New Insights into Development of Transglutaminase 2 Inhibitors as Pharmaceutical Lead Compounds

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Prospective Benefits of Therapeutic Approach through TGase 2 Inhibition In Clinical Side
1.2. Is the Active Site of TGase 2 the Unique Target for Inhibition of Enzyme Activity?
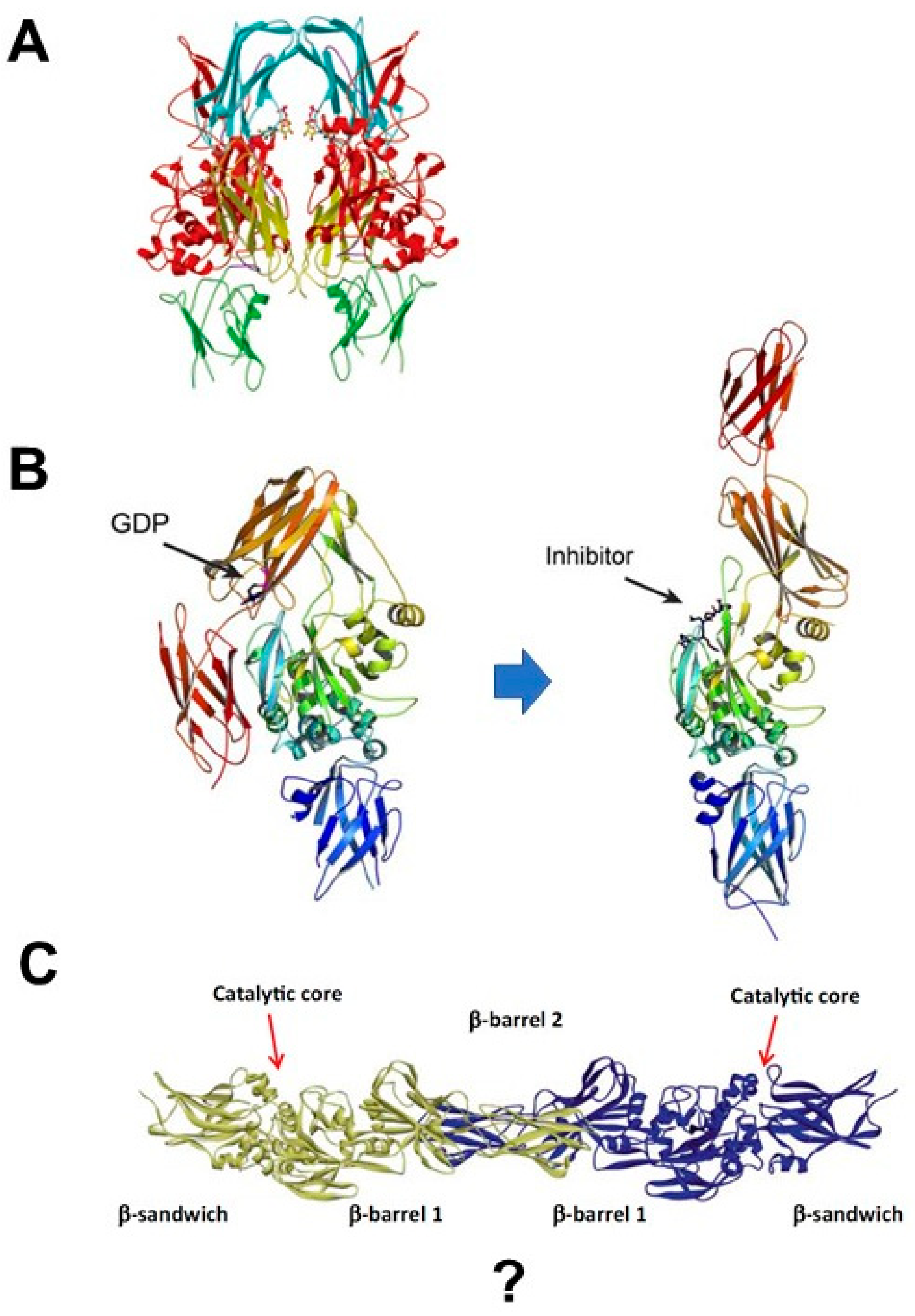
1.3. Is TGase 2 active as a Monomer?
1.4. Does TGase 2 Change Conformation by Activation?
2. Dynamics of TGase 2 Conformation and Activity
2.1. Proposal: The TGase 2 Dimer Is the Active Enzyme
2.2. TGase 2 Dimer Formation and Activity
2.3. Multiple Complexes of TGase 2 in the ECM
2.4. Triple Complex of TGase 2 in Cancer
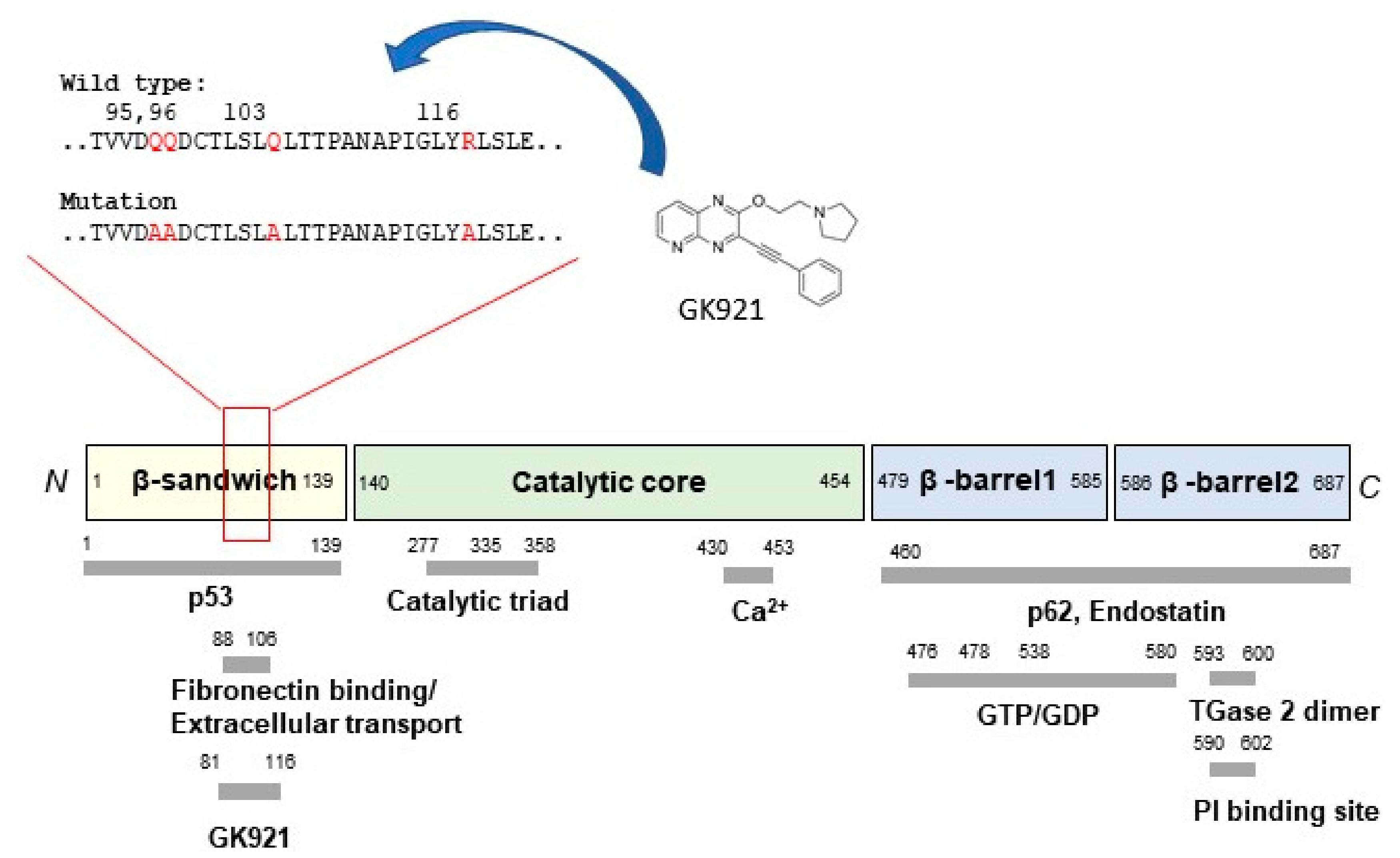
3. Discovery of Allosteric Site of TGase 2
4. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef] [PubMed]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Lentini, A.; Abbruzzese, A.; Caraglia, M.; Marra, M.; Beninati, S. Protein-polyamine conjugation by transglutaminase in cancer cell differentiation: Review article. Amino Acids 2004, 26, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Balklava, Z.; Verderio, E.; Collighan, R.; Gross, S.; Adams, J.; Griffin, M. Analysis of tissue transglutaminase function in the migration of swiss 3T3 fibroblasts: The active-state conformation of the enzyme does not affect cell motility but is important for its secretion. J. Biol. Chem. 2002, 277, 16567–16575. [Google Scholar] [CrossRef] [PubMed]
- Verderio, E.A.; Johnson, T.; Griffin, M. Tissue transglutaminase in normal and abnormal wound healing: Review article. Amino Acids 2004, 26, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Nanda, N.; Iismaa, S.E.; Owens, W.A.; Husain, A.; Mackay, F.; Graham, R.M. Targeted inactivation of Gh/tissue transglutaminase II. J. Biol. Chem. 2001, 276, 20673–20678. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Sarang, Z.; Molnar, P.; Nemeth, T.; Piacentini, M.; Mastroberardino, P.G.; Falasca, L.; Aeschlimann, D.; Kovacs, J.; Kiss, I.; et al. Transglutaminase 2−/− mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 7812–7817. [Google Scholar] [CrossRef] [PubMed]
- Bernassola, F.; Federici, M.; Corazzari, M.; Terrinoni, A.; Hribal, M.L.; De Laurenzi, V.; Ranalli, M.; Massa, O.; Sesti, G.; McLean, W.H.; et al. Role of transglutaminase 2 in glucose tolerance: Knockout mice studies and a putative mutation in a mody patient. FASEB J. 2002, 16, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Kerr, C. Transglutaminase is a tumor cell and cancer stem cell survival factor. Mol. Carcinog. 2015, 54, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Cerione, R.A.; Antonyak, M. Tissue transglutaminase and its role in human cancer progression. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 78, 247–293. [Google Scholar] [PubMed]
