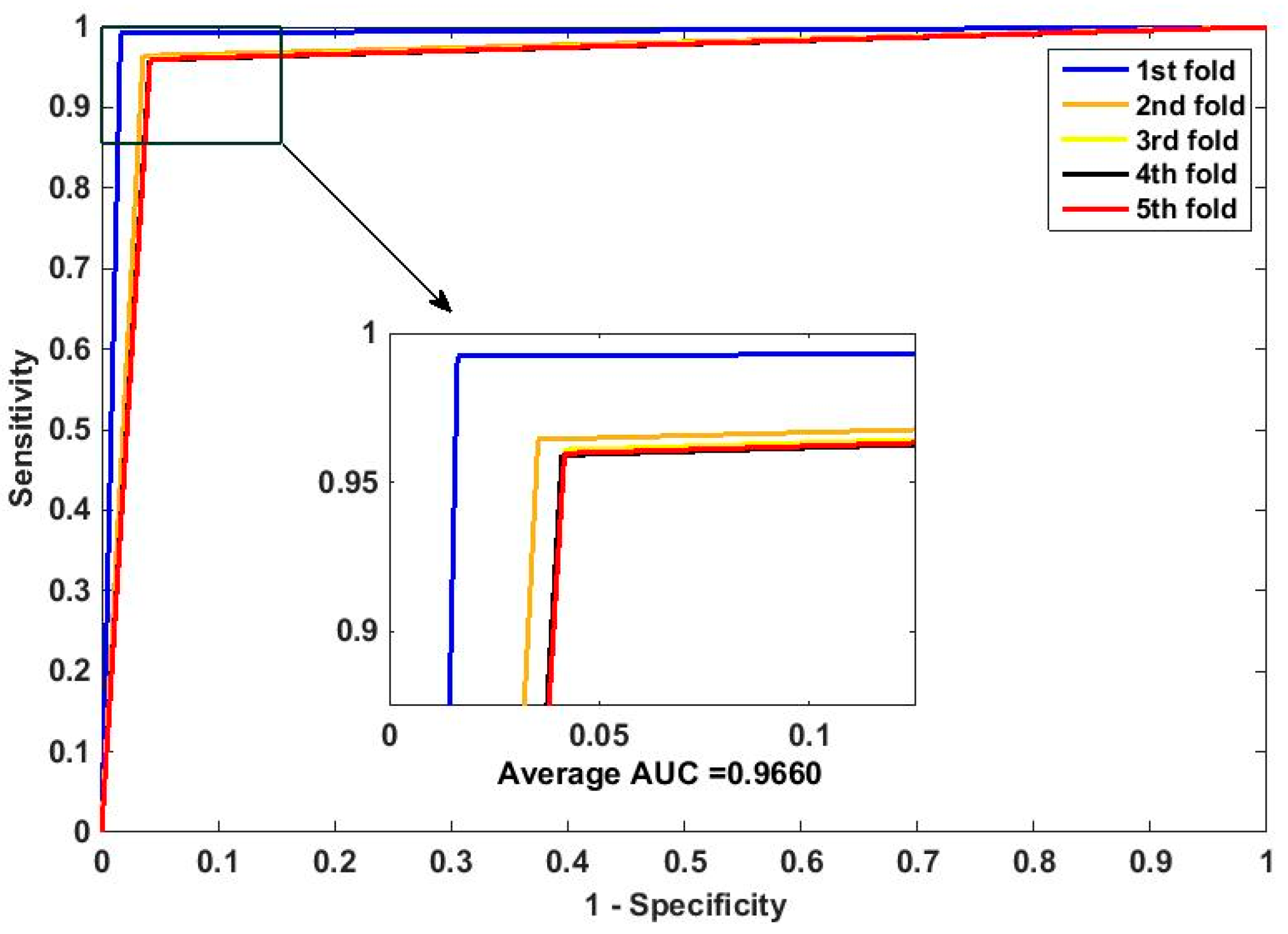
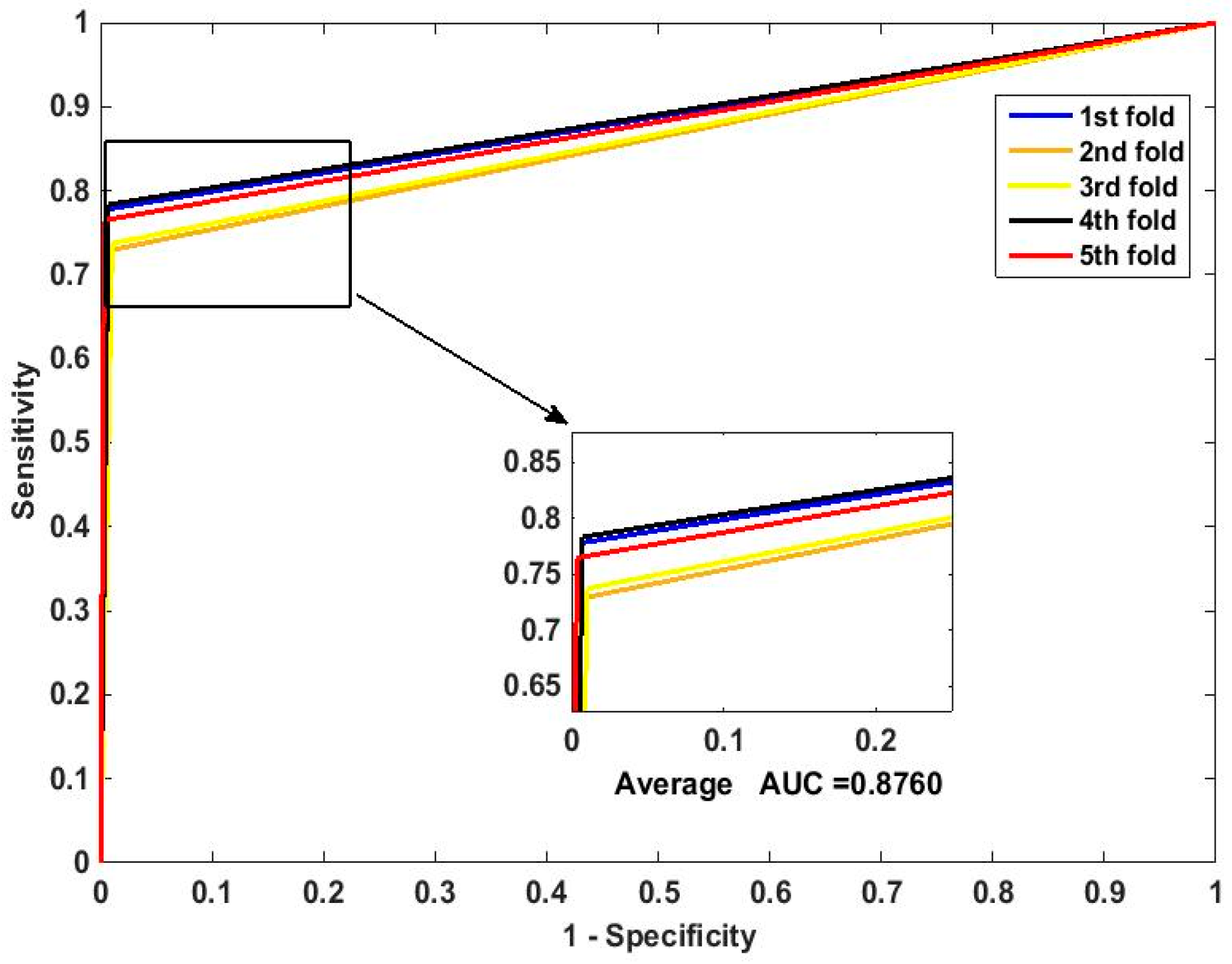
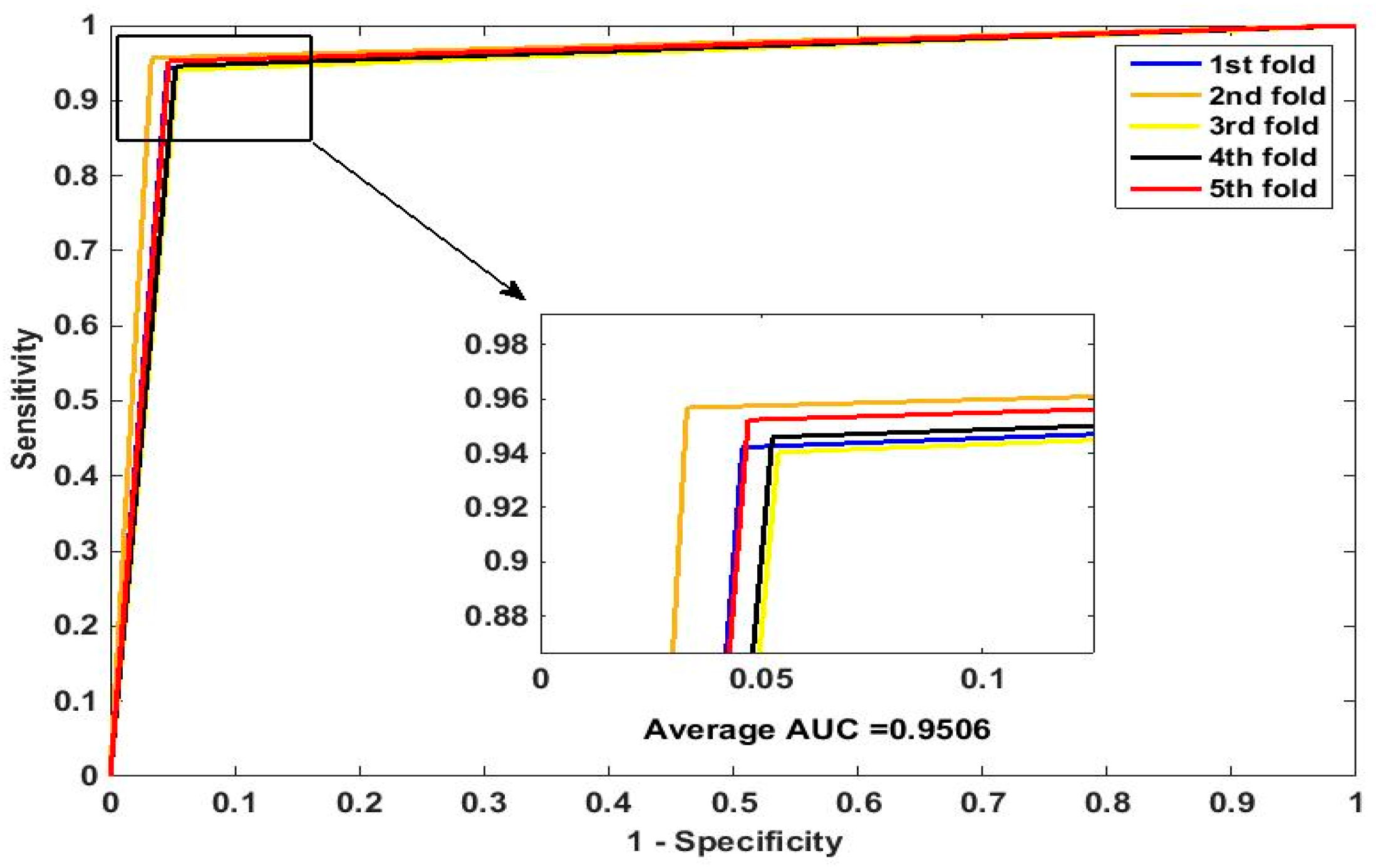
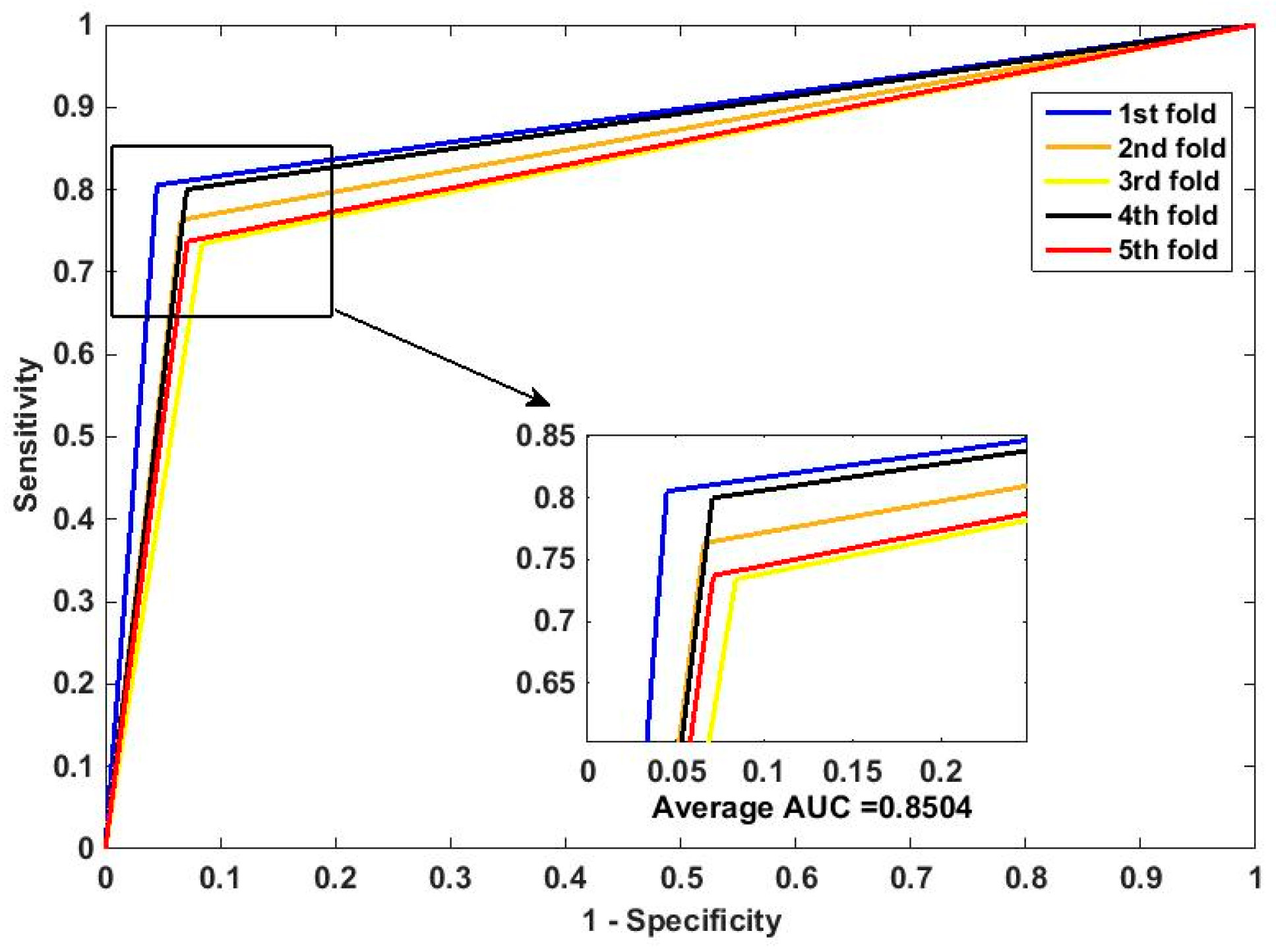
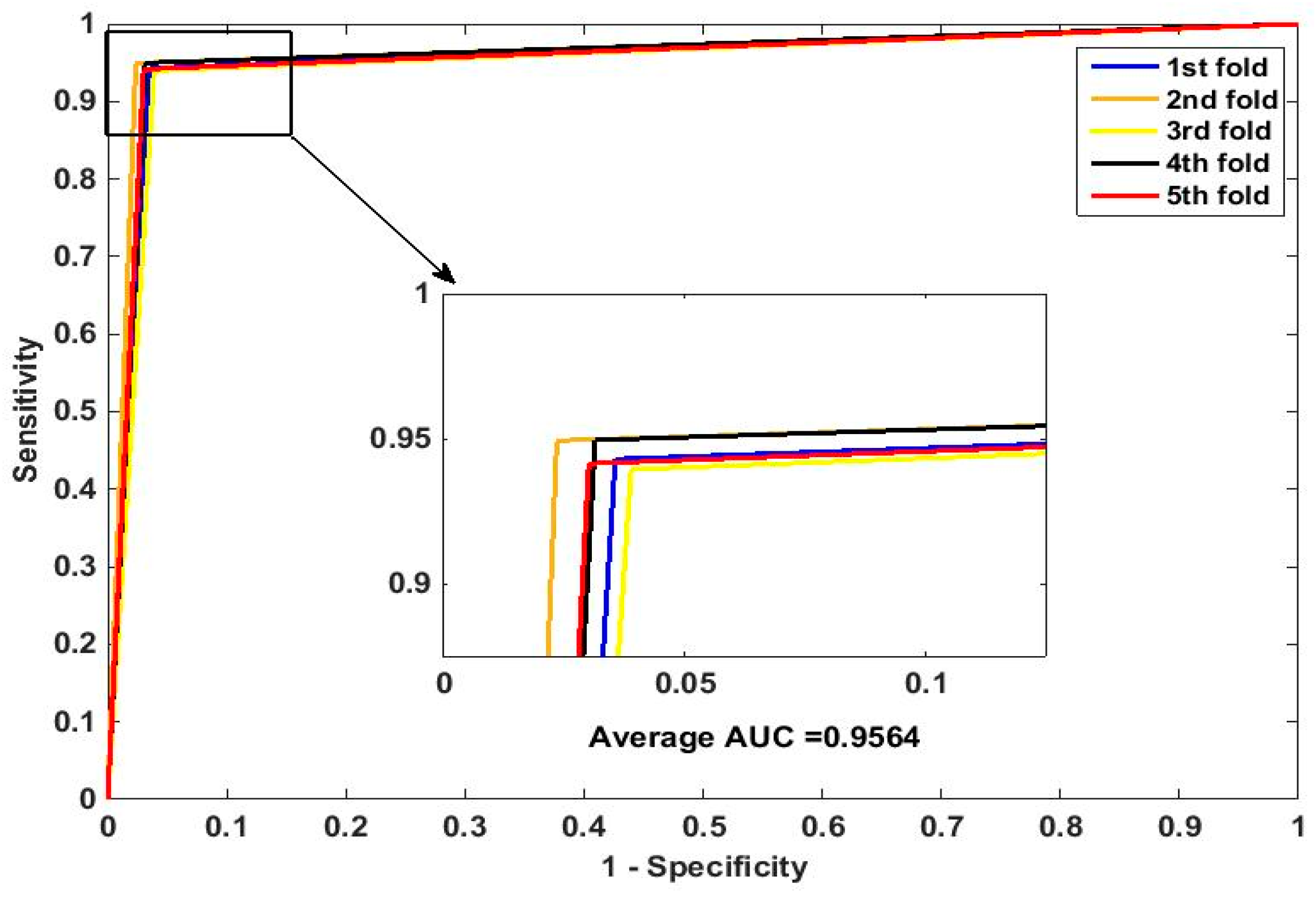
