


A Comparative Assessment of High-Throughput Quantitative Polymerase Chain Reaction versus Shotgun Metagenomic Sequencing in Sediment Resistome Profiling

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area and Sample Collection
2.2. DNA Isolation
2.3. SmartChipTM HT-qPCR Analysis
2.4. Shotgun Metagenomic Sequencing
2.5. Statistical Analysis
3. Results
3.1. HT-qPCR Analysis
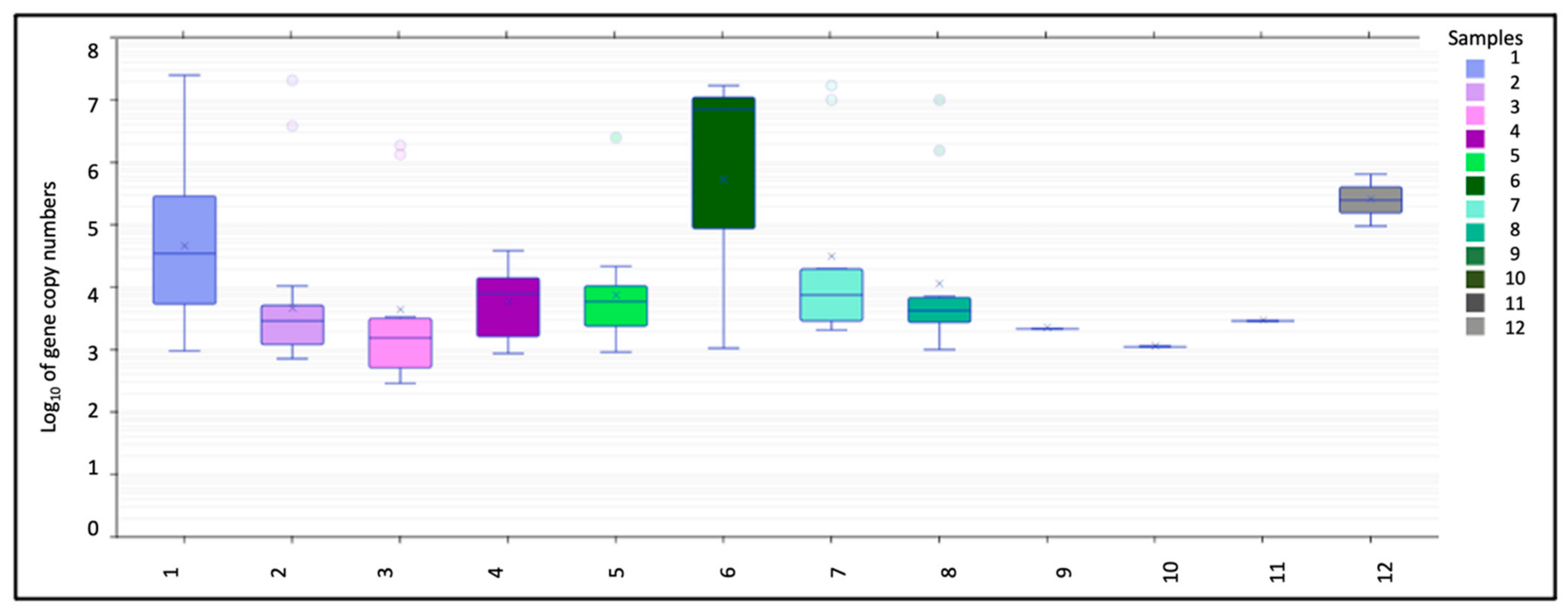
3.1.1. Bacterial Gene Copies
3.1.2. Antibiotic Resistance Genes and Drug Classes
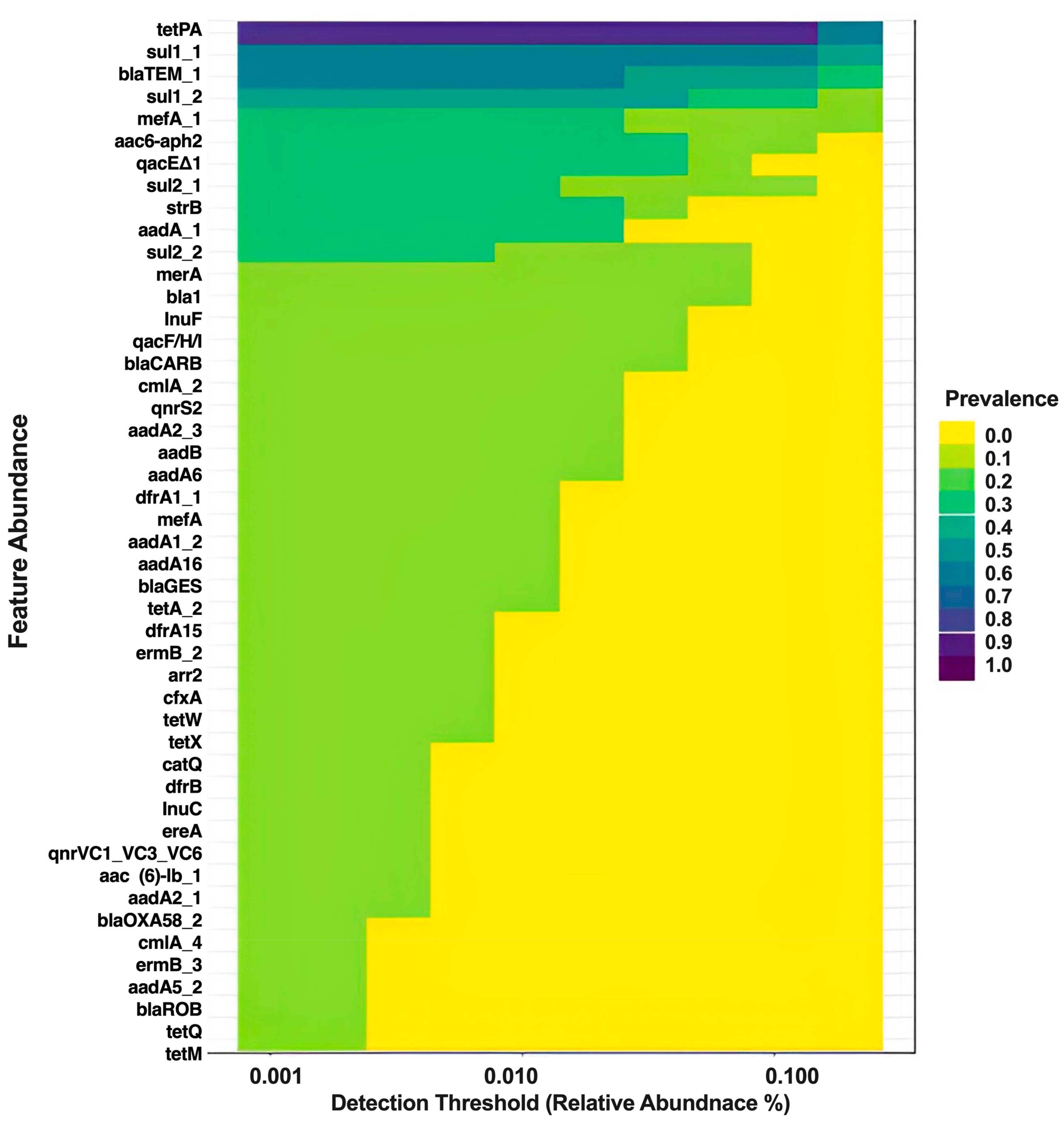
3.1.3. Core Resistome
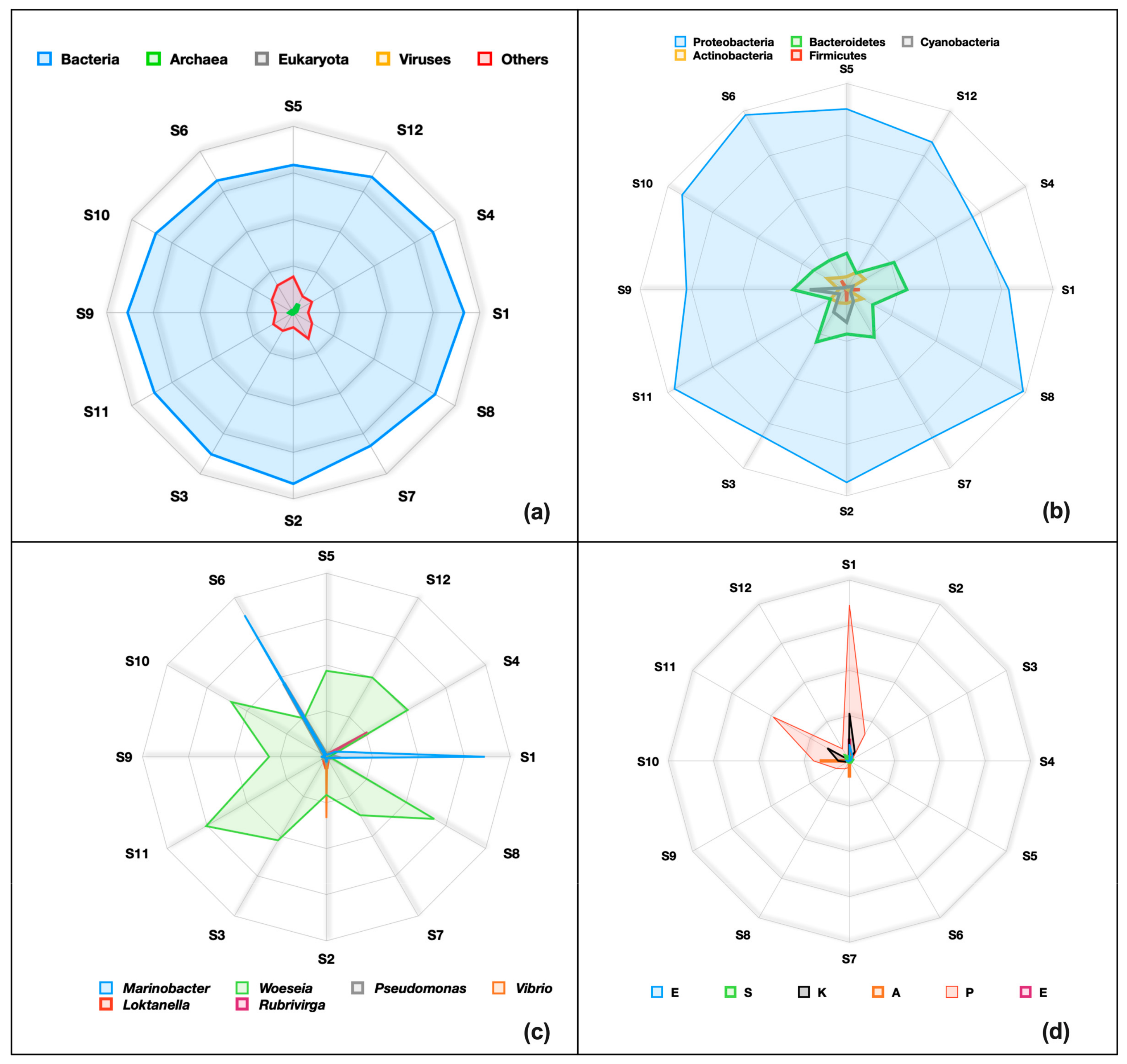
3.1.4. Taxonomic Profiles
3.2. Comparative Analysis of HT-qPCR with Shotgun Metagenomic Sequencing
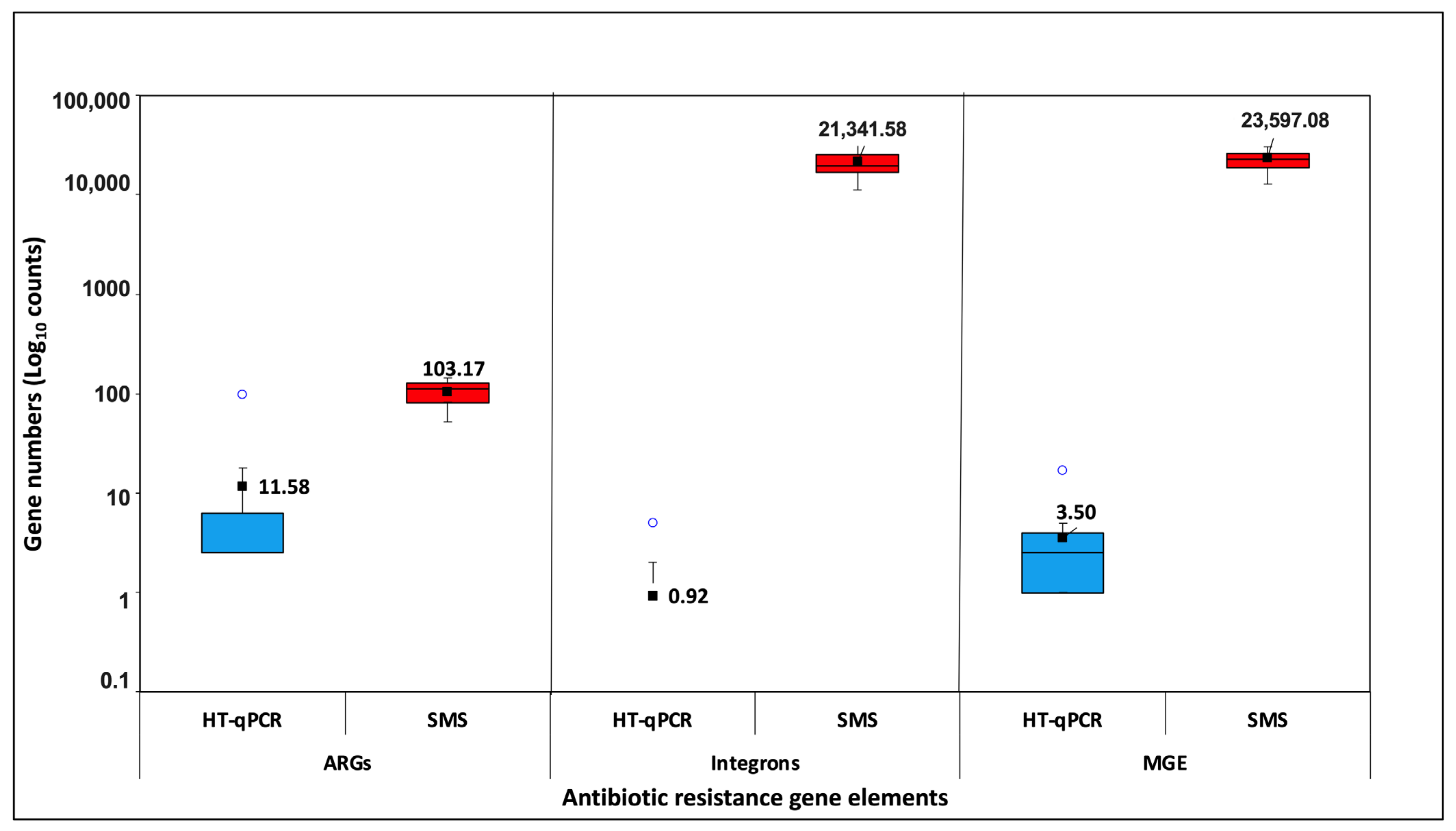
3.2.1. Gene Numbers/Richness
3.2.2. Core Resistomes
3.2.3. Drug Classes
3.2.4. Taxonomic Profiling via Shotgun Metagenomic Sequencing
4. Discussion
4.1. Target Genes and Primers in HT-qPCR
4.2. PCR Biases and Artefacts
4.3. Low Abundance
4.4. Costing and Skills
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, D.-W.; Cha, C.-J. Antibiotic resistome from the One-Health perspective: Understanding and controlling antimicrobial resistance transmission. Exp. Mol. Med. 2021, 53, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.; The staggering death toll of drug-resistant bacteria. Nature 2022. Available online: https://www.nature.com/articles/d41586-022-00228-x (accessed on 9 October 2023).
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Asokan, G.V.; Vanitha, A. WHO global priority pathogens list on antibiotic resistance: An urgent need for action to in tegrate One Health data. Perspect. Public Health 2018, 138, 87–88. [Google Scholar]
- Berendonk, T.U.; Manaia, C.M.; Merlin, C.; Fatta-Kassinos, D.; Cytryn, E.; Walsh, F.; Bürgmann, H.; Sørum, H.; Norström, M.; Pons, M.-N. Tackling antibiotic resistance: The environmental framework. Nat. Rev. Microbiol. 2015, 13, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Hernando-Amado, S.; Coque, T.M.; Baquero, F.; Martínez, J.L. Defining and combating antibiotic resistance from One Health and Global Health perspectives. Nat. Microbiol. 