Traces of Human-Mediated Selection in the Gene Pool of Red Deer Populations


 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals Collection and Data Mining
2.2. Overall Heterozygosity
2.3. Population Structure
2.4. Genetic Admixture and Gene Flow
3. Results
3.1. Overall Heterozygosity
3.2. Population Structure
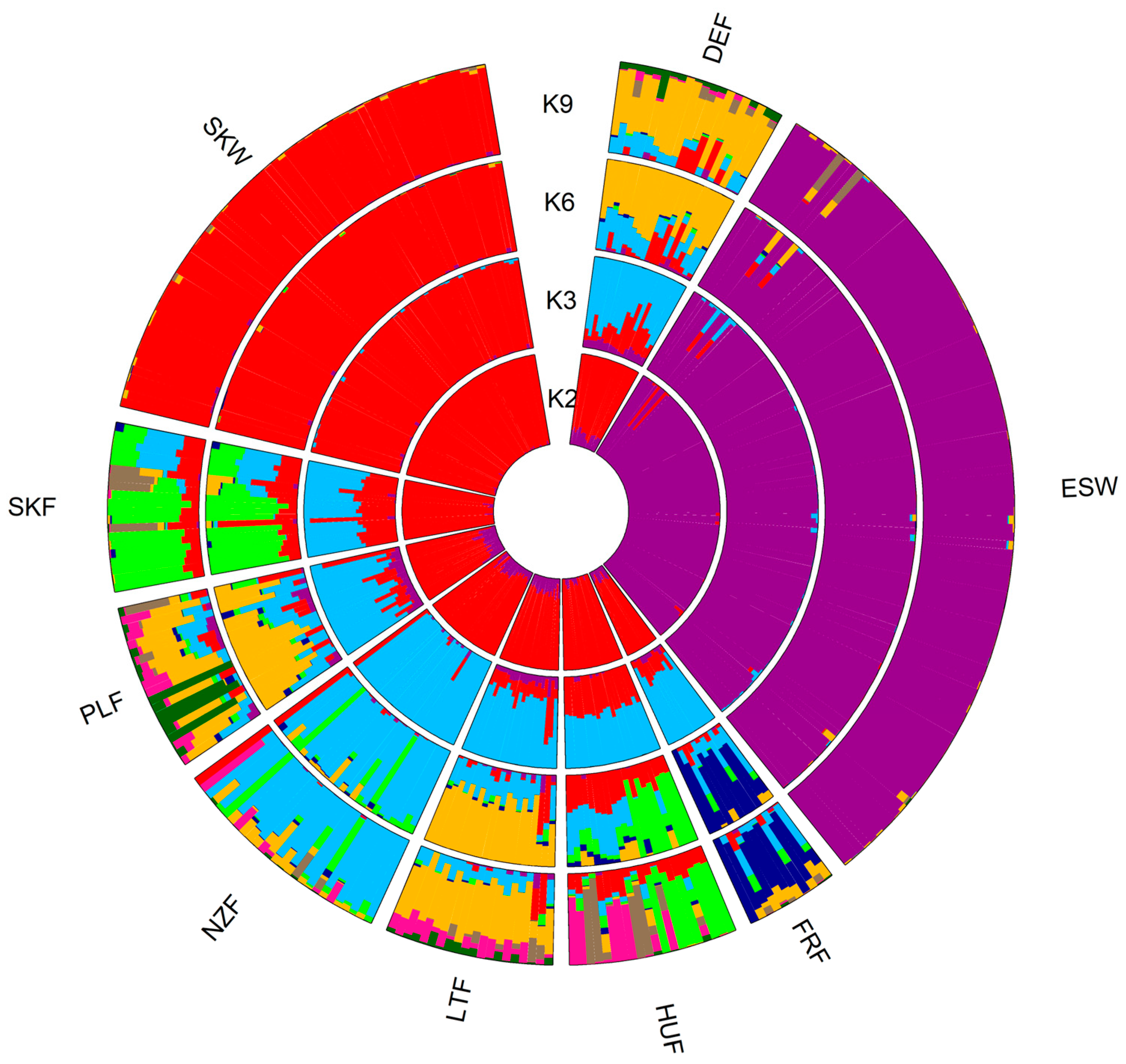
3.3. Genetic Admixture and Gene Flow
4. Discussion
4.1. Data Mining
4.2. Overall Heterozygosity
4.3. Population Structure
4.4. Genetic Admixture and Gene Flow
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Apollonio, M.; Andersen, R.; Putman, R. European Ungulates and Their Management in the 21st Century; Cambridge University Press: New York, NY, USA, 2010. [Google Scholar]
- Ludt, C.J.; Schroeder, W.; Rottmann, O.; Kuehn, R. Mitochondrial DNA phylogeography of red deer (Cervus elaphus). Mol. Phylogenet. Evol. 2004, 31, 1064–1083. [Google Scholar] [CrossRef] [PubMed]
- Sommer, R.S.; Zachos, F.E.; Street, M.; Jöris, O.; Skog, A.; Benecke, N. Late quaternary distribution dynamics and phylogeography of the red deer (Cervus elaphus) in Europe. Quat. Sci. Rev. 2008, 27, 714–733. [Google Scholar] [CrossRef]
- Skog, A.; Zachos, F.E.; Rueness, E.K.; Fuelner, P.G.D.; Mysterud, A.; Langvatn, R.; Lorenzini, R.; Hmwe, S.S.; Lehoczky, I.; Hartl, G.B.; et al. Phylogeography of red deer (Cervus elaphus) in Europe. J. Biogeogr. 2009, 36, 66–77. [Google Scholar] [CrossRef]
- Rey-Iglesia, A.; Grandal-d’Anglade, A.; Campos, P.F.; Hansen, A.J. Mitochondrial DNA of pre-last glacial maximum red deer from NW Spain suggests a more complex phylogeographical history for the species. Ecol. Evol. 2017, 7, 10690–10700. [Google Scholar] [CrossRef] [PubMed]
- Carranza, J.; Salinas, M.; Andrés, D.; Pérez-González, J. Iberian red deer: Paraphyletic nature at mtDNA but nuclear markers support its genetic identity. Ecol. Evol. 2016, 6, 905–922. [Google Scholar] [CrossRef] [Green Version]
- Zachos, F.E.; Frantz, A.C.; Kuehn, R.; Bertouille, S.; Colyn, M.; Niedziałkowska, M.; Pérez-González, J.; Skog, A.; Sprem, N.; Flamand, M.-C. Genetic Structure and Effective Population Sizes in European Red Deer (Cervus elaphus) at a Continental Scale: Insights from Microsatellite DNA. J. Hered. 2016, 107, 318–326. [Google Scholar] [CrossRef] [Green Version]
- Hartl, G.B.; Zachos, F.; Nadlinger, K. Genetic diversity in European red deer (Cervus elaphus L.): Anthropogenic influences on natural populations. C R Biol. 2003, 326, 37–42. [Google Scholar] [CrossRef]
- Linnell, J.D.C.; Zachos, F.E. Status and distribution patterns of European ungulates: Genetics, population history and conservation. In Ungulate Management in Europe: Problems and Practices; Apollonio, M., Andersen, R., Putman, R., Eds.; Cambridge University Press: Cambridge, UK, 2011; pp. 12–53. ISBN 9780521760591. [Google Scholar]
