
Sorption of Lithium on Bentonite, Kaolin and Zeolite

Abstract
:1. Introduction
2. Materials and Methods
2.1. Clay Minerals and Zeolite Used

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Material | Mineral Composition | |
|---|---|---|
| Mineral | % by Weight | |
| Bentonite (from Morocco, IBECCO) Obtained from the Federal Institute for Geosciences and Natural Resources (BGR) in Hannover (Germany). | Montmorillonite | 80.3 |
| Plagioclase | 12.1 | |
| Orthoclase | 5.5 | |
| Cristobalite | 5.5 | |
| Quartz | 0.9 | |
| Kaolin (from Ukraine) Supplied from the Ukrainian Kaolin Company. | Kaolinite | 87 |
| Quartz | 3 | |
| Potassium feldspar | 3 | |
| Mixed layer | 7 | |
| Zeolite (from Turkey) Supplied from the company ZEOnatura. | Clinoptilolite | 88–95 |
| Feldspars | 3–5 | |
| Montmorillonite | 2–5 | |
| Muscovite | 0–3 | |
| Cristobalite | 0–2 | |
2.2. Salt Solutions Used
2.3. Desorption and Leaching of Li from the Minerals
2.4. Sorption Experiments
2.5. Li Analysis with ICP-MS
3. Results
3.1. Li Release from the Untreated Solids
| Solid | Li Release (µeq/100 G) By Agitation With | |||||
|---|---|---|---|---|---|---|
| 0.1 M HNO3 | 1M HNO3 | NaCl at pH 6 | NaCl at pH 8 | |||
| Min. Release | Max. Release | Min. Release | Max. Release | |||
| Bentonite | 4.7 ± 0.5 | 4.8 ± 0.5 | 1.4 ± 0.1 | 50 ±5 | 0.5 ± 0.05 | 93 ± 9 |
| Kaolin | 2.5 ± 0.3 | 1.5 ± 0.2 | 0.4 ± 0.04 | 19 ± 2 | 4.3 ± 0.4 | 7.8 ± 0.8 |
| Zeolite | 11.5 ± 1.2 | 18.3 ± 1.8 | 7.7 ± 0.8 | 45 ± 5 | 6.5 ± 0.7 | 31 ± 3 |
3.2. Absolute Li Sorption
| - 55 ± 6 meq/100 g | i.e., 3800 ± 380 ppm | for bentonite |
| - 18 ± 1 meq/100 g | i.e., 1300 ± 130 ppm | for kaolin |
| - 56 ± 6 meq/100 g | i.e., 3900 ± 390 ppm | for zeolite |