- Mehta, K. Biological and therapeutic significance of tissue transglutaminase in pancreatic cancer. Amino Acids 2009, 36, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Jeitner, T.M.; Steinert, P.M. Transglutaminases in disease. Neurochem. Int. 2002, 40, 85–103. [Google Scholar] [CrossRef]
- Muma, N.A. Transglutaminase is linked to neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Ronchetti, R.D.; Quezado, M.M.; Kim, S.Y.; Mouradian, M.M. Tissue transglutaminase-induced aggregation of α-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 2003, 100, 2047–2052. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y. Transglutaminase 2: A new paradigm for NF-κB involvement in disease. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 78, 161–195. [Google Scholar] [PubMed]
- Lee, J.; Kim, Y.S.; Choi, D.H.; Bang, M.S.; Han, T.R.; Joh, T.H.; Kim, S.Y. Transglutaminase 2 induces nuclear factor-κB activation via a novel pathway in BV-2 microglia. J. Biol. Chem. 2004, 279, 53725–53735. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Choi, Y.B.; Han, B.G.; Park, S.Y.; Jeon, Y.; Kim, D.H.; Ahn, E.R.; Shin, J.E.; Lee, B.I.; Lee, H.; et al. Cancer cells promote survival through depletion of the von Hippel–Lindau tumor suppressor by protein crosslinking. Oncogene 2011, 30, 4780–4790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, B.M.; Kim, D.S.; Kim, K.H.; Yoo, B.C.; Kim, S.H.; Gong, Y.D.; Kim, S.Y. Transglutaminase 2 inhibition found to induce p53 mediated apoptosis in renal cell carcinoma. FASEB J. 2013, 27, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Hwang, H.; Im, C.Y.; Kim, S.Y. Recent progress in the development of transglutaminase 2 (TGase2) inhibitors. J. Med. Chem. 2017, 60, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Nyabam, S.; Wang, Z.; Thibault, T.; Oluseyi, A.; Basar, R.; Marshall, L.; Griffin, M. A novel regulatory role for tissue transglutaminase in epithelial-mesenchymal transition in cystic fibrosis. Biochim. Biophys. Acta 2016, 1863, 2234–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannaccone, M.; Serretiello, E.; De Vivo, G.; Martin, A.; Stefanile, A.; Titta, F.; Gentile, V. Transglutaminase inhibition as a possible therapeutical approach to protect cells from death in neurodegenerative diseases. Recent Pat. CNS Drug Discov. 2013, 8, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Borrell-Pages, M.; Canals, J.M.; Cordelieres, F.P.; Parker, J.A.; Pineda, J.R.; Grange, G.; Bryson, E.A.; Guillermier, M.; Hirsch, E.; Hantraye, P.; et al. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J. Clin. Investig. 2006, 116, 1410–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prime, M.E.; Andersen, O.A.; Barker, J.J.; Brooks, M.A.; Cheng, R.K.; Toogood-Johnson, I.; Courtney, S.M.; Brookfield, F.A.; Yarnold, C.J.; Marston, R.W.; et al. Discovery and structure–activity relationship of potent and selective covalent inhibitors of transglutaminase 2 for Huntington’s disease. J. Med. Chem. 2012, 55, 1021–1046. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kang, J.H.; Lee, W.K.; Kim, S.G.; Lee, J.S.; Lee, S.H.; Park, J.B.; Kim, K.H.; Gong, Y.D.; Hwang, K.Y.; et al. Allosteric inhibition site of transglutaminase 2 is unveiled in the N terminus. Amino Acids 2018, 50, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Kim, S.J.; Kim, N.; Hong, D.; Choi, Y.B.; Lee, S.H.; Gong, Y.D.; Kim, S.Y. Transglutaminase 2 inhibitor abrogates renal cell carcinoma in xenograft models. J. Cancer Res. Clin. Oncol. 2014, 140, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.; McNeil, N.M.R.; Albert, M.R.; Ta, V.; Adhikary, G.; Bourgeois, K.; Eckert, R.L.; Keillor, J.W. Structure–activity relationships of potent, targeted covalent inhibitors that abolish both the transamidation and GTP binding activities of human tissue transglutaminase. J. Med. Chem. 2017, 60, 7910–7927. [Google Scholar] [CrossRef] [PubMed]
- Wodtke, R.; Hauser, C.; Ruiz-Gomez, G.; Jackel, E.; Bauer, D.; Lohse, M.; Wong, A.; Pufe, J.; Ludwig, F.A.; Fischer, S.; et al. Nε-acryloyllysine piperazides as irreversible inhibitors of transglutaminase 2: Synthesis, structure-activity relationships, and pharmacokinetic profiling. J. Med. Chem. 2018, 61, 4528–4560. [Google Scholar] [CrossRef] [PubMed]
- Lortat-Jacob, H.; Burhan, I.; Scarpellini, A.; Thomas, A.; Imberty, A.; Vives, R.R.; Johnson, T.; Gutierrez, A.; Verderio, E.A. Transglutaminase-2 interaction with heparin: Identification of a heparin binding site that regulates cell adhesion to fibronectin-transglutaminase-2 matrix. J. Biol. Chem. 2012, 287, 18005–18017. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Lee, W.K.; Lee, S.H.; Jin, K.S.; Kim, K.H.; Lee, Y.; Song, M.; Kim, S.Y. Inter-molecular crosslinking activity is engendered by the dimeric form of transglutaminase 2. Amino Acids 2017, 49, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Lee, J.S.; Hong, D.; Lee, S.H.; Kim, N.; Lee, W.K.; Sung, T.W.; Gong, Y.D.; Kim, S.Y. Renal cell carcinoma escapes death by p53 depletion through transglutaminase 2-chaperoned autophagy. Cell Death Dis. 2016, 7, e2163. [Google Scholar] [CrossRef] [PubMed]