- Sykes, N.; Putman, R. Management of Ungulates in 21st century: How far have we come? In Behaviour and Management of European Ungulates; Putman, R., Apollonio, M., Eds.; Whittle Publishing: Dunbeath, UK, 2014; pp. 267–289. [Google Scholar]
- Serrano, M.P.; Maggiolino, A.; Pateiro, M.; Landete-Castillejos, T.; Domínguez, R.; García, A.; Franco, D.; Gallego, L.; De Palo, P.; Lorenzo, J.M. Carcass characteristics and meat quality of deer. In More than Beef, Pork and Chicken—The Production, Processing and Quality Traits of Other Sources of Meat for Human Diet; Lorenzo, J.M., Munekata, P.E.S., Barba, F.J., Toldrá, F., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 227–268. ISBN 978-3-030-05484-7. [Google Scholar]
- Barton, N.H. The role of hybridisation in evolution. Mol. Ecol. 2008, 10, 551–568. [Google Scholar] [CrossRef]
- Frankham, R. Conservation genetics. Annu. Rev. Genet. 1995, 29, 305–327. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Leary, R.F.; Spruell, P.; Wenburg, J.K. The problems with hybrids: Setting conservation guidelines. Trends Ecol. Evol. 2001, 16, 613–622. [Google Scholar] [CrossRef]
- Haanes, H.; Røed, K.H.; Mysterud, A.; Langvatn, R.; Rosef, O. Consequences for genetic diversity and population performance of introducing continental red deer into the northern distribution range. Conserv. Genet. 2010, 11, 1653–1665. [Google Scholar] [CrossRef]
- Hartl, G.B.; Nadlinger, K.; Apollonio, M.; Markov, G.; Findo, S.; Klein, F.; Lang, G.; Markowski, J. Extensive mitochondrial-DNA differentiation among European Red deer (Cervus elaphus) populations: Implications for conservation and management. Z. Für Säugetierkunde 1995, 60, 41–52. [Google Scholar]
- Zachos, F.; Hartl, G.B.; Apollonio, M.; Reutershan, T. On the phylogeographic origin of the Corsican red deer (Cervus elaphus corsicanus): Evidence from microsatellites and mitochondrial DNA. Mammal. Biol. 2003, 68, 284–298. [Google Scholar] [CrossRef]
- Bartoš, L. White Red Deer in Deer Parks Žehušice and Žleby, Central Bohemia, Czech Republic. In The 6th International Deer Biology Congress; Research Institue of Animal Production: Prague, Czech Republic, 2006. [Google Scholar]
- Whitehead, G.K. The Whitehead Encyclopedia of Deer; Swan Hill Press: Shrewsbury, UK, 1993. [Google Scholar]
- Polziehn, R.O.; Strobeck, C. A phylogenetic comparison of red deer and wapiti using mitochondrial DNA. Mol. Phylogenet. Evol. 2002, 22, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Foote, G.R.; Otte, M. Could pre-Last Glacial Maximum humans have existed in North America undetected? An interregional approach to the question. Curr. Anthropol. 2001, 42, 563–566. [Google Scholar] [CrossRef]