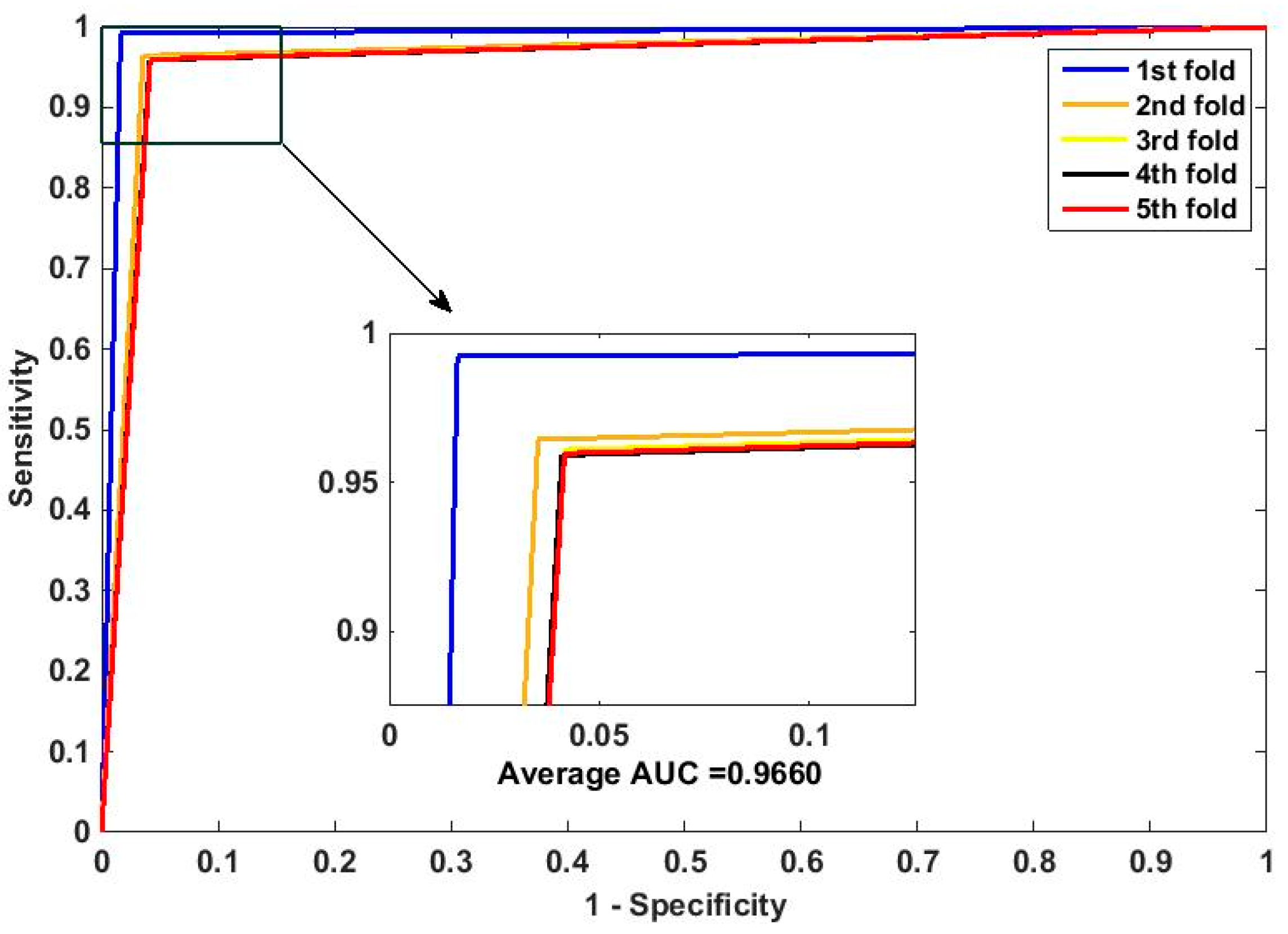
In this paper, we propose an RPEC-based approach for detecting PPIs by combining a protein sequence with its evolutionary information. Firstly, a position-specific scoring matrix (PSSM) is used to express the amino acid sequence. Secondly, three 400 dimensional feature vectors are extracted from the PSSM matrix by using DCT, FFT, and SVD, respectively, and each protein sequence is described as a 2400 size of eigen vector. Then, an 1000 dimensionally reduced feature vector is obtained via PCA. Finally, the RP ensemble model is built by employing the feature matrix of the protein pairs as an input to predict PPIs. Our method is estimated on the PPI datasets of Yeast, Human, and H. pylori and yields higher prediction accuracy of 95.64%, 96.59% and 87.62%, respectively. When compared with the SVM classifier fed into the same feature vector, the accuracies of our method are increased by 0.58%, 1.4%, and 2.57%, respectively. We also compare our method with other approaches. The obtained outcomes prove that our method is much better at predicting PPIs than those of previous works.