2019, 4, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Wermuth, H.R.; Calhoun, C.; Hall, G.A. Antibiotics. In Continuing Education Activity; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Pournajaf, A.; Ardebili, A.; Goudarzi, L.; Khodabandeh, M.; Narimani, T.; Abbaszadeh, H. PCR-based identification of methicillin–resistant Staphylococcus aureus strains and their antibiotic resistance profiles. Asian Pac. J. Trop. Biomed. 2014, 4, S293–S297. [Google Scholar] [CrossRef]
- Roe, C.C.; Vazquez, A.J.; Esposito, E.P.; Zarrilli, R.; Sahl, J.W. Diversity, virulence, and antimicrobial resistance in isolates from the newly emerging Klebsiella pneumoniae ST101 lineage. Front. Microbiol. 2019, 10, 542. [Google Scholar] [CrossRef]
- Hu, Y.; Zhu, K.; Jin, D.; Shen, W.; Liu, C.; Zhou, H.; Zhang, R. Evaluation of IR Biotyper for carbapenem-resistant Pseudomonas aeruginosa typing and its application potential for the investigation of nosocomial infection. Front. Microbiol. 2023, 14, 1068872. [Google Scholar] [CrossRef]
- Benveniste, R.; Davies, J. Aminoglycoside antibiotic-inactivating enzymes in actinomycetes similar to those present in clinical isolates of antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA 1973, 70, 2276–2280. [Google Scholar] [CrossRef]
- Ocheretina, O.; Escuyer, V.E.; Mabou, M.-M.; Royal-Mardi, G.; Collins, S.; Vilbrun, S.C.; Pape, J.W.; Fitzgerald, D.W. Correlation between genotypic and phenotypic testing for resistance to rifampin in Mycobacterium tuberculosis clinical isolates in Haiti: Investigation of cases with discrepant susceptibility results. PLoS ONE 2014, 9, e90569. [Google Scholar] [CrossRef]
- Ryu, S.-H.; Park, S.-G.; Choi, S.-M.; Hwang, Y.-O.; Ham, H.-J.; Kim, S.-U.; Lee, Y.-K.; Kim, M.-S.; Park, G.-Y.; Kim, K.-S. Antimicrobial resistance and resistance genes in Escherichia coli strains isolated from commercial fish and seafood. Int. J. Food Microbiol. 2012, 152, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Burakoff, A.; Brown, K.; Knutsen, J.; Hopewell, C.; Rowe, S.; Bennett, C.; Cronquist, A. Outbreak of fluoroquinolone-resistant Campylobacter jejuni infections associated with raw milk consumption from a herdshare dairy—Colorado, 2016. Morb. Mortal. Wkly. Rep. 2018, 67, 146. [Google Scholar] [CrossRef] [PubMed]