- Mysterud, A. Effects of selective harvesting on ungulate populations. In Behaviour and Management of European Ungulates; Putman, R., Apollonio, M., Eds.; Whittle Publishing: Dunbeath, UK, 2014; pp. 46–77. [Google Scholar]
- Milner, J.M.; Bonenfant, C.; Mysterud, A.; Gaillard, J.-M.; Csányi, S.; Stenseth, N.C. Temporal and spatial development of red deer harvesting in Europe: Biological and cultural factors. J. Appl. Ecol. 2006, 43, 721–734. [Google Scholar] [CrossRef]
- Niedziałkowska, M.; Jędrzejewska, B.; Honnen, A.-C.; Otto, T.; Sidorovich, V.E.; Perzanowski, K.; Skog, A.; Hartl, G.B.; Borowik, T.; Bunevich, A.N.; et al. Molecular biogeography of red deer Cervus elaphus from eastern Europe: Insights from mitochondrial DNA sequences. Acta Theriol. 2011, 56, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Landete-Castillejos, T.; Gallego, L.; Estévez, J.A.; García, A.J.; Fierro, Y. Fencing of game estates in Spain considered as management unit. In Enclosures: A Dead-End? Influence on Game Biology, Conservation and Hunting; Chapman, H.G., Hecker, K., Eds.; International Council for Game and Wildlife Conservation—CIC: Sopron, Hungary, 2010; pp. 68–79. [Google Scholar]
- Zachos, F.E.; Hartl, G.B. Phylogeography, population genetics and conservation of the European red deer Cervus elaphus. Mamm. Rev. 2011, 41, 138–150. [Google Scholar] [CrossRef]
- Chardonnet, P.H.; Des Clers, B.; Fisher, J.R.; Gerhold, R.; Jori, F.; Lamarque, F. The value of wildlife. Rev. Sci. Tech. Loie 2002, 21, 15–51. [Google Scholar] [CrossRef]
- Archer, J.A.; Ward, J.F.; Newman, S.-A.N.; Cruickshank, G.J.; Pearse, A.J. Implementing genetic evaluation in the New Zealand deer industry. Proc. Assoc. Adv. Anim. Breed. Genet. 2005, 16, 4–7. [Google Scholar]
- Rowe, S.J.; Clarke, S.M.; Van Stijn, T.C.; Hyndman, D.L.; Ward, J.F.; McEwan, K.M.; Dodds, K.G.; McEwan, J.C.; Newman, S.-A.N.; Asher, G.W. Brief communication: Developing genomic tools in the New Zealand deer industry. Proc. N. Z. Soc. Anim. Prod. 2015, 75, 91–93. [Google Scholar]
- Dodds, K.G.; Newman, S.-A.N.; Clarke, S.M.; Brauning, R.; Hess, A.S.; Bilton, T.P.; Chappell, A.J.; McEwan, J.C.; Van Stijn, T.C.; Bates, M.; et al. Genomic tools for use in the New Zealand deer industry. In Proceedings of the 23rd Conference of the Association for the Advancement of Animal Breeding and Genetics (AAABG), Armidale, Australia, 27 October–1 November 2019. [Google Scholar]
