Retrospective Analysis of Archived Pyrazinamide Resistant Mycobacterium tuberculosis Complex Isolates from Uganda—Evidence of Interspecies Transmission

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Institutional Review Board (IRB) and Ethical Approvals
2.2. Study Site
2.3. Study Samples
2.4. Sampling, Culture, and Isolation of Mycobacteria
2.5. Molecular Testing to Confirm MTC
2.6. DNA Extraction/DNA Preparation for PCR
2.7. Genomic Deletions (Regions of Difference) Analysis
2.8. Genomic Analysis—Sequencing, Assembly, and Annotation of Eight Genomes
3. Results
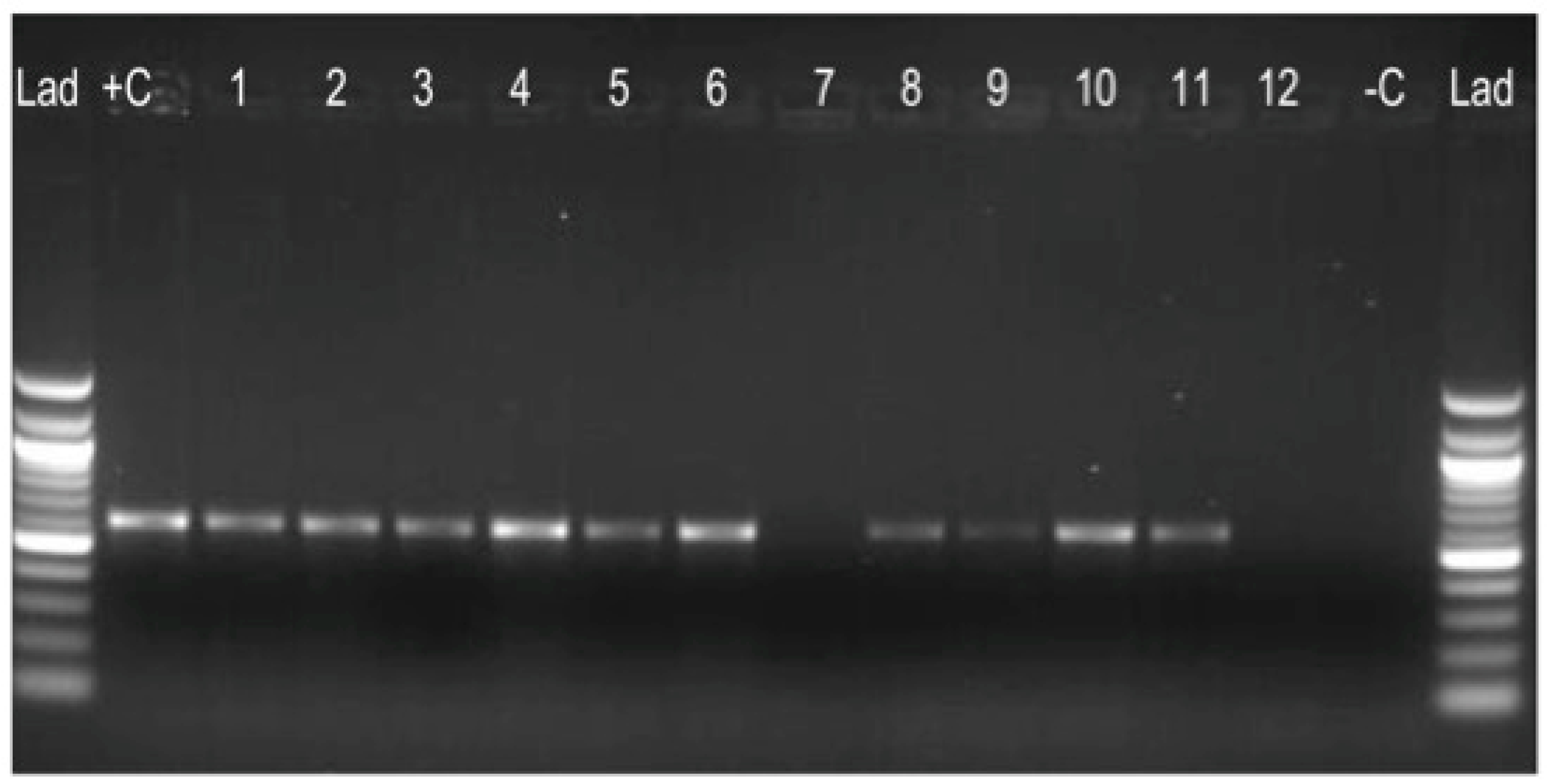
3.1. Culture Results
3.2. Regions of Difference (RD) Analysis: RD9, RD4, RD12, and RD1
3.3. Phenotypic Drug Susceptibility Profiles
3.4. Mycobacterium Bovis from Human Host Among Sequenced Genomes
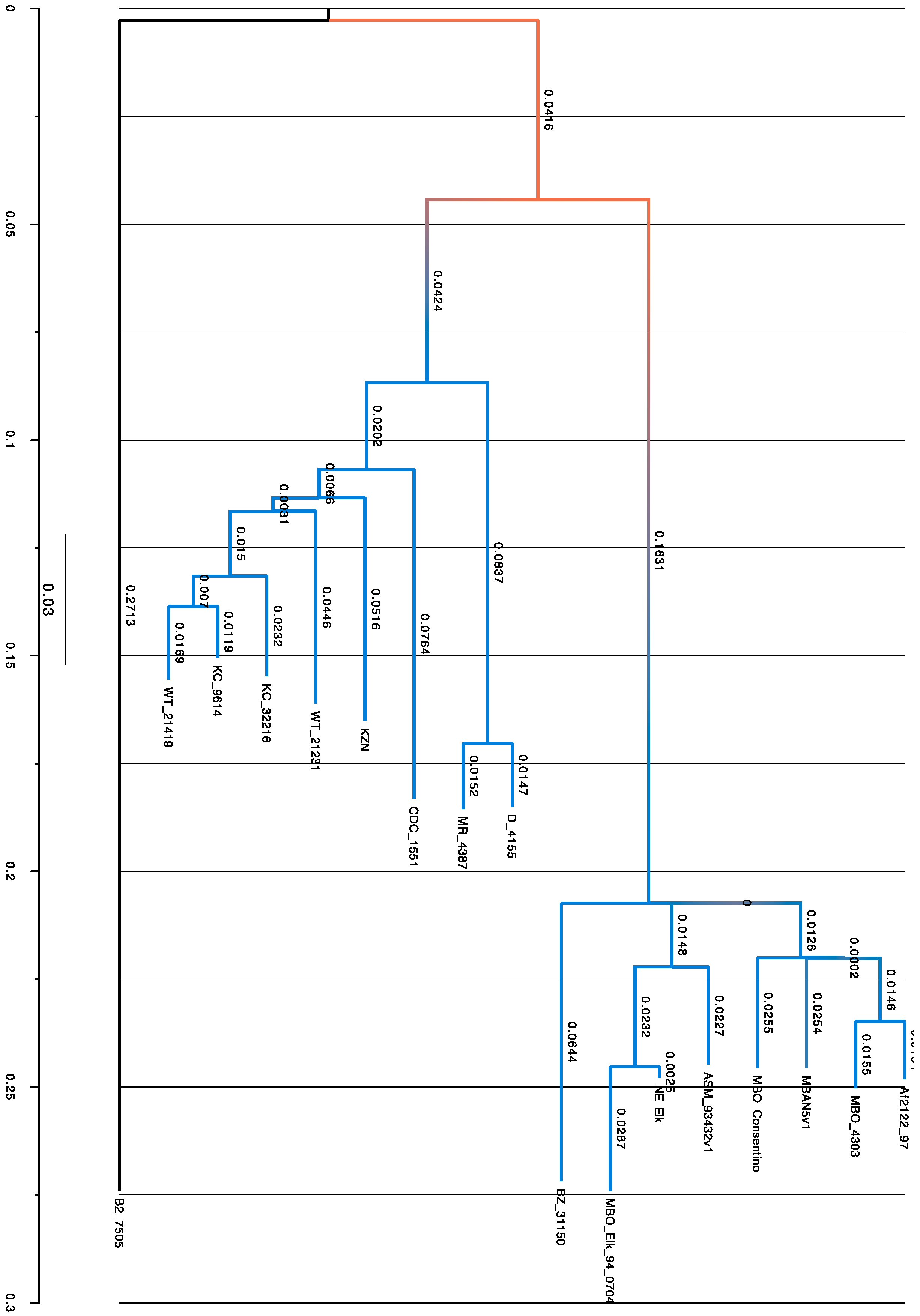
3.5. SNPs Obtained by Phylogenetic Analysis
3.6. Genome Analysis of Multi-Drug Resistance Mutations
4. Discussion
4.1. Mycobacterium Bovis in PZA Resistant Isolates
4.2. Whole Genome Sequencing (WGS) and Analysis
4.3. Phylogenetic Analysis
4.4. Antibiotic Resistance Genes of Uganda Isolates
4.5. Application of WGS/SNP Chip in the Field
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., III; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Banuls, A.L.; Sanou, A.; Anh, N.T.; Godreuil, S. Mycobacterium tuberculosis: Ecology and evolution of a human bacterium. J. Med. Microbiol. 2015, 64, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Dos Vultos, T.; Mestre, O.; Rauzier, J.; Golec, M.; Rastogi, N.; Rasolofo, V.; Tonjum, T.; Sola, C.; Matic, I.; Gicquel, B. Evolution and diversity of clonal bacteria: The paradigm of Mycobacterium tuberculosis. PLoS ONE 2008, 3, e1538. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.C.; Brisse, S.; Brosch, R.; Fabre, M.; Omais, B.; Marmiesse, M.; Supply, P.; Vincent, V. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog. 2005, 1, e5. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, L.M.; Daborn, C.J. The epidemiology of Mycobacterium bovis infections in animals and man: A review. Tuber. Lung Dis. 1995, 76, 1–46. [Google Scholar] [CrossRef]