- Gambino, D.; Sciortino, S.; Migliore, S.; Galuppo, L.; Puleio, R.; Dara, S.; Vicari, D.; Seminara, S.; Gargano, V. Preliminary Results on the Prevalence of Salmonella spp. in Marine Animals Stranded in Sicilian Coasts: Antibiotic Susceptibility Profile and ARGs Detection in the Isolated Strains. Pathogens 2021, 10, 930. [Google Scholar] [CrossRef]
- Al-Sarawi, H.A.; Najem, A.B.; Lyons, B.P.; Uddin, S.; Al-Sarawi, M.A. Antimicrobial Resistance in Escherichia coli Isolated from Marine Sediment Samples from Kuwait Bay. Sustainability 2022, 14, 11325. [Google Scholar] [CrossRef]
- Koczura, R.; Mokracka, J.; Jabłońska, L.; Gozdecka, E.; Kubek, M.; Kaznowski, A. Antimicrobial resistance of integron-harboring Escherichia coli isolates from clinical samples, wastewater treatment plant and river water. Sci. Total Environ. 2012, 414, 680–685. [Google Scholar] [CrossRef]
- Kotlarska, E.; Łuczkiewicz, A.; Pisowacka, M.; Burzyński, A. Antibiotic resistance and prevalence of class 1 and 2 integrons in Escherichia coli isolated from two wastewater treatment plants, and their receiving waters (Gulf of Gdansk, Baltic Sea, Poland). Environ. Sci. Pollut. Res. 2015, 22, 2018–2030. [Google Scholar] [CrossRef]
- Perry, J.A.; Wright, G.D. The antibiotic resistance “mobilome”: Searching for the link between environment and clinic. Front. Microbiol. 2013, 4, 138. [Google Scholar] [CrossRef]
- Perry, J.A.; Wright, G.D. Forces shaping the antibiotic resistome. BioEssays 2014, 36, 1179–1184. [Google Scholar] [CrossRef]
- Wu, J.; Wang, J.; Li, Z.; Guo, S.; Li, K.; Xu, P.; Ok, Y.S.; Jones, D.L.; Zou, J. Antibiotics and antibiotic resistance genes in agricultural soils: A systematic analysis. Crit. Rev. Environ. Sci. Technol. 2023, 53, 847–864. [Google Scholar] [CrossRef]
- Qiao, M.; Ying, G.-G.; Singer, A.C.; Zhu, Y.-G. Review of antibiotic resistance in China and its environment. Environ. Int. 2018, 110, 160–172. [Google Scholar] [CrossRef]