- Clarke, S.M.; Henry, H.M.; Dodds, K.G.; Jowett, T.W.D.; Manley, T.R.; Anderson, R.M.; McEwan, J.C. A high throughput single nucleotide polymorphism multiplex assay for parentage assignment in New Zealand sheep. PLoS ONE 2014, 9, e93392. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; Anderson, R.M.; Van Stijn, T.C.; Kristjánsson, T.; Clarke, S.M. Construction of relatedness matrices using genotyping-by-sequencing data. BMC Genomics 2015, 16, 1047. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, F.; Lipka, A.E.; Glaubitz, J.; Elshire, R.; Cherney, J.H.; Casler, M.D.; Buckler, E.S.; Costich, D.E. Switchgrass genomic diversity, ploidy, and evolution: Novel insights from a network-based SNP discovery protocol. PLoS Genet. 2013, 9, e1003215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Wright, S. Breeding structure of populations in relation to speciation. Am. Nat. 1940, 74, 232–248. [Google Scholar] [CrossRef]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [Green Version]
- Neuditschko, M.; Khatkar, M.S.; Raadsma, H.W. NetView: A high-definition network-visualisation approach to detect fine-scale population structures from genome-wide patterns of variation. PLoS ONE 2012, 7, e48375. [Google Scholar] [CrossRef] [Green Version]
- Pembleton, L.W.; Cogan, N.O.I.; Forster, J.W. StAMPP: An R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 2013, 13, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Collins, C. A Tutorial for Discriminant Analysis of Principal Components (DAPC) Using Adegenet 2.0.0; MRC Centre for Outbreak Analysis and Modelling: London, UK, 2015. [Google Scholar]
- Steinig, E.J.; Neuditschko, M.; Khatkar, M.S.; Raadsma, H.W.; Zenger, K.R. Netview p: A network visualisation tool to unravel complex population structure using genome-wide SNPs. Mol. Ecol. Resour. 2016, 16, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, G.A.; Rannala, B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 2003, 163, 1177–1191. [Google Scholar] [CrossRef]
- Wickland, D.P.; Battu, G.; Hudson, K.A.; Diers, B.W.; Hudson, M.E. A comparison of genotyping-by-sequencing analysis methods on low-coverage crop datasets shows advantages of a new workflow, GB-eaSy. BMC Bioinform. 2017, 18, 586. [Google Scholar] [CrossRef]
- Wang, N.; Yuan, Y.; Wang, H.; Yu, D.; Liu, Y.; Zhang, A.; Gowda, M.; Nair, S.K.; Hao, Z.; Lu, Y.; et al. Applications of genotyping-by-sequencing (GBS) in maize genetics and breeding. Sci. Rep. 2020, 10, 16308. [Google Scholar] [CrossRef]
- Friel, J.; Bombarely, A.; Fornell, C.D.; Luque, F.; Fernández-Ocaña, A.M. Comparative Analysis of Genotyping by Sequencing and Whole-Genome Sequencing Methods in Diversity Studies of Olea Europaea L. Plants 2021, 10, 2514. [Google Scholar] [CrossRef]