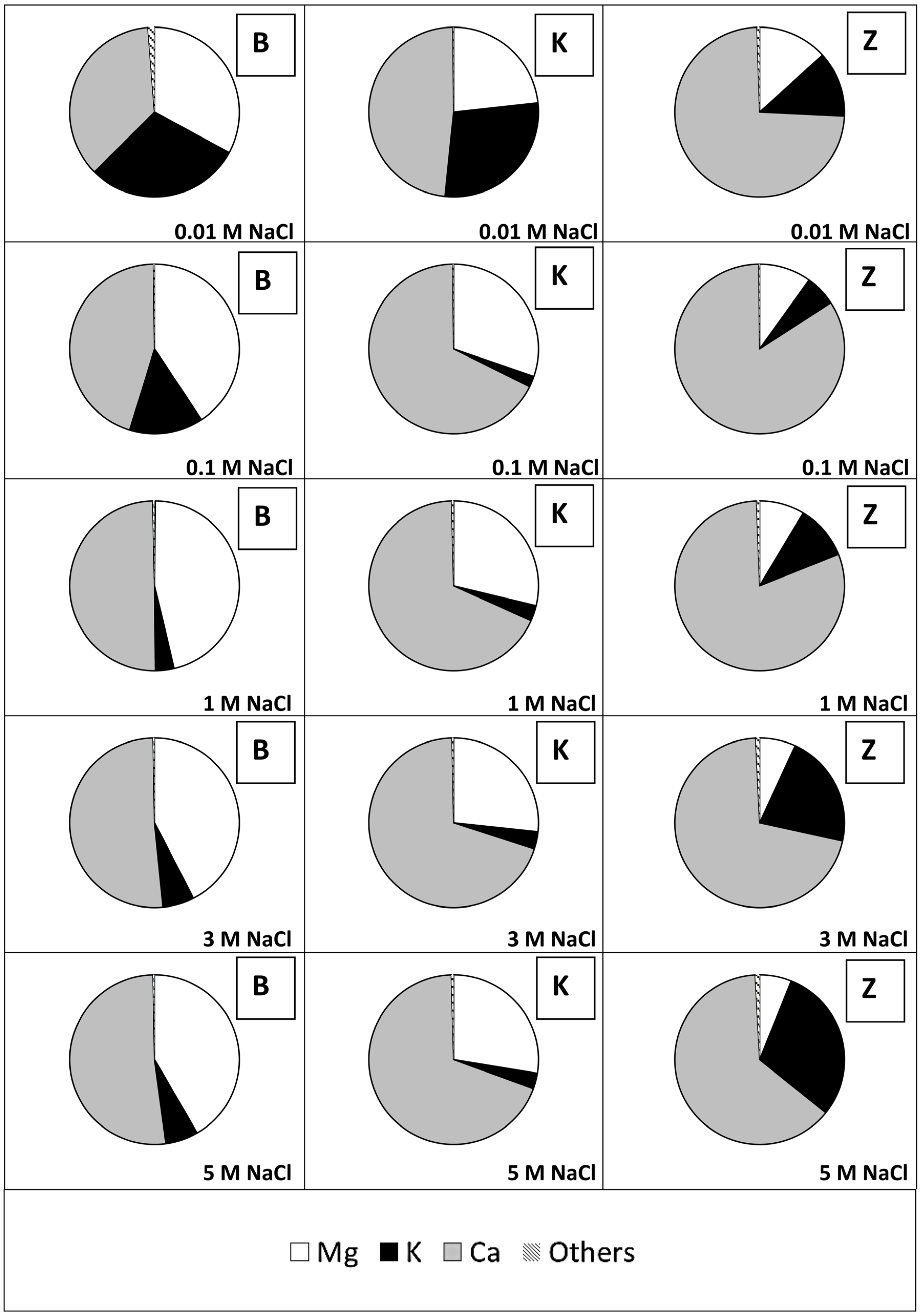
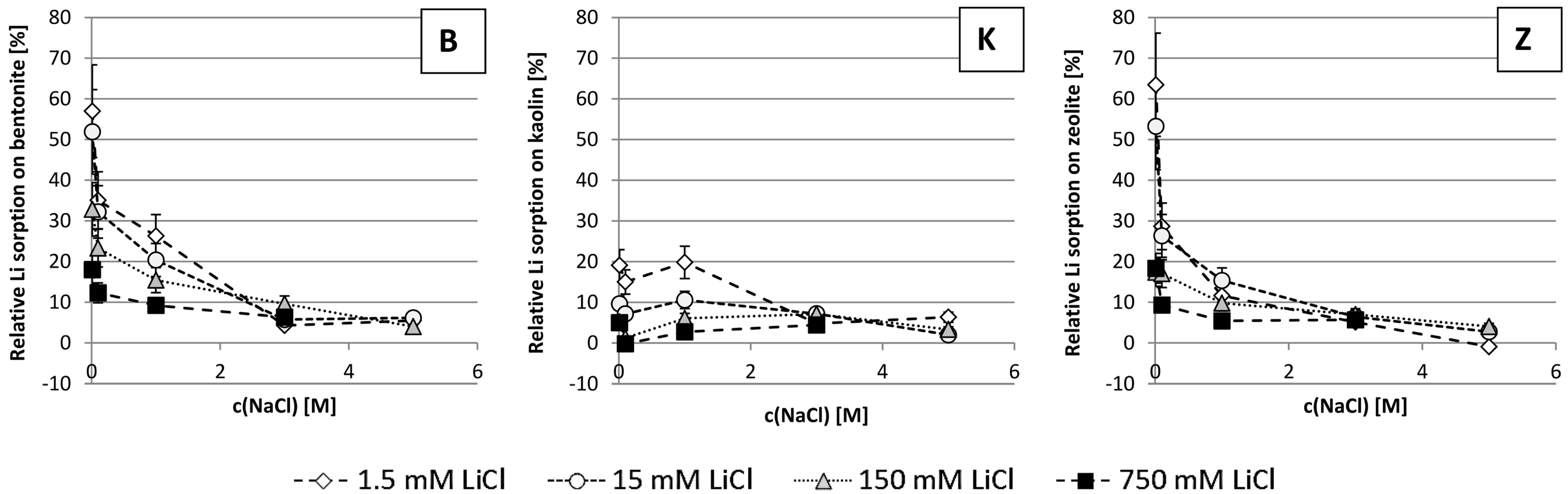
3.3. Relative Li Sorption
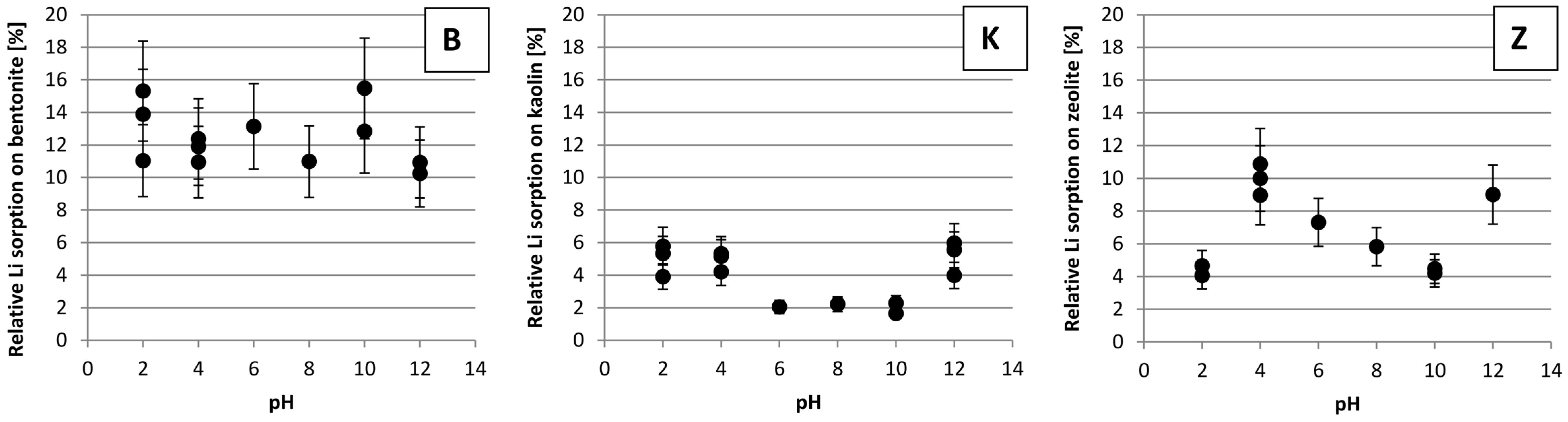
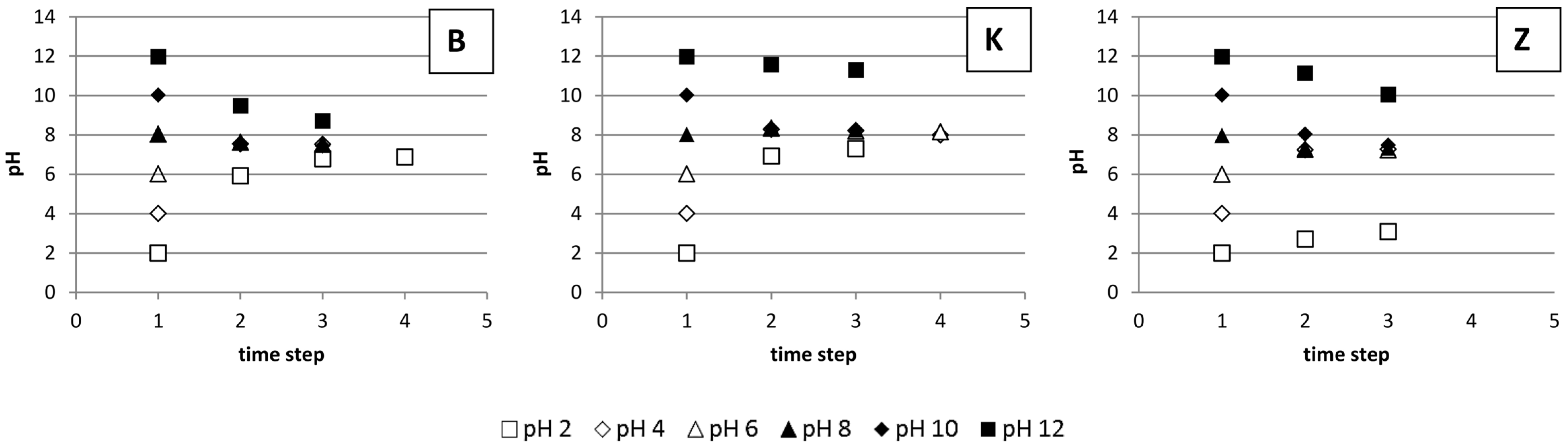
3.4. The Impact of pH on Li Sorption

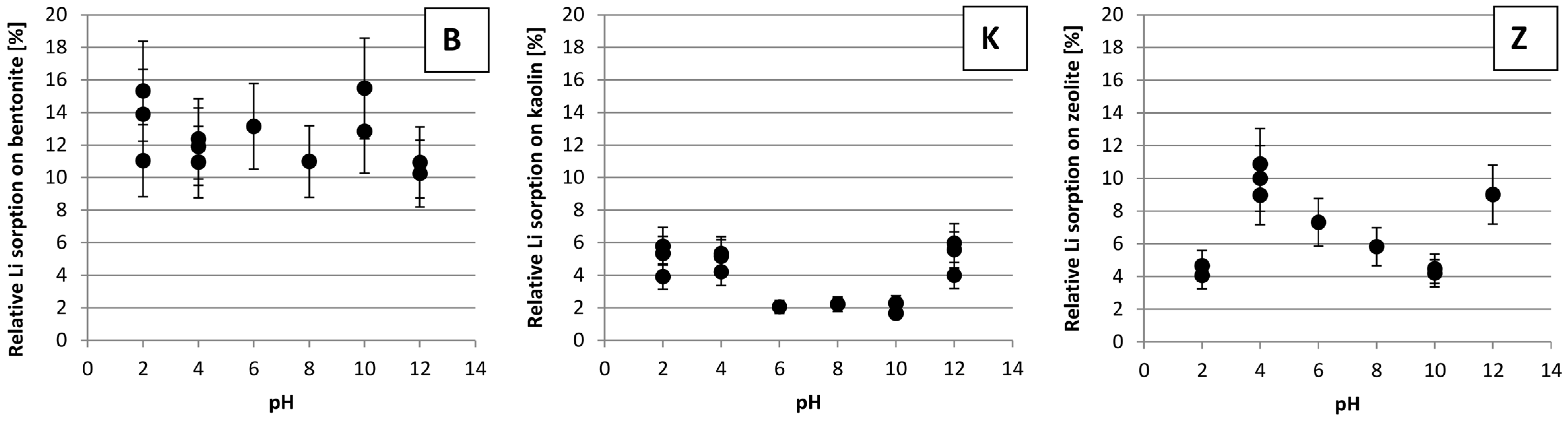
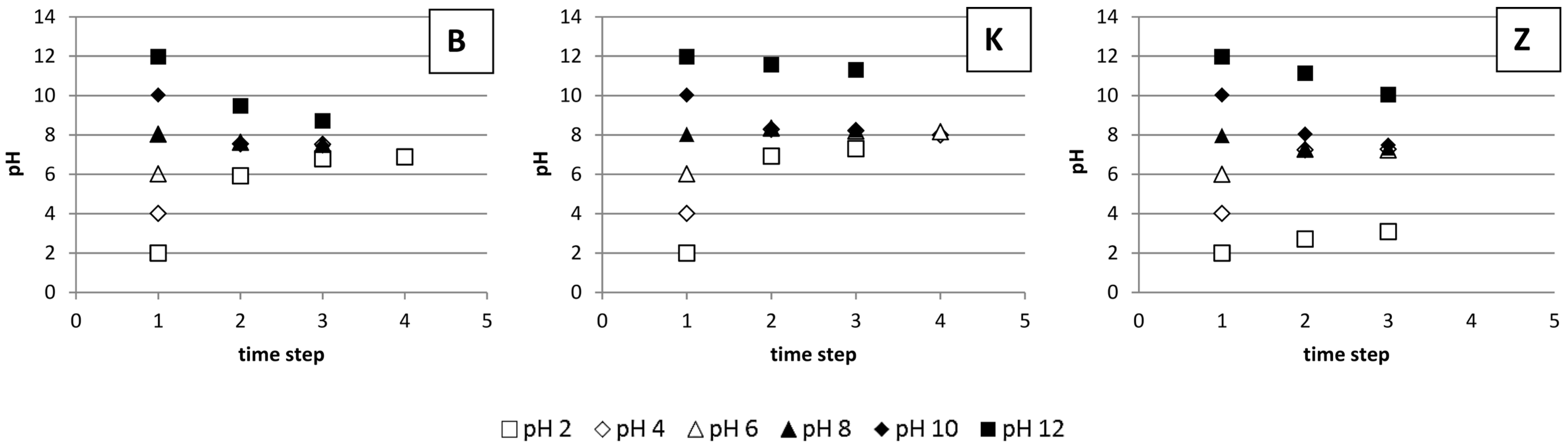
4. Discussion
4.1. Li Release from the Untreated Solids
4.2. Absolute Li Sorption
- -
- 70–100 meq/100 g for smectites (the mineral group to which montmorillonite belongs)
- -
- 3–15 meq/100 g for kaolinite (the main constituent of kaolin)
- -
- 220–620 meq/100 g for zeolites (depending on the type of zeolite)
4.3. Relative Li Sorption
- -
- Mineral composition (especially Ca, Mg, K content)
- -
- Structure of the minerals and how and where Ca, Mg and K are bound
- -
- The valency and consequently the binding strength of Ca, Mg and K