2. Materials and Methods
2.1. Position-Specific Scoring Matrix
A position-specific scoring matrix (
PSSM) can be applied to search distantly related proteins. It emerged from a group of sequences formerly arranged by structural or similarity [
34]. There are many methods of calculating distances and metric spaces [
35,
36]. Here, some research on
PSSM methods and its relation to amino acids is discussed. Liu et al. [
37] discovered that the
PSSM profile has been shown to provide a useful source of information for improving the prediction performance of the protein structural class. Wang et al. [
38] proposes two fusion feature representations of DipPSSM and PseAAPSSM to integrate
PSSM with DipC and PseAAC, respectively. In our method, we predict PPIs based on
PSSM. Thus, each protein can be converted to a
PSSM by using the Position-Specific Iterated Basic Local Alignment Search Tool (PSI-BLAST) [
39].
The
PSSM can be described as follows:
where
. With a size of
PSSM,
L represents the length of an amino acid, and 20 is the number of amino acids. In our research, we achieved the experiment datasets by using PSI-BLAST to use a
PSSM for PPI detection. In order to achieve a wide and analogous sequences, the parameter e-value of PSI-BLAST was adjusted to 0.001 and opted for three iterations, and the other values in PSI-BLAST were defaults. Finally, the
PSSM from a certain protein sequence was expressed as a
matrix, where
M denotes the quantity of residues, and 20 indicates the number of amino acids.
2.2. Discrete Cosine Transform
Discrete cosine transform (DCT) is a classical orthogonal transformation, which was first proposed in the 1970s by Ahmed [
40]. It is used in image compression processing with lossy signals because of its strong compaction performance. DCT has better energy aggregation than others. It can convert spatial signals into frequency domains and thus work well in de-correlation. DCT can be defined as follows:
where
The N × 20 PSSM is the input signal, which is xi ∈ Rn. Here, we can obtain 400 coefficients as the protein feature vector after the DCT feature descriptor. At the end, we can obtain a feature vector whose dimension is 800 from each protein pair via DCT.
2.3. Fast Fourier Transform
Fast Fourier transform (FFT) is a feature extraction method. The simplified energy function for FFT algorithms evaluate in the space of protein partner mutual orientations. The mesh displacements of one protein centroid in regard to another protein centroid can represent the translational space [
41].
Here we describe the simplified energy scoring function, as PPIs are defined on a mesh and indicate
M correlation functions all possible values of
l,
m, and
n (assuming that one protein is the ligand and the other is the receptor):
where
Lp(
a,
b,
c) and
Rp(
a,
b,
c) are the integral part of the related function defined with protein interactions on the ligand and the receptor, respectively. We can thus use
M forward and an inverse fast Fourier transform to calculate the expression efficiently, and the forward and inverse fast Fourier transform can be denoted by
FT and
IFT, respectively:
where
and
N3 are grid sizes of the three coordinates. If
, the complexity of this method is
O(
N3 log(
N3)). Then, we can use FFT to compute the related function of
Lp with the pre-calculated function of
Rp. The final sum offers the scoring values of function for all probable conversions of the ligands. Finally, we obtained and sorted the results from different rotations.
2.4. Singular Value Decomposition
It is a challenge for bioinformatics to explore effective methods for analyzing global gene expression data. Singular value decomposition (SVD) is a common technique for multivariate data analysis [
42]. We assumed that
M is the size of an
m ×
n matrix. The decomposition for SVD can be expressed as follows:
where
U means an
m ×
m unitary matrix,
S indicates a positive semi-definite
m ×
n diagonal matrix,
V represents an
n ×
n unitary matrix, and
V* is a conjugate transposed of
V. As a result, we can obtain the singular value of the matrix with proteins. The columns of
U form the base vector of orthogonal input or analysis for
M and are called left singular vectors. Rows of
V* form the base vector of orthogonal output for
M and are called right singular vectors. Thus, the diagonal element values of
S are singular.