- Gurgul, A.; Miksza-Cybulska, A.; Szmatoła, T.; Jasielczuk, I.; Piestrzyńska-Kajtoch, A.; Fornal, A.; Semik-Gurgul, E.; Bugno-Poniewierska, M. Genotyping-by-sequencing performance in selected livestock species. Genomics 2019, 111, 186–195. [Google Scholar] [CrossRef]
- Gorjanc, G.; Cleveland, M.A.; Houston, R.D.; Hickey, J.M. Potential of genotyping-by-sequencing for genomic selection in livestock populations. Genet. Sel. Evol. 2015, 47, 12. [Google Scholar] [CrossRef] [PubMed]
- Bedhiaf-Romdhani, S.; Baazaoui, I.; Dodds, K.G.; Brauning, R.; Anderson, R.M.; Van Stijn, T.C.; McCulloch, A.F.; McEwan, J.C. Efficiency of genotyping by sequencing in inferring genomic relatedness and molecular insights into fat tail selection in Tunisian sheep. Anim. Genet. 2023, 54, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Beissinger, T.M.; Hirsch, C.N.; Sekhon, R.S.; Foerster, J.M.; Johnson, J.M.; Muttoni, G.; Vaillancourt, B.; Buell, C.R.; Kaeppler, S.M.; de Leon, N. Marker density and read depth for genotyping populations using genotyping-by-sequencing. Genetics 2013, 193, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef] [Green Version]
- Brouard, J.-S.; Boyle, B.; Ibeagha-Awemu, E.M.; Bissonnette, N. Low-depth genotyping-by-sequencing (GBS) in a bovine population: Strategies to maximize the selection of high quality genotypes and the accuracy of imputation. BMC Genet. 2017, 18, 32. [Google Scholar] [CrossRef] [Green Version]
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; van Stijn, T.C.; Rowe, S.J.; McEwan, K.M.; Clarke, S.M. Exclusion and Genomic Relatedness Methods for Assignment of Parentage Using Genotyping-by-Sequencing Data. G3 Genes Genomes Genet. 2019, 9, 3239–3247. [Google Scholar] [CrossRef] [Green Version]
- Palaiokostas, C.; Clarke, S.M.; Jeuthe, H.; Brauning, R.; Bilton, T.P.; Dodds, K.G.; McEwan, J.C.; De Koning, D.-J. Application of Low Coverage Genotyping by Sequencing in Selectively Bred Arctic Charr (Salvelinus Alpinus). G3 Genes Genomes Genet. 2020, 10, 2069–2078. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Cole, J.W.; Grond-Ginsbach, C. Departure from Hardy Weinberg equilibrium and genotyping error. Front. Genet. 2017, 8, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearman, W.S.; Urban, L.; Alexander, A. Commonly used Hardy–Weinberg equilibrium filtering schemes impact population structure inferences using RADseq data. Mol. Ecol. Resour. 2022, 22, 2599–2613. [Google Scholar] [CrossRef] [PubMed]
- Fardo, D.W.; Becker, K.D.; Bertram, L.; Tanzi, R.E.; Lange, C. Recovering unused information in genome-wide association studies: The benefit of analyzing SNPs out of Hardy-Weinberg equilibrium. Eur. J. Hum. Genet. 2009, 17, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.C. Mamíferos de España II; Planeta: Barcelona, Spain, 1998. [Google Scholar]