- Faye, C.; Inforzato, A.; Bignon, M.; Hartmann, D.J.; Muller, L.; Ballut, L.; Olsen, B.R.; Day, A.J.; Ricard-Blum, S. Transglutaminase-2: A new endostatin partner in the extracellular matrix of endothelial cells. Biochem. J. 2010, 427, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.Y.; Streets, A.J.; Watson, P.F.; Huang, L.; Verderio, E.A.; Johnson, T.S. A crucial sequence for transglutaminase type 2 extracellular trafficking in renal tubular epithelial cells lies in its N-terminal β-sandwich domain. J. Biol. Chem. 2011, 286, 27825–27835. [Google Scholar] [CrossRef] [PubMed]
- Laki, K.; Lorand, L. On the solubility of fibrin clots. Science 1948, 108, 280. [Google Scholar] [CrossRef] [PubMed]
- Folk, J.E.; Cole, P.W. Mechanism of action of guinea pig liver transglutaminase. I. Purification and properties of the enzyme: Identification of a functional cysteine essential for activity. J. Biol. Chem. 1966, 241, 5518–5525. [Google Scholar] [PubMed]
- Folk, J.E.; Cole, P.W. Structural requirements of specific substrates for guinea pig liver transglutaminase. J. Biol. Chem. 1965, 240, 2951–2960. [Google Scholar] [PubMed]
- Sarkar, N.K.; Clarke, D.D.; Waelsch, H. An enzymically catalyzed incorporation of amines into proteins. Biochim. Biophys. Acta 1957, 25, 451–452. [Google Scholar] [CrossRef]
- Komaromi, I.; Bagoly, Z.; Muszbek, L. Factor XIII: Novel structural and functional aspects. J. Thromb. Haemost. 2011, 9, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Ishikawa, K.; Yokoyama, K.; Ohtsuka, T.; Nio, N.; Suzuki, E. Crystal structure of red sea bream transglutaminase. J. Biol. Chem. 2001, 276, 12055–12059. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cerione, R.A.; Clardy, J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc. Natl. Acad. Sci. USA 2002, 99, 2743–2747. [Google Scholar] [CrossRef] [PubMed]
- Pinkas, D.M.; Strop, P.; Brunger, A.T.; Khosla, C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007, 5, e327. [Google Scholar] [CrossRef] [PubMed]
- Birckbichler, P.J.; Orr, G.R.; Carter, H.A.; Patterson, M.K., Jr. Catalytic formation of ε-(γ-glutamyl)lysine in guinea pig liver transglutaminase. Biochem. Biophys. Res. Commun. 1977, 78, 1–7. [Google Scholar] [CrossRef]
- Akimov, S.S.; Krylov, D.; Fleischman, L.F.; Belkin, A.M. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 2000, 148, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Lee, S.H.; Kim, S.Y. Discovery of a novel target for renal cell carcinoma: Transglutaminase 2. Cell Death Dis. 2016, 7, e2200. [Google Scholar] [CrossRef] [PubMed]