- -
- The ionic strength of the solution
4.4. The Impact of pH on Li Sorption
5. Summary and Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Stumm, W.; Morgan, J.J. Aquatic Chemistry; Wiley: New York, NY, USA, 1996. [Google Scholar]
- Garrett, D.E. Handbook of Lithium and Natural Calcium Chloride; Elsevier: Amsterdam, The Netherlands, 2004. [Google Scholar]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements; Elsevier Butterworth-Heinemann: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Cannon, H.L.; Harms, T.F.; Hamilton, J.C. Lithium in Unconsolidated Sediments and Plants of the Basin and Range Province, Southern California and Nevada; U.S. Government Printing Office: Washington, DC, USA, 1975.
- Millot, R.; Vigier, N.; Gaillardet, J. Behaviour of lithium and its isotopes during weathering in the Mackenzie Basin, Canada. Geochim. Cosmochim. Acta 2010, 74, 3897–3912. [Google Scholar] [CrossRef] [Green Version]
- Brigatti, M.F.; Kile, D.E.; Poppi, M. Crystal structure and crystal chemistry of lithium-bearing muscovite-2M1. Can. Mineral. 2001, 39, 1171–1180. [Google Scholar] [CrossRef]
- Takamatsu, N.; Imahashi, M.; Shimodaira, K.; Kamiya, H. The dissolution of lithium minerals in salt solutions. Geochem. J. 1983, 17, 153–160. [Google Scholar] [CrossRef]
- Risacher, F.; Fritz, B. Quaternary geochemical evolution of the salars of Uyuni and Coipasa, Central Altiplano, Bolivia. Chem. Geol. 1991, 90, 211–231. [Google Scholar] [CrossRef]
- Sieland, R. Hydraulic Investigations of the Salar de Uyuni, Bolivia. Available online: http://tu-freiberg.de/sites/default/files/media/institut-fuer-geologie-718/pdf/fog_volume_37.pdf (accessed on 23 October 2014).