- Crestanello, B.; Pecchioli, E.; Vernesi, C.; Mona, S.; Martínková, N.; Janiga, M.; Hauffe, H.C.; Bertorelle, G. The genetic impact of translocations and habitat fragmentation in chamois (Rupicapra) spp. J. Hered. 2009, 100, 691–708. [Google Scholar] [CrossRef] [Green Version]
- Pilot, M.; Greco, C.; vonHoldt, B.M.; Jędrzejewska, B.; Randi, E.; Jędrzejewski, W.; Sidorovich, V.E.; Ostrander, E.A.; Wayne, R.K. Genome-wide signatures of population bottlenecks and diversifying selection in European wolves. Heredity 2014, 112, 428–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabangin, M.E.; Woo, J.G.; Martin, L.J. The effect of minor allele frequency on the likelihood of obtaining false positives. BMC Proc. 2009, 3, S41. [Google Scholar] [CrossRef] [PubMed]
- Pertoldi, C.; Tokarska, M.; Wójcik, J.M.; Kawałko, A.; Randi, E.; Kristensen, T.N.; Loeschcke, V.; Coltman, D.; Wilson, G.A.; Gregersen, V.R.; et al. Phylogenetic relationships among the European and American bison and seven cattle breeds reconstructed using the BovineSNP50 Illumina Genotyping BeadChip. Acta. Theriol. 2010, 55, 97–108. [Google Scholar] [CrossRef]
- Haynes, G.D.; Latch, E.K. Identification of novel single nucleotide polymorphisms (SNPs) in deer (Odocoileus spp.) using the BovineSNP50 BeadChip. PLoS ONE 2012, 7, e36536. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.J.; Song, L.J.; Wu, F.J.; Liang, X.W.; Yang, B.Z.; Wathes, D.C.; Pollott, G.E.; Cheng, Z.; Shi, D.S.; Liu, Q.Y.; et al. Investigation of transferability of BovineSNP50 BeadChip from cattle to water buffalo for genome wide association study. Mol. Biol. Rep. 2013, 40, 743–750. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Biscarini, F.; Auzino, B.; Ragatzu, M.; Spaterna, A.; Ciampolini, R. Genome-wide diversity and runs of homozygosity in the "Braque Français, type Pyrénées" dog breed. BMC Res. Notes 2018, 11, 13. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Zhang, T.; Liu, Y.; Wang, Z.; Xu, L.; Zhu, B.; Gao, X.; Zhang, L.; Gao, H.; Liu, G.E.; et al. Genome-Wide Assessment of Runs of Homozygosity in Chinese Wagyu Beef Cattle. Animals 2020, 10, 1425. [Google Scholar] [CrossRef]
- Vostry, L.; Vostra-Vydrova, H.; Citek, J.; Gorjanc, G.; Curik, I. Association of inbreeding and regional equine leucocyte antigen homozygosity with the prevalence of insect bite hypersensitivity in Old Kladruber horse. Anim. Genet. 2021, 52, 422–430. [Google Scholar] [CrossRef]