- Mootapally, C.; Nathani, N.M.; Poriya, P.; Beleem, I.; Dabhi, J.C.; Gadhvi, I.R.; Joshi, C.G. Antibiotic Resistome Biomarkers associated to the pelagic Sediments of the Gulfs of Kathiawar peninsula and Arabian Sea. Sci. Rep. 2019, 9, 17281. [Google Scholar] [CrossRef] [PubMed]
- Nathani, N.M.; Mootapally, C.; Dave, B.P. Antibiotic resistance genes allied to the pelagic sediment microbiome in the Gulf of Khambhat and Arabian Sea. Sci. Total Environ. 2019, 653, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Waseem, H.; ur Rehman, H.S.; Ali, J.; Iqbal, M.J.; Ali, M.I. Global trends in ARGs measured by HT-qPCR platforms. In Antibiotics and Antimicrobial Resistance Genes in the Environment; Elsevier: Amsterdam, The Netherlands, 2020; pp. 206–222. [Google Scholar]
- Lai, F.Y.; Muziasari, W.; Virta, M.; Wiberg, K.; Ahrens, L. Profiles of environmental antibiotic resistomes in the urban aquatic recipients of Sweden using high-throughput quantitative PCR analysis. Environ. Pollut. 2021, 287, 117651. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, P.; Guo, Y.; Liu, L.; Yang, J. The impacts of different high-throughput profiling approaches on the understanding of bacterial antibiotic resistance genes in a freshwater reservoir. Sci. Total Environ. 2019, 693, 133585. [Google Scholar] [CrossRef]
- Waseem, H.; Jameel, S.; Ali, J.; Saleem Ur Rehman, H.; Tauseef, I.; Farooq, U.; Jamal, A.; Ali, M.I. Contributions and challenges of high throughput qPCR for determining antimicrobial resistance in the environment: A critical review. Molecules 2019, 24, 163. [Google Scholar] [CrossRef]
- Wu, D.; Jin, L.; Xie, J.; Liu, H.; Zhao, J.; Ye, D.; Li, X.-d. Inhalable antibiotic resistomes emitted from hospitals: Metagenomic insights into bacterial hosts, clinical relevance, and environmental risks. Microbiome 2022, 10, 19. [Google Scholar] [CrossRef]
- Li, X.; Wu, Z.; Dang, C.; Zhang, M.; Zhao, B.; Cheng, Z.; Chen, L.; Zhong, Z.; Ye, Y.; Xia, Y. A metagenomic-based method to study hospital air dust resistome. Chem. Eng. J. 2021, 406, 126854. [Google Scholar] [CrossRef]
