Genome-Wide Analysis of Allele-Specific Expression Patterns in Seventeen Tissues of Korean Cattle (Hanwoo)

 , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection
2.3. Whole-Denome Resequencing and RNA-Sequencing
2.4. Identification of Imprinted Genes
2.5. Functional Annotation Analysis
2.6. Experimental Validation of Monoallelically Expressed Genes Using Direct Sequencing
3. Results
3.1. Identification of Heterozygous SNPs Based on NGS Sequencing Data
3.2. Identification of Monoallelic Expression in Korean Cattle (Hanwoo)
3.3. Tissue-Specific Expression Patterns of Monoallelically Expressed Genes
3.4. Functional Annotation of MAE Genes
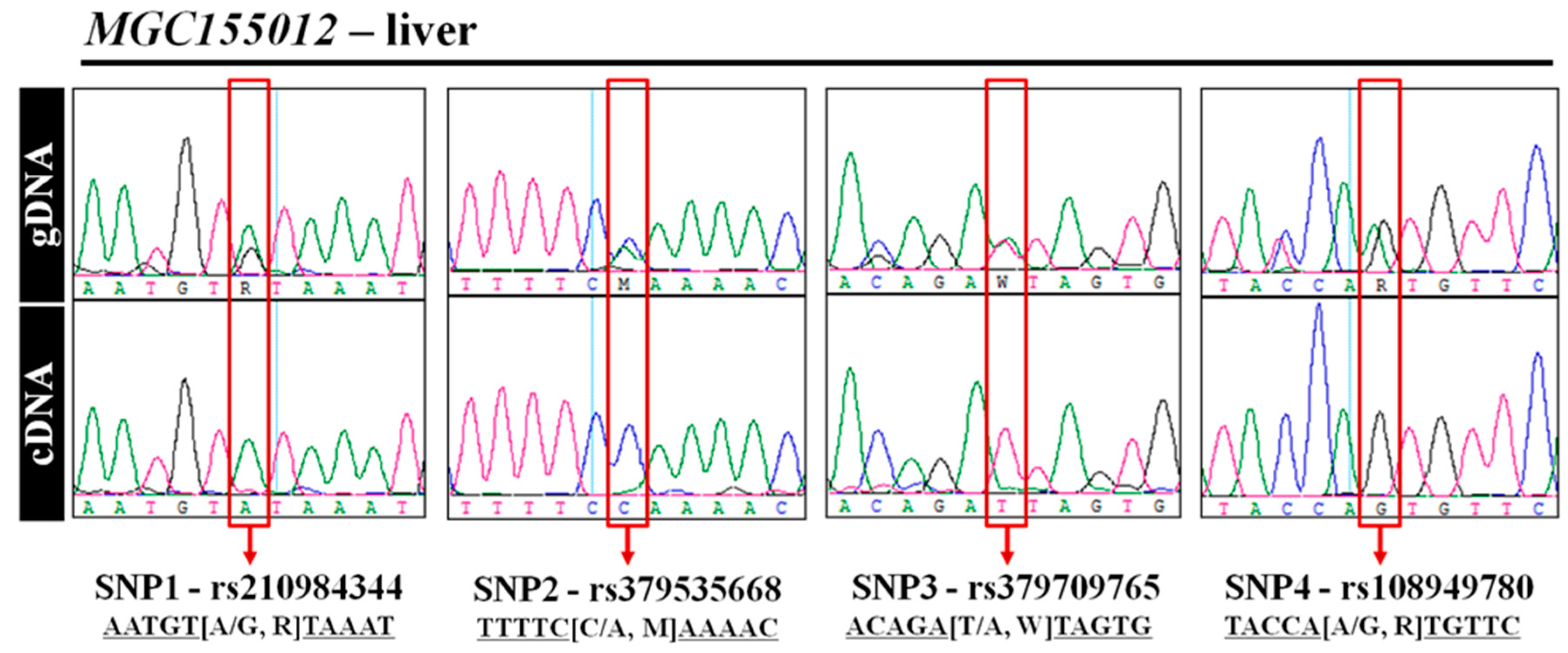
3.5. Experimental Validation of MAE Gene Using Direct Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- MacEachern, S.; Muir, W.M.; Crosby, S.D.; Cheng, H. Genome-wide identification and quantification of cis-and trans-regulated genes responding to Marek’s disease virus infection via analysis of allele-specific expression. Front. Genet. 2012, 2, 113. [Google Scholar] [CrossRef] [PubMed]
- Aberle, K.S.; Hamann, H.; Drögemüller, C.; Distl, O. Genetic diversity in German draught horse breeds compared with a group of primitive, riding and wild horses by means of microsatellite DNA markers. Anim. Genet. 2004, 35, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Khatib, H.; Schutzkus, V.; Chang, Y.; Rosa, G. Pattern of expression of the uterine milk protein gene and its association with productive life in dairy cattle. J. Dairy Sci. 2007, 90, 2427–2433. [Google Scholar] [CrossRef] [PubMed]
- Esteve-Codina, A.; Kofler, R.; Palmieri, N.; Bussotti, G.; Notredame, C.; Pérez-Enciso, M. Exploring the gonad transcriptome of two extreme male pigs with RNA-seq. BMC Genom. 2011, 12, 552. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Viltrop, T.; Tiirats, A.; Rajashekar, B.; Reimann, E.; Kõks, S.; Rull, K.; Milani, L.; Acharya, G.; Basnet, P. Using RNA sequencing for identifying gene imprinting and random monoallelic expression in human placenta. Epigenetics 2014, 9, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Gregg, C.; Zhang, J.; Weissbourd, B.; Luo, S.; Schroth, G.P.; Haig, D.; Dulac, C. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 2010, 329, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Frésard, L.; Leroux, S.; Servin, B.; Gourichon, D.; Dehais, P.; Cristobal, M.S.; Marsaud, N.; Vignoles, F.; Bed’Hom, B.; Coville, J.-L. Transcriptome-wide investigation of genomic imprinting in chicken. Nucleic Acids Res. 2014, 42, 3768–3782. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hagen, D.E.; Wang, J.; Elsik, C.G.; Ji, T.; Siqueira, L.G.; Hansen, P.J.; Rivera, R.M. Global assessment of imprinted gene expression in the bovine conceptus by next generation sequencing. Epigenetics 2016, 11, 501–516. [Google Scholar] [CrossRef]