- Michel, A.L.; Muller, B.; Van Helden, P.D. Mycobacterium bovis at the animal-human interface: A problem, or not? Vet. Microbiol. 2010, 140, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.M. The cost of disease eradication. Smallpox and bovine tuberculosis. Ann. N. Y. Acad. Sci. 1999, 894, 83–91. [Google Scholar] [CrossRef]
- Pesciaroli, M.; Alvarez, J.; Boniotti, M.B.; Cagiola, M.; Di Marco, V.; Marianelli, C.; Pacciarini, M.; Pasquali, P. Tuberculosis in domestic animal species. Res. Vet. Sci. 2014, 97, S78–S85. [Google Scholar] [CrossRef]
- El-Sayed, A.; El-Shannat, S.; Kamel, M.; Castaneda-Vazquez, M.A.; Castaneda-Vazquez, H. Molecular Epidemiology of Mycobacterium bovis in Humans and Cattle. Zoonoses. Public Health 2016, 63, 251–264. [Google Scholar] [CrossRef]
- Daborn, C.J.; Grange, J.M.; Kazwala, R.R. The bovine tuberculosis cycle—An African perspective. J. Appl. Bacteriol. 1996, 25, 27S–32S. [Google Scholar]
- Cosivi, O.; Grange, J.M.; Daborn, C.J.; Raviglione, M.C.; Fujikura, T.; Cousins, D.; Robinson, R.A.; Huchzermeyer, H.F.A.K.; De Kantor, I.; Meslin, F.X. Zoonotic tuberculosis due to Mycobacterium bovis in developing countries. Emerg. Infect. Dis. 1998, 4, 59–70. [Google Scholar] [CrossRef]
- Berg, S.; Schelling, E.; Hailu, E.; Firdessa, R.; Gumi, B.; Erenso, G.; Gadisa, E.; Mengistu, A.; Habtamu, M.; Hussein, J.; et al. Investigation of the high rates of extrapulmonary tuberculosis in Ethiopia reveals no single driving factor and minimal evidence for zoonotic transmission of Mycobacterium bovis infection. BMC Infect. Dis. 2015, 15, 112. [Google Scholar] [CrossRef]
- Wedlock, D.N.; Skinner, M.A.; De Lisle, G.W.; Buddle, B.M. Control of Mycobacterium bovis infections and the risk to human populations. Microbes. Infect. 2002, 4, 471–480. [Google Scholar] [CrossRef]
- Muller, B.; Durr, S.; Alonso, S.; Hattendorf, J.; Laisse, C.J.; Parsons, S.D.; van Helden, P.D.; Zinsstag, J. Zoonotic Mycobacterium bovis-induced tuberculosis in humans. Emerg. Infect. Dis. 2013, 19, 899–908. [Google Scholar] [CrossRef]