- Kamathewatta, K.I.; Bushell, R.N.; Young, N.D.; Stevenson, M.A.; Billman-Jacobe, H.; Browning, G.F.; Marenda, M.S. Exploration of antibiotic resistance risks in a veterinary teaching hospital with Oxford Nanopore long read sequencing. PLoS ONE 2019, 14, e0217600. [Google Scholar] [CrossRef]
- Chng, K.R.; Li, C.; Bertrand, D.; Ng, A.H.Q.; Kwah, J.S.; Low, H.M.; Tong, C.; Natrajan, M.; Zhang, M.H.; Xu, L.; et al. Cartography of opportunistic pathogens and antibiotic resistance genes in a tertiary hospital environment. Nat. Med. 2020, 26, 941–951. [Google Scholar] [CrossRef]
- Cui, H.; Zhang, C.; Zhao, K.; Liu, J.; Pu, J.; Kong, Y.; Dong, S.; Chen, L.; Zhao, Y.; Chen, Y. Effects of different laying periods on airborne bacterial diversity and antibiotic resistance genes in layer hen houses. Int. J. Hyg. Environ. Health 2023, 251, 114173. [Google Scholar] [CrossRef]
- Li, L.; Wang, Q.; Bi, W.; Hou, J.; Xue, Y.; Mao, D.; Das, R.; Luo, Y.; Li, X. Municipal solid waste treatment system increases ambient airborne bacteria and antibiotic resistance genes. Environ. Sci. Technol. 2020, 54, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Jiang, L.; Bi, X.; Cheng, L.; Zheng, X.; Wang, X.; Zhou, X. Submicron aerosols share potential pathogens and antibiotic resistomes with wastewater or sludge. Sci. Total Environ. 2022, 821, 153521. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Jin, L.; Wu, D.; Pruden, A.; Li, X. Inhalable antibiotic resistome from wastewater treatment plants to urban areas: Bacterial hosts, dissemination risks, and source contributions. Environ. Sci. Technol. 2022, 56, 7040–7051. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Wu, Y.; Huang, W.; Wu, X.; Lv, J.; Liu, P.; Bu, L.; Bai, Z.; Chen, S.; Feng, W. Characteristics of and variation in airborne ARGs among urban hospitals and adjacent urban and suburban communities: A metagenomic approach. Environ. Int. 2020, 139, 105625. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Q.; Chen, Z.; Mao, D.; Luo, Y. Significant higher airborne antibiotic resistance genes and the associated inhalation risk in the indoor than the outdoor. Environ. Pollut. 2021, 268, 115620. [Google Scholar] [CrossRef] [PubMed]