- Han, B.G.; Cho, J.W.; Cho, Y.D.; Jeong, K.C.; Kim, S.Y.; Lee, B.I. Crystal structure of human transglutaminase 2 in complex with adenosine triphosphate. Int. J. Biol. Macromol. 2010, 47, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Keillor, J.W.; Apperley, K.Y.; Akbar, A. Inhibitors of tissue transglutaminase. Trends Pharmacol. Sci. 2015, 36, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Csősz, É.; Meskó, B.; Fésüs, L. Transdab wiki: The interactive transglutaminase substrate database on web 2.0 surface. Amino acids 2009, 36, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Spraggon, G.; Everse, S.J.; Doolittle, R.F. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature 1997, 389, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Francis, C.W.; Nagaswami, C.; Marder, V.J. Determination of the topology of factor XIIIa-induced fibrin γ-chain cross-links by electron microscopy of ligated fragments. J. Biol. Chem. 1993, 268, 26618–26624. [Google Scholar] [PubMed]
- Purves, L.; Purves, M.; Brandt, W. Cleavage of fibrin-derived D-dimer into monomers by endopeptidase from puff adder venom (Bitis arietans) acting at cross-linked sites of the γ-chain. Sequence of carboxy-terminal cyanogen bromide γ-chain fragments. Biochemistry 1987, 26, 4640–4646. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Stamnaes, J.; Iversen, R.; du Pre, M.F.; Chen, X.; Sollid, L.M. Enhanced B-cell receptor recognition of the autoantigen transglutaminase 2 by efficient catalytic self-multimerization. PLoS ONE 2015, 10, e0134922. [Google Scholar] [CrossRef] [PubMed]
- Barsigian, C.; Stern, A.M.; Martinez, J. Tissue (type II) transglutaminase covalently incorporates itself, fibrinogen, or fibronectin into high molecular weight complexes on the extracellular surface of isolated hepatocytes. Use of 2-[(2-oxopropyl)thio] imidazolium derivatives as cellular transglutaminase inactivators. J. Biol. Chem. 1991, 266, 22501–22509. [Google Scholar] [PubMed]
- Park, S.S.; Kim, D.S.; Park, K.S.; Song, H.J.; Kim, S.Y. Proteomic analysis of high-molecular-weight protein polymers in a doxorubicin-resistant breast-cancer cell line. Proteom. Clin. Appl. 2007, 1, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Shirley, B.A.; McNutt, M.; Gajiwala, K. Forces contributing to the conformational stability of proteins. FASEB J. 1996, 10, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Zemskov, E.A.; Mikhailenko, I.; Hsia, R.C.; Zaritskaya, L.; Belkin, A.M. Unconventional secretion of tissue transglutaminase involves phospholipid-dependent delivery into recycling endosomes. PLoS ONE 2011, 6, e19414. [Google Scholar] [CrossRef] [PubMed]