- Kazwala, R.R.; Daborn, C.J.; Sharp, J.M.; Kambarage, D.M.; Jiwa, S.F.; Mbembati, N.A. Isolation of Mycobacterium bovis from human cases of cervical adenitis in Tanzania: A cause for concern? Int. J. Tuberc. Lung Dis. 2001, 5, 87–91. [Google Scholar]
- World Health Organization. Tuberculosis; WHO: Geneva, Switzerland, 2015; Volume 2016. [Google Scholar]
- American Thoracic Society; Centers for Disease Control. Mycobacterioses and the acquired immunodeficiency syndrome. Am. Rev. Respir. Dis. 1987, 136, 492–496. [Google Scholar] [CrossRef]
- World Health Organization. Uganda TB Profile; WHO: Geneva, Switzerland, 2016; Volume 2016. [Google Scholar]
- World Health Organization. TB Diagnostics and Lab Strengthening; WHO: Geneva, Switzerland, 2016; Volume 2016. [Google Scholar]
- Oloya, J.; Muma, J.B.; Opuda-Asibo, J.; Djonne, B.; Kazwala, R.; Skjerve, E. Risk factors for herd-level bovine-tuberculosis seropositivity in transhumant cattle in Uganda. Prev. Vet. Med. 2007, 80, 318–329. [Google Scholar] [CrossRef]
- Oloya, J.; Opuda-Asibo, J.; Djonne, B.; Muma, J.B.; Matope, G.; Kazwala, R.; Skjerve, E. Responses to tuberculin among Zebu cattle in the transhumance regions of Karamoja and Nakasongola district of Uganda. Trop. Anim. Health Prod. 2006, 38, 275–283. [Google Scholar] [CrossRef]
- Oloya, J.; Kazwala, R.; Lund, A.; Opuda-Asibo, J.; Demelash, B.; Skjerve, E.; Johansen, T.B.; Djonne, B. Characterisation of mycobacteria isolated from slaughter cattle in pastoral regions of Uganda. BMC Microbiol. 2007, 7, 95. [Google Scholar] [CrossRef]
- Gallivan, M.; Shah, N.; Flood, J. Epidemiology of human Mycobacterium bovis disease, California, USA, 2003–2011. Emerg. Infect. Dis. 2015, 21, 435–443. [Google Scholar] [CrossRef]
- Huard, R.C.; Lazzarini, L.C.; Butler, W.R.; Van Soolingen, D.; Ho, J.L. PCR-based method to differentiate the subspecies of the Mycobacterium tuberculosis complex on the basis of genomic deletions. J. Clin. Microbiol. 2003, 41, 1637–1650. [Google Scholar] [CrossRef]
- Joint Clinical Research Centre. 2016 Annual Report. Available online: https://www.jcrc.org.ug/resources/reports/annual-report-2016-sep-2017 (accessed on 26 July 2019).