- Wilkins, J.F. Genomic imprinting and methylation: Epigenetic canalization and conflict. Trends Genet. 2005, 21, 356–365. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef]
- O’Doherty, A.M.; MacHugh, D.E.; Spillane, C.; Magee, D.A. Genomic imprinting effects on complex traits in domesticated animal species. Front. Genet. 2015, 6, 156. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Chen, Y.; Liang, J.; Li, L.; Wu, T.; Tian, X.C.; Zhang, S. Developmental stage-specific imprinting of IPL in domestic pigs (Sus scrofa). Biomed Res. Int. 2010, 2010, 527539. [Google Scholar]
- Dindot, S.V.; Kent, K.C.; Evers, B.; Loskutoff, N.; Womack, J.; Piedrahita, J.A. Conservation of genomic imprinting at the XIST, IGF2, and GTL2 loci in the bovine. Mamm. Genome 2004, 15, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Curchoe, C.; Zhang, S.; Bin, Y.; Zhang, X.; Yang, L.; Feng, D.; O’Neill, M.; Tian, X.C. Promoter-specific expression of the imprinted IGF2 gene in cattle (Bos taurus). Biol. Reprod. 2005, 73, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Koo, C.-Y.; Muir, K.W.; Lam, E.W.-F. FOXM1: From cancer initiation to progression and treatment. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2012, 1819, 28–37. [Google Scholar] [CrossRef]
- Tian, X. Genomic imprinting in farm animals. Annu. Rev. Anim. Biosci. 2014, 2, 23–40. [Google Scholar] [CrossRef]
- Abramowitz, L.K.; Bartolomei, M.S. Genomic imprinting: Recognition and marking of imprinted loci. Curr. Opin. Genet. Dev. 2012, 22, 72–78. [Google Scholar] [CrossRef]
- Kelsey, G.; Feil, R. New insights into establishment and maintenance of DNA methylation imprints in mammals. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20110336. [Google Scholar] [CrossRef]
- Lee, S.-H.; Park, B.-H.; Sharma, A.; Dang, C.-G.; Lee, S.-S.; Choi, T.-J.; Choy, Y.-H.; Kim, H.-C.; Jeon, K.-J.; Kim, S.-D. Hanwoo cattle: Origin, domestication, breeding strategies and genomic selection. J. Anim. Sci. Technol. 2014, 56, 2. [Google Scholar] [CrossRef] [PubMed]
- Savova, V.; Patsenker, J.; Vigneau, S.; Gimelbrant, A.A. dbMAE: The database of autosomal monoallelic expression. Nucleic Acids Res. 2015, 44, D753–D756. [Google Scholar] [CrossRef] [PubMed]
- Zwemer, L.M.; Zak, A.; Thompson, B.R.; Kirby, A.; Daly, M.J.; Chess, A.; Gimelbrant, A.A. Autosomal monoallelic expression in the mouse. Genome Biol. 2012, 13, R10. [Google Scholar] [CrossRef] [PubMed]