- Meiri, M.; Kosintsev, P.; Conroy, K.; Meiri, S.; Barnes, I.; Lister, A. Subspecies dynamics in space and time: A study of the red deer complex using ancient and modern DNA and morphology. J. Biogeogr. 2018, 45, 367–380. [Google Scholar] [CrossRef]
- Queirós, J.; Acevedo, P.; Santos, J.P.V.; Barasona, J.; Beltran-Beck, B.; González-Barrio, D.; Armenteros, J.A.; Diez-Delgado, I.; Boadella, M.; de Mera, I.F.; et al. Red deer in Iberia: Molecular ecological studies in a southern refugium and inferences on European postglacial colonisation history. PLoS ONE 2019, 14, e0210282. [Google Scholar] [CrossRef] [Green Version]
- Meiri, M.; Lister, A.M.; Higham, T.F.G.; Stewart, J.R.; Straus, L.G.; Obermaier, H.; Morales, M.R.G.; Marín-Arroyo, A.B.; Barnes, I. Late-glacial recolonisation and phylogeography of European red deer (Cervus elaphus L.). Mol. Ecol. 2013, 22, 4711–4722. [Google Scholar] [CrossRef] [PubMed]
- Frantz, A.C.; Zachos, F.E.; Bertouille, S.; Eloy, M.-C.; Colyn, M.; Flamand, M.-C. Using genetic tools to estimate the prevalence of non-native red deer (Cervus elaphus) in a Western European population. Ecol. Evol. 2017, 7, 7650–7660. [Google Scholar] [CrossRef] [PubMed]
- Basarab, J.A.; Crowley, J.J.; Abo-Ismail, M.K.; Manafiazar, G.M.; Akanno, E.C.; Baron, V.S.; Plastow, G. Genomic retained heterosis effects on fertility and lifetime productivity in beef heifers. Can. J. Anim. Sci. 2018, 98, 642–655. [Google Scholar] [CrossRef]
- Kuehn, R.; Schroeder, W.; Pirchner, F.; Rottmann, O. Genetic diversity, gene flow and drift in Bavarian red deer populations (Cervus elaphus). Conserv. Genet. 2003, 4, 157–166. [Google Scholar] [CrossRef]
- Niedziałkowska, M.; Fontaine, M.C.; Jędrzejewska, B. Factors shaping gene flow in red deer (Cervus elaphus) in seminatural landscapes of central Europe. Can. J. Zool. 2012, 90, 150–162. [Google Scholar] [CrossRef]
- Edelhoff, H.; Zachos, F.E.; Fickel, J.; Epps, C.W.; Balkenhol, N. Genetic analysis of red deer (Cervus elaphus) administrative management units in a human-dominated landscape. Conserv. Genet. 2020, 21, 261–276. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Population | Abbreviation | No. of Animals | |
|---|---|---|---|
| Germany | farmed | DEF | 20 |
| Spain | wild | ESW | 102 |
| France | farmed | FRF | 11 |
| Hungary | farmed | HUF | 20 |
| Latvia | farmed | LTF | 20 |
| New Zealand | farmed | NZF | 27 |
| Poland | farmed | PLF | 20 |
| Slovakia | farmed | SKF | 20 |
| Slovakia | wild | SKW | 62 |
| Population | HO ± SD (SE) | He ± SD (SE) | MAF ± SD (SE) | FHOM ± SD (SE) |
|---|---|---|---|---|
| DEF | 0.145 ± 0.158 (0.001) | 0.176 ± 0.162 (0.001) | 0.119 ± 0.131 (0.001) | 0.127 ± 0.211 (0.047) |
| ESW | 0.072 ± 0.102 (0.001) | 0.154 ± 0.168 (0.001) | 0.105 ± 0.133 (0.001) | 0.359 ± 0.111 (0.011) |