- Adamis, Z.; Williams, R.B. Bentonite, Kaolin, and Selected Clay Minerals; World Health Organization: Geneva, Switzerland, 2005. [Google Scholar]
- Zeolites. Available online: http://cool.conservation-us.org/byorg/abbey/an/an20/an20-7/an20-702.html (accessed on 23 October 2014).
- Bachmaf, S.; Merkel, B. Sorption of uranium (VI) at the clay mineral–water interface. Environ. Earth Sci. 2011, 63, 925–935. [Google Scholar] [CrossRef]
- Scheffer, F.; Schachtschabel, P.H.-P.; Thiele-Brune, S. Lehrbuch der Bodenkunde; Spektrum Akad.-Verl.: Heidelberg, Germany, 2010. [Google Scholar]
- Starkey, H.C. The Role of Clays in Fixing Lithium; U.S. Government Printing Office: Washington, DC, USA, 1982.
- Carroll, D. Ion exchange in clays and other minerals. Geol. Soc. Am. Bull. 1959, 70, 749–779. [Google Scholar] [CrossRef]
- Cation-Exchange Capacity of Zeolites. Available online: http://www.gsaresources.com/cationexchange.htm (accessed on 24 October 2014).
- Deberitz, J. Lithium; Verl. Moderne Industrie: Landsberg/Lech, Germany, 1993. [Google Scholar]
- Chang, F.-R.C.; Skipper, N.T.; Sposito, G. Monte Carlo and Molecular Dynamics Simulations of Interfacial Structure in Lithium-Montmorillonite Hydrates. Langmuir 1997, 13, 2074–2082. [Google Scholar] [CrossRef]
- Baik, M.H.; Lee, S.Y. Colloidal stability of bentonite clay considering surface charge properties as a function of pH and ionic strength. J. Ind. Eng. Chem. 2010, 16, 837–841. [Google Scholar] [CrossRef]
- Rex, R.W. Authigenic kaolinite and mica as evidence for phase equilibria at low temperatures. Clays Clay Miner. 1965, 13, 95–104. [Google Scholar] [CrossRef]
- Alba, M.D.; Castro, M.A.; Chain, P.; Orta, M.M.; Pazos, M.C.; Pavon, E. Hydrothermal Stability of layered silicates in neutral and acidic media. Clays Clay Miner. 2010, 58, 501–514. [Google Scholar] [CrossRef]
- Dohnalová, Ž.; Svoboda, L.; Šulcová, P. Characterization of kaolin dispersion using acoustic and electroacoustic spectroscopy. J. Min. Metall. Sect. B Metall. 2008, 44, 63–72. [Google Scholar] [CrossRef]
- Jodin, M.C.; Gaboriaud, F.; Humbert, B. Limitations of potentiometric studies to determine the surface charge of gibbsite gamma-Al(OH)3 particles. J. Colloid Interface Sci. 2005, 287, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J. Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoyer, M.; Kummer, N.-A.; Merkel, B. Sorption of Lithium on Bentonite, Kaolin and Zeolite. Geosciences 2015, 5, 127-140. https://doi.org/10.3390/geosciences5020127
Hoyer M, Kummer N-A, Merkel B. Sorption of Lithium on Bentonite, Kaolin and Zeolite. Geosciences. 2015; 5(2):127-140. https://doi.org/10.3390/geosciences5020127
Chicago/Turabian StyleHoyer, Mandy, Nicolai-Alexeji Kummer, and Broder Merkel. 2015. "Sorption of Lithium on Bentonite, Kaolin and Zeolite" Geosciences 5, no. 2: 127-140. https://doi.org/10.3390/geosciences5020127