- Lü, F.; Wang, W.; Hu, T.; Duan, H.; Shao, L.; Zhang, H.; He, P. Release of airborne antibiotic resistance genes from municipal solid waste transfer stations. Sustain. Environ. Res. 2022, 32, 28. [Google Scholar] [CrossRef]
- Yang, F.; Gao, Y.; Zhao, H.; Li, J.; Cheng, X.; Meng, L.; Dong, P.; Yang, H.; Chen, S.; Zhu, J. Revealing the distribution characteristics of antibiotic resistance genes and bacterial communities in animal-aerosol-human in a chicken farm: From One-Health perspective. Ecotoxicol. Environ. Saf. 2021, 224, 112687. [Google Scholar] [CrossRef]
- Gevao, B.; Uddin, S.; Krishnan, D.; Rajagopalan, S.; Habibi, N. Antibiotics in Wastewater: Baseline of the Influent and Effluent Streams in Kuwait. Toxics 2022, 10, 174. [Google Scholar] [CrossRef]
- Gevao, B.; Uddin, S.; Dupont, S. Baseline concentrations of pharmaceuticals in Kuwait’s coastal marine environment. Mar. Pollut. Bull. 2021, 173, 113040. [Google Scholar] [CrossRef]
- Habibi, N.; Uddin, S.; Lyons, B.; Al-Sarawi, H.A.; Behbehani, M.; Shajan, A.; Razzack, N.A.; Zakir, F.; Alam, F. Antibiotic Resistance Genes Associated with Marine Surface Sediments: A Baseline from the Shores of Kuwait. Sustainability 2022, 14, 8029. [Google Scholar] [CrossRef]
- Muziasari, W.I.; Pitkänen, L.K.; Sørum, H.; Stedtfeld, R.D.; Tiedje, J.M.; Virta, M. Corrigendum: The resistome of farmed fish feces contributes to the enrichment of antibiotic resistance genes in sediments below Baltic Sea fish farms. Front. Microbiol. 2017, 8, 1491. [Google Scholar] [CrossRef] [PubMed]
- Habibi, N.; Uddin, S.; Al-Sarawi, H.; Aldhameer, A.; Shajan, A.; Zakir, F.; Abdul Razzack, N.; Alam, F. Metagenomes from Coastal Sediments of Kuwait: Insights into the Microbiome, Metabolic Functions and Resistome. Microorganisms 2023, 11, 531. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, A.; Junges, R.; Chen, T.; Petersen, F.C. ResistoXplorer: A web-based tool for visual, statistical and exploratory data analysis of resistome data. NAR Genom. Bioinform. 2021, 3, lqab018. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. Statistics Kingdom. 2017. Available online: https://www.statskingdom.com (accessed on 17 January 2023).