| FRF | 0.151 ± 0.197 (0.001) | 0.158 ± 0.177 (0.001) | 0.113 ± 0.144 (0.001) | 0.036 ± 0.139 (0.042) |
| HUF | 0.097 ± 0.128 (0.001) | 0.169 ± 0.166 (0.001) | 0.115 ± 0.132 (0.001) | 0.295 ± 0.177 (0.040) |
| LTF | 0.111 ± 0.137 (0.001) | 0.171 ± 0.164 (0.001) | 0.116 ± 0.132 (0.001) | 0.242 ± 0.183 (0.041) |
| NZF | 0.114 ± 0.147 (0.001) | 0.165 ± 0.176 (0.001) | 0.116 ± 0.142 (0.001) | 0.242 ± 0.226 (0.043) |
| PLF | 0.119 ± 0.143 (0.001) | 0.170 ± 0.165 (0.001) | 0.116 ± 0.132 (0.001) | 0.219 ± 0.223 (0.050) |
| SKF | 0.114 ± 0.147 (0.001) | 0.166 ± 0.167 (0.001) | 0.114 ± 0.134 (0.001) | 0.253 ± 0.226 (0.046) |
| SKW | 0.182 ± 0.158 (0.001) | 0.187 ± 0.143 (0.001) | 0.122 ± 0.118 (0.001) | 0.035 ± 0.069 (0.009) |
| DEF | ESW | FRF | HUF | LTF | NZF | PLF | SKF | SKW | |
|---|---|---|---|---|---|---|---|---|---|
| DEF | 0.103 | 0.076 | 0.050 | 0.025 | 0.058 | 0.027 | 0.066 | 0.074 | |
| ESW | 0.027 | 0.141 | 0.119 | 0.101 | 0.138 | 0.101 | 0.134 | 0.121 | |
| FRF | 0.027 | 0.039 | 0.085 | 0.072 | 0.087 | 0.074 | 0.099 | 0.112 | |
| HUF | 0.020 | 0.032 | 0.031 | 0.045 | 0.065 | 0.047 | 0.015 | 0.061 | |
| LTF | 0.014 | 0.027 | 0.027 | 0.020 | 0.055 | 0.020 | 0.062 | 0.074 | |
| NZF | 0.020 | 0.035 | 0.029 | 0.023 | 0.020 | 0.053 | 0.069 | 0.125 | |
| PLF | 0.014 | 0.027 | 0.027 | 0.020 | 0.013 | 0.019 | 0.064 | 0.080 | |
| SKF | 0.023 | 0.035 | 0.033 | 0.013 | 0.023 | 0.023 | 0.023 | 0.074 | |
| SKW | 0.023 | 0.030 | 0.035 | 0.020 | 0.023 | 0.036 | 0.025 | 0.023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moravčíková, N.; Kasarda, R.; Židek, R.; McEwan, J.C.; Brauning, R.; Landete-Castillejos, T.; Chonco, L.; Ciberej, J.; Pokorádi, J. Traces of Human-Mediated Selection in the Gene Pool of Red Deer Populations. Animals 2023, 13, 2525. https://doi.org/10.3390/ani13152525
Moravčíková N, Kasarda R, Židek R, McEwan JC, Brauning R, Landete-Castillejos T, Chonco L, Ciberej J, Pokorádi J. Traces of Human-Mediated Selection in the Gene Pool of Red Deer Populations. Animals. 2023; 13(15):2525. https://doi.org/10.3390/ani13152525
Chicago/Turabian StyleMoravčíková, Nina, Radovan Kasarda, Radoslav Židek, John Colin McEwan, Rudiger Brauning, Tomás Landete-Castillejos, Louis Chonco, Juraj Ciberej, and Jaroslav Pokorádi. 2023. "Traces of Human-Mediated Selection in the Gene Pool of Red Deer Populations" Animals 13, no. 15: 2525. https://doi.org/10.3390/ani13152525
APA StyleMoravčíková, N., Kasarda, R., Židek, R., McEwan, J. C., Brauning, R., Landete-Castillejos, T., Chonco, L., Ciberej, J., & Pokorádi, J. (2023). Traces of Human-Mediated Selection in the Gene Pool of Red Deer Populations. Animals, 13(15), 2525. https://doi.org/10.3390/ani13152525