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989; Volume 2. [Google Scholar]
- Sambrook, J.; Russell, D.W. Storage of bacterial cultures growing on solid medium. CSH Protoc. 2006, 2006, pdb.prot4453. [Google Scholar] [CrossRef]
- Huard, R.C.; Fabre, M.; De Haas, P.; Lazzarini, L.C.; Van Soolingen, D.; Cousins, D.; Ho, J.L. Novel genetic polymorphisms that further delineate the phylogeny of the Mycobacterium tuberculosis complex. J. Bacteriol. 2006, 188, 4271–4287. [Google Scholar] [CrossRef]
- Pym, A.S.; Brodin, P.; Brosch, R.; Huerre, M.; Cole, S.T. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol. Microbiol. 2002, 46, 709–717. [Google Scholar] [CrossRef]
- Thorne, N.; Borrell, S.; Evans, J.; Magee, J.; Garcia De Viedma, D.; Bishop, C.; Gonzalez-Martin, J.; Gharbia, S.; Arnold, C. IS6110-based global phylogeny of Mycobacterium tuberculosis. Infect. Genet. Evol. 2011, 11, 132–138. [Google Scholar] [CrossRef]
- Brosch, R.; Gordon, S.V.; Marmiesse, M.; Brodin, P.; Buchrieser, C.; Eiglmeier, K.; Garnier, T.; Gutierrez, C.; Hewinson, G.; Kremer, K.; et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci. USA 2002, 99, 3684–3689. [Google Scholar] [CrossRef]
- Kamerbeek, J.; Schouls, L.; Kolk, A.; Van Agterveld, M.; Van Soolingen, D.; Kuijper, S.; Bunschoten, A.; Molhuizen, H.; Shaw, R.; Goyal, M.; et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 1997, 35, 907–914. [Google Scholar] [Green Version]
- Van Soolingen, D.; Hermans, P.W.; De Haas, P.E.; Soll, D.R.; Van Embden, J.D. Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: Evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J. Clin. Microbiol. 1991, 29, 2578–2586. [Google Scholar]
- Asiimwe, B.B.; Asiimwe, J.; Kallenius, G.; Ashaba, F.K.; Ghebremichael, S.; Joloba, M.; Koivula, T. Molecular characterisation of Mycobacterium bovis isolates from cattle carcases at a city slaughterhouse in Uganda. Vet. Rec. 2009, 164, 655–658. [Google Scholar] [CrossRef]
- Tuberculosis. Broad Institute. 2016. Available online: https://www.broadinstitute.org/infectious-disease-and-microbiome/tuberculosis (accessed on 26 July 2019).