2.5. Principal Component Analysis
Principal component analysis (PCA) is a data-dimensionally reduced method. PCA is widely used for data analysis, and the variable interacting with information from the dimensionally reduced dataset can persist [
43,
44]. It embeds samples in high-dimensional space into low-dimensional space, and the dimensionally reduced data represents the original data as closely as possible. The PCA of a data matrix determines the main information from a matrix according to a complementary group of scores and loading diagrams.
Furthermore, PCA converts primitive variables into a linear combination set, the principal components (PCs), which catch the data variables, are linearly independent, and are weighted in decreasing order of variance coverage [
45]. This can reduce the data dimension directly by discarding low variability characteristic elements. In this way, all original
α-dimensional datasets can be optimally implanted in a feature space with lower dimension.
The concept and calculation of PCA technology is simple. It can be expressed as follows: Given , where Aij denotes the feature value of the j-th sample with the i-th feature. Firstly, α-dimensional means that the full datasets with vector μj and α × α covariance matrix are calculated. Secondly, the feature vectors and feature values are calculated and sorted according to decreasing feature values. These feature vectors can be expressed as F1 with feature value λ1, as F2 with feature value λ2, and so on. The largest k feature vectors can then be obtained. This can be done by observing the frequency content of feature vectors. The maximum feature values are equivalent to the dimensions which is the large variance of the dataset. Finally, we can construct an α × k matrix X, whose rows denote the number of samples, and k makes up the feature vectors. Afterwards, the lower dimensional feature space’s number of k (k < α) was transformed by N = XTM(A). It is thus shown that the representation can minimize the square error criterion.
2.6. Random Projection Ensemble Classifier
Machine learning has been extensively applied in many fields. In mathematical statistics, RP is a method for reducing the dimensionality of a series of points that lie in Euclidean space. Compared with other methods, the RP method is simpler and has less output error. It has been successfully used in the reconstruction of dispersion signals, facial recognition, and textual and visual information retrieval. Now we introduce the RP algorithm in detail.
Let be a series of column vectors in primitive data space with high dimension. , n is the high dimension, and N denotes the number of columns. Dimensionality reduction embeds the vectors into a space Rq, which has lower dimension than Rn, where q << n. The output results are column vectors in the space with lower dimension.
where
q is close to the intrinsic dimensionality of
[
46,
47]. The embedded vectors refer to the vectors in the set
.
Here, if we want to employ the RP model to reduce the dimensionality of , a random vector set must be structured first, where . There are two choices in structuring the random basis:
- (1)
The vectors are spread on the -dimensional unit spherical surface.
- (2)
The components of conform to Bernoulli +1/+2 distribution and the vectors are standardized such that for .
We generated a
matrix
R, where
q consists of the vectors in
.
n was mentioned in the previous paragraph. The projecting result
can be obtained by
In our method, RP is used to construct a training set on which the classifiers will be trained. The use of RP lays the foundation for our ensemble model.
Now, we illustrate the theory of our ensemble method. A training set
is given in Equation (9). We generate a
G whose degree is
, and
G comes from the column vectors in
.
We then structure , whose size is , where and are introduced in the preceding paragraph, k means the size of ensemble classifiers. The columns are standardized so as to the norm is 1.
In the ensemble classifiers, we constructed the training sets
by means of projecting
G onto
.
They are then fed into the base classifier, and the outcomes are a group of classifiers . For the sake of classifying a fresh set with classifier , firstly, will be inlaid into the target space . It can be obtained by embedding into .
where
is the result of embedding. The classification of
can be garnered by
. In this ensemble algorithm, the RP ensemble classifier will use the classification result of all classifiers
of
to decide the final result with a majority voting scheme.
In this study, the 1000 coefficients were divided into 100 non-overlapping blocks. We chose the projection from a block of size 10 that obtained the smallest test error with the leave-one-out test error estimate. We used the k-Nearest Neighbor (KNN) as a base classifier, where k = seq(1, 25, by = 3). Prior probability of interaction pairs in the calculated training samples dataset was considered as the voting parameter.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}