- Magee, D.; Spillane, C.; Berkowicz, E.; Sikora, K.; MacHugh, D. Imprinted loci in domestic livestock species as epigenomic targets for artificial selection of complex traits. Anim. Genet. 2014, 45, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Zaitoun, I.; Khatib, H. Assessment of genomic imprinting of SLC38A4, NNAT, NAP1L5, and H19 in cattle. BMC Genet. 2006, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Khatib, H. Imprinting of Nesp55 gene in cattle. Mamm. Genome 2004, 15, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Ramsköld, D.; Reinius, B.; Sandberg, R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 2014, 343, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Qadri, I.; Hu, L.-J.; Iwahashi, M.; Al-Zuabi, S.; Quattrochi, L.C.; Simon, F.R. Interaction of hepatocyte nuclear factors in transcriptional regulation of tissue specific hormonal expression of human multidrug resistance-associated protein 2 (abcc2). Toxicol. Appl. Pharmacol. 2009, 234, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Teh, M.-T.; Gemenetzidis, E.; Patel, D.; Tariq, R.; Nadir, A.; Bahta, A.W.; Waseem, A.; Hutchison, I.L. FOXM1 induces a global methylation signature that mimics the cancer epigenome in head and neck squamous cell carcinoma. PLoS ONE 2012, 7, e34329. [Google Scholar] [CrossRef]
- Luedi, P.P.; Dietrich, F.S.; Weidman, J.R.; Bosko, J.M.; Jirtle, R.L.; Hartemink, A.J. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007, 17, 1723–1730. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Tissues | Tested SNPs 1 | Tested Genes | MAE Genes 2 | MAE% | |||
|---|---|---|---|---|---|---|---|
| PE | ME | UN | Total | ||||
| Pituitary gland | 679 | 467 | 6 | 3 | 15 | 24 | 5.14% |
| Hypothalamus | 823 | 561 | 5 | 3 | 19 | 27 | 4.81% |
| Spinal cord | 467 | 327 | 4 | 2 | 12 | 18 | 5.50% |
| Lymph node | 1104 | 654 | 4 | 5 | 23 | 32 | 4.89% |
| Spleen | 1131 | 676 | 4 | 3 | 23 | 30 | 4.44% |
| Liver | 852 | 515 | 6 | 5 | 15 | 26 | 5.05% |
| Large intestine | 1000 | 594 | 6 | 2 | 20 | 28 | 4.71% |
| Small intestine | 1042 | 613 | 7 | 3 | 20 | 30 | 4.89% |
| Heart | 593 | 388 | 4 | 2 | 17 | 23 | 5.93% |
| Loin muscle | 508 | 340 | 4 | 2 | 13 | 19 | 5.59% |
| Femoral muscle | 593 | 397 | 4 | 2 | 15 | 21 | 5.29% |
| Kidney | 946 | 589 | 7 | 3 | 22 | 32 | 5.43% |
| Adrenal gland | 853 | 531 | 4 | 3 | 17 | 24 | 4.52% |
| Blood | 823 | 511 | 4 | 3 | 17 | 24 | 4.70% |
| Lung | 1047 | 644 | 5 | 2 | 20 | 27 | 4.19% |
| Cerebrum | 988 | 666 | 5 | 4 | 17 | 26 | 3.90% |
| Cerebellum | 1043 | 699 | 6 | 4 | 18 | 28 | 4.01% |
| Gene Symbol | Gene Description | BTA | Status | Cell Localization | Class |
|---|---|---|---|---|---|
| Transcriptional regulation | |||||
| FOXD2 | forkhead box D2 | 3 | UN | Nucleus | TF |
| FOXM1 | forkhead box M1 | 5 | UN | Nucleus | TF |
| HTATSF1 | HIV-1 Tat specific factor 1 | 12 | ME | Nucleus | TF |
| SCRT1 | scratch family zinc finger 1 | 14 | PE | Nucleus | TF |
| NKX6-2 | NK6 homeobox 2 | 18 | UN | Nucleus | TF |
| TRIP6 | thyroid hormone receptor interactor 6 | 25 | UN | Cytoplasm | - |