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.-M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef]
- Doster, E.; Lakin, S.M.; Dean, C.J.; Wolfe, C.; Young, J.G.; Boucher, C.; Belk, K.E.; Noyes, N.R.; Morley, P.S. MEGARes 2.0: A database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Res. 2020, 48, D561–D569. [Google Scholar] [CrossRef]
- Carattoli, A.; Hasman, H. PlasmidFinder and in silico pMLST: Identification and typing of plasmid replicons in whole-genome sequencing (WGS). In Horizontal Gene Transfer; Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2020; pp. 285–294. [Google Scholar]
- Moura, A.; Soares, M.; Pereira, C.; Leitão, N.; Henriques, I.; Correia, A. INTEGRALL: A database and search engine for integrons, integrases and gene cassettes. Bioinformatics 2009, 25, 1096–1098. [Google Scholar] [CrossRef]
- Siguier, P.; Pérochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34 (Suppl. S1), D32–D36. [Google Scholar] [CrossRef]
- Acinas, S.G.; Sarma-Rupavtarm, R.; Klepac-Ceraj, V.; Polz, M.F. PCR-induced sequence artifacts and bias: Insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 2005, 71, 8966–8969. [Google Scholar] [CrossRef]
- Frostegård, Å.; Courtois, S.; Ramisse, V.; Clerc, S.; Bernillon, D.; Le Gall, F.; Jeannin, P.; Nesme, X.; Simonet, P. Quantification of bias related to the extraction of DNA directly from soils. Appl. Environ. Microbiol. 1999, 65, 5409–5420. [Google Scholar] [CrossRef] [PubMed]
- Steffan, R.J.; Goksøyr, J.; Bej, A.; Atlas, R. Recovery of DNA from soils and sediments. Appl. Environ. Microbiol. 1988, 54, 2908–2915. [Google Scholar] [CrossRef] [PubMed]
- Luna, G.M.; Dell’Anno, A.; Danovaro, R. DNA extraction procedure: A critical issue for bacterial diversity assessment in marine sediments. Environ. Microbiol. 2006, 8, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Habibi, N.; Behbehani, M.; Uddin, S.; Al-Salameen, F.; Shajan, A.; Zakir, F. A safe and effective sample collection method for assessment of SARS-CoV-2 in aerosol samples. In Environmental Resilience and Transformation in Times of COVID-19; Elsevier: Amsterdam, The Netherlands, 2021; pp. 173–178. [Google Scholar]
- García-Arroyo, L.; Prim, N.; Martí, N.; Roig, M.C.; Navarro, F.; Rabella, N. Benefits and drawbacks of molecular techniques for diagnosis of viral respiratory infections. Experience with two multiplex PCR assays. J. Med. Virol. 2016, 88, 45–50. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Habibi, N.; Uddin, S.; Behbehani, M.; Kishk, M.; Abdul Razzack, N.; Zakir, F.; Shajan, A. Antibiotic Resistance Genes in Aerosols: Baseline from Kuwait. Int. J. Mol. Sci. 2023, 24, 6756. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sulphonamides | Tetracycline | Beta-Lactam | Other | Aminoglycoside | Quinolone | MLSB | Trimethoprim | Phenicol | Integrons | MGEs |
|---|---|---|---|---|---|---|---|---|---|---|
| sul1_1 | tetPA | blaTEM_1 | merA | aac6-aph2 | qnrS2 | lnuF | dfrA1_1 | cmlA_2 | Intl_3 | tnpA_1 |
| sul1_2 | tetA_2 | bla1 | qacE∆1_3 | aadA6 | qnrVC1_VC3_VC6 | mefA_1 | dfrA15 | catQ | intlI_2 | tnpA_6 |