- Antonyak, M.A.; Li, B.; Boroughs, L.K.; Johnson, J.L.; Druso, J.E.; Bryant, K.L.; Holowka, D.A.; Cerione, R.A. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4852–4857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkin, A.M. Extracellular TG2: Emerging functions and regulation. FEBS J. 2011, 278, 4704–4716. [Google Scholar] [CrossRef] [PubMed]
- Zemskov, E.A.; Janiak, A.; Hang, J.; Waghray, A.; Belkin, A.M. The role of tissue transglutaminase in cell-matrix interactions. Front. Biosci. 2006, 11, 1057–1076. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Lee, C.H.; Lee, S.H.; Kim, S.Y. Increased expression of transglutaminase 2 drives glycolytic metabolism in renal carcinoma cells. Amino Acids 2014, 46, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, M.; Chen, D.; Gao, W.; Guan, J.L.; Komatsu, M.; Yin, X.M. Autophagy induced by calcium phosphate precipitates involves endoplasmic reticulum membranes in autophagosome biogenesis. PLoS ONE 2012, 7, e52347. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Oh, Y.T.; Kim, J.Y.; Kim, S.S.; Choi, E.; Kim, T.H.; Hong, J.H.; Chang, N.; Cho, H.J.; Sa, J.K.; et al. Transglutaminase 2 inhibition reverses mesenchymal transdifferentiation of glioma stem cells by regulating C/EBPβ signaling. Cancer Res. 2017, 77, 4973–4984. [Google Scholar] [CrossRef] [PubMed]
- Hang, J.; Zemskov, E.A.; Lorand, L.; Belkin, A.M. Identification of a novel recognition sequence for fibronectin within the NH2-terminal β-sandwich domain of tissue transglutaminase. J. Biol. Chem. 2005, 280, 23675–23683. [Google Scholar] [CrossRef] [PubMed]
- Iversen, R.; Mysling, S.; Hnida, K.; Jorgensen, T.J.; Sollid, L.M. Activity-regulating structural changes and autoantibody epitopes in transglutaminase 2 assessed by hydrogen/deuterium exchange. Proc. Natl. Acad. Sci. USA 2014, 111, 17146–17151. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-Y. New Insights into Development of Transglutaminase 2 Inhibitors as Pharmaceutical Lead Compounds. Med. Sci. 2018, 6, 87. https://doi.org/10.3390/medsci6040087
Kim S-Y. New Insights into Development of Transglutaminase 2 Inhibitors as Pharmaceutical Lead Compounds. Medical Sciences. 2018; 6(4):87. https://doi.org/10.3390/medsci6040087
Chicago/Turabian StyleKim, Soo-Youl. 2018. "New Insights into Development of Transglutaminase 2 Inhibitors as Pharmaceutical Lead Compounds" Medical Sciences 6, no. 4: 87. https://doi.org/10.3390/medsci6040087
APA StyleKim, S.-Y. (2018). New Insights into Development of Transglutaminase 2 Inhibitors as Pharmaceutical Lead Compounds. Medical Sciences, 6(4), 87. https://doi.org/10.3390/medsci6040087