| UBN1 | ubinuclein 1 | 25 | UN | Nucleus | TF |
| Translational Regulation | |||||
| PDE5A | phosphodiesterase 5A, cGMP-specific | 6 | UN | . | - |
| MAN1B1 | mannosidase, alpha, class 1B, member 1 | 11 | UN | ER membrane | - |
| WBSCR17 | Williams-Beuren syndrome chromosome region 17 | 17 | UN | Golgi apparatus | - |
| GALNT1 | polypeptide N-acetylgalactosaminyltransferase 1 | 24 | UN | Golgi apparatus | - |
| Metabolic Process | |||||
| GLYAT | glycine-N-acyltransferase | 15 | ME | Mitochondrion | - |
| PDPR | pyruvate dehydrogenase phosphatase regulatory subunit | 18 | UN | Mitochondrion | - |
| KIF19 | kinesin family member 19 | 19 | UN | Cytoplasm | - |
| DCXR | dicarbonyl/L-xylulose reductase | 19 | UN | Membrane | - |
| GSTA1 | glutathione S-transferase alpha 1 | 23 | PE | Cytoplasm | - |
| ABCC2 | ATP-binding cassette, sub-family C (CFTR/MRP), member 2 | 26 | UN | Apical cell membrane | Transporter |
| Immune Response | |||||
| TNFSF12 | tumor necrosis factor (ligand) superfamily, member 12 | 19 | UN | Cell membrane | Ligand |
| CRCP | CGRP receptor component | 25 | UN | Nucleus | Receptor |
| ITGAD | integrin, alpha D | 25 | UN | Membrane | Receptor |
| Cell Cycle | |||||
| HIST1H4D | histone cluster 1, H4d | 3 | UN | Nucleus | - |
| SSPO | SCO-spondin | 4 | ME | Extracellular space | - |
| CHRNA10 | cholinergic receptor, nicotinic,alpha 10 (neuronal) | 15 | UN | Cell junction | - |
| CTDNEP1 | CTD nuclear envelope phosphatase 1 | 19 | PE | ER membrane | Protein phosphatase |
| DSG1 | desmoglein 1 | 24 | ME | Cell membrane | - |
| DCLRE1A | DNA cross-link repair 1A | 26 | ME | Nucleus | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, K.-S.; Chang, S.-S.; Choi, B.-H.; Lee, S.-H.; Lee, K.-T.; Chai, H.-H.; Park, J.-E.; Park, W.; Lim, D. Genome-Wide Analysis of Allele-Specific Expression Patterns in Seventeen Tissues of Korean Cattle (Hanwoo). Animals 2019, 9, 727. https://doi.org/10.3390/ani9100727
Lim K-S, Chang S-S, Choi B-H, Lee S-H, Lee K-T, Chai H-H, Park J-E, Park W, Lim D. Genome-Wide Analysis of Allele-Specific Expression Patterns in Seventeen Tissues of Korean Cattle (Hanwoo). Animals. 2019; 9(10):727. https://doi.org/10.3390/ani9100727
Chicago/Turabian StyleLim, Kyu-Sang, Sun-Sik Chang, Bong-Hwan Choi, Seung-Hwan Lee, Kyung-Tai Lee, Han-Ha Chai, Jong-Eun Park, Woncheoul Park, and Dajeong Lim. 2019. "Genome-Wide Analysis of Allele-Specific Expression Patterns in Seventeen Tissues of Korean Cattle (Hanwoo)" Animals 9, no. 10: 727. https://doi.org/10.3390/ani9100727
APA StyleLim, K.-S., Chang, S.-S., Choi, B.-H., Lee, S.-H., Lee, K.-T., Chai, H.-H., Park, J.-E., Park, W., & Lim, D. (2019). Genome-Wide Analysis of Allele-Specific Expression Patterns in Seventeen Tissues of Korean Cattle (Hanwoo). Animals, 9(10), 727. https://doi.org/10.3390/ani9100727