| sul2_1 | tetX | blaCARB | qacE∆1_1 | aadB | qnrS_1 | mefA | dfrB | cmlA_4 | intlI_1 | tnpA_2 |
| sul2_2 | tetW | blaGES | arr2 | aadA2_3 | qnrB_2 | ermB_2 | dfra17 | catB3 | intl3 | ISPps |
| sul4 | tetM | cfxA | crAss64 | aadA_1 | qnrB | ereA | dfrA27 | catB8 | Intl3_2 | ISI247_2 |
| tetQ | blaOXA58_2 | crAss56 | strB | qnrD | lnuC | dfrA12 | catB2 | tnpA_3 | ||
| tetG | blaROB | sat4 | aadA16 | ermB_3 | mdtL | IncP_oriT | ||||
| tetO_2 | blaVEB | mcr1 | aadA1_2 | mefB | floR_1 | tnpA_5 | ||||
| tet32 | cphA_1 | aadA2_1 | ermF | ISI247_1 | ||||||
| tet39 | blaMIR | aac(6’)-Ib_1 | erm42 | tnpA_4 | ||||||
| tet44 | cphA_2 | aadA5_2 | mphA | IS613 | ||||||
| tetL_2 | blaCMY_2 | aadA10 | lsaC | IncN_rep | ||||||
| tetE | blaSHV11 | aac(6’)-II | msrE | Tp614 | ||||||
| blaOXA10_1 | aacC2 | ISAba3 | ||||||||
| blaCMY_1 | aadA7 | IncQ_oriT | ||||||||
| blaMOX/blaCMY | aphA3_1 | Orf37- | ||||||||
| blaBEL-nonmobile | IS26 | |||||||||
| ampC_4 | Tn5 | |||||||||
| blaCMY2 | ||||||||||
| ampC_1 | ||||||||||
| ampC_6 | ||||||||||
| blaSHV_2 | ||||||||||
| blaACT_2 | ||||||||||
| blaPER_1 | ||||||||||
| blaACT |
| Gene | Site 1 | Site 2 | Site 3 | Site 4 | Site 5 | Site 6 | Site 7 | Site 8 | Site 9 | Site 10 | Site 11 | Site 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bacteroidetes | 0.23 | 0.44 | 0.19 | 0.37 | - | 1.27 | 0.13 | 0.84 | - | - | - | 1.99 |
| Firmicutes | 0.14 | 0.08 | 0.27 | 0.11 | 0.14 | 0.55 | 0.23 | 0.13 | - | - | - | 0.29 |
| Acinetobacter baumannii | - | - | - | - | - | - | - | - | - | - | - | - |
| Campylobacter | - | - | - | - | - | - | - | - | - | - | - | - |
| Enterococci | - | - | - | - | - | - | - | - | - | - | - | - |
| Klebsiella. pneumoniae | - | - | - | - | - | - | - | - | - | - | - | - |
| Psuedomonas aeruginosa | - | - | - | - | - | - | - | - | - | - | - | - |
| Staphylococci | - | - | - | - | - | - | - | - | - | - | - | - |
| 16S rRNA | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habibi, N.; Uddin, S.; Behbehani, M.; Al-Sarawi, H.A.; Kishk, M.; Al-Zakri, W.; AbdulRazzack, N.; Shajan, A.; Zakir, F. A Comparative Assessment of High-Throughput Quantitative Polymerase Chain Reaction versus Shotgun Metagenomic Sequencing in Sediment Resistome Profiling. Appl. Sci. 2023, 13, 11229. https://doi.org/10.3390/app132011229
Habibi N, Uddin S, Behbehani M, Al-Sarawi HA, Kishk M, Al-Zakri W, AbdulRazzack N, Shajan A, Zakir F. A Comparative Assessment of High-Throughput Quantitative Polymerase Chain Reaction versus Shotgun Metagenomic Sequencing in Sediment Resistome Profiling. Applied Sciences. 2023; 13(20):11229. https://doi.org/10.3390/app132011229
Chicago/Turabian StyleHabibi, Nazima, Saif Uddin, Montaha Behbehani, Hanan A. Al-Sarawi, Mohamed Kishk, Waleed Al-Zakri, Nasreem AbdulRazzack, Anisha Shajan, and Farhana Zakir. 2023. "A Comparative Assessment of High-Throughput Quantitative Polymerase Chain Reaction versus Shotgun Metagenomic Sequencing in Sediment Resistome Profiling" Applied Sciences 13, no. 20: 11229. https://doi.org/10.3390/app132011229