- Olive, D. TB-ARC M.Bovis.1. 2014, Volume 2016. Available online: https://olive.broadinstitute.org/projects/Mycobacterium%20tuberculosis%20Comparative (accessed on 26 July 2019).
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome. Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. Progressivemauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. Basic Local Alignment Search Tool; National Center for Biotechnology Information: Bethesda, MD, USA, 2016; Volume 2016.
- National Center for Biotechnology Information. TB-ARC-M. Bovis; National Center for Biotechnology Information: Bethesda, MD, USA, 2016; Volume 2016.
- National Center for Biotechnology Information. Bioproject; National Center for Biotechnology Information: Bethesda, MD, USA, 2016; Volume 2016.
- Wanzala, S.I.; Nakavuma, J.; Travis, D.A.; Kia, P.; Ogwang, S.; Sreevatsan, S. Draft Genome Sequences of Mycobacterium bovis BZ 31150 and Mycobacterium bovis B2 7505, Pathogenic Bacteria Isolated from Archived Captive Animal Bronchial Washes and Human Sputum Samples in Uganda. Genome. Announc. 2015, 3, e01102–e01115. [Google Scholar] [CrossRef]
- Sreevatsan, S.; Pan, X.; Stockbauer, K.E.; Connell, N.D.; Kreiswirth, B.N.; Whittam, T.S.; Musser, J.M. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc. Natl. Acad. Sci. USA 1997, 94, 9869–9874. [Google Scholar] [CrossRef]
- Sreevatsan, S.; Pan, X.; Zhang, Y.; Kreiswirth, B.N.; Musser, J.M. Mutations associated with pyrazinamide resistance in pncA of Mycobacterium tuberculosis complex organisms. Antimicrob. Agents Chemother. 1997, 41, 636–640. [Google Scholar] [CrossRef] [Green Version]
- Sreevatsan, S.; Escalante, P.; Pan, X.; Gillies, D.A., II; Siddiqui, S.; Khalaf, C.N.; Kreiswirth, B.N.; Bifani, P.; Adams, L.G.; Ficht, T.; et al. Identification of a polymorphic nucleotide in oxyR specific for Mycobacterium bovis. J. Clin. Microbiol. 1996, 34, 2007–2010. [Google Scholar] [Green Version]
- Seifert, M.; Catanzaro, D.; Catanzaro, A.; Rodwell, T.C. Genetic mutations associated with isoniazid resistance in Mycobacterium tuberculosis: A systematic review. PLoS ONE 2015, 10, e0119628. [Google Scholar] [CrossRef]
- Brammacharry, U.; Muthaiah, M. Characterization of rpsL Gene Mutations in Streptomycin-Resistant Mycobacterium tuberculosis Isolates. Am. J. Microbiol. Res. 2014, 2, 80–85. [Google Scholar] [CrossRef]
- Vilcheze, C.; Jacobs, W.R., Jr. Resistance to Isoniazid and Ethionamide in Mycobacterium tuberculosis: Genes, Mutations, and Causalities. Microbiol. Spectr. 2014, 2, 431–453. [Google Scholar] [CrossRef]
- Gagneux, S. Host-pathogen coevolution in human tuberculosis. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 850–859. [Google Scholar] [CrossRef]
- Sekiguchi, J.; Miyoshi-Akiyama, T.; Augustynowicz-Kopec, E.; Zwolska, Z.; Kirikae, F.; Toyota, E.; Kobayashi, I.; Morita, K.; Kudo, K.; Kato, S.; et al. Detection of multidrug resistance in Mycobacterium tuberculosis. J. Clin. Microbiol. 2007, 45, 179–192. [Google Scholar] [CrossRef]
- Dominguez, J.; Boettger, E.C.; Cirillo, D.; Cobelens, F.; Eisenach, K.D.; Gagneux, S.; Hillemann, D.; Horsburgh, R.; Molina-Moya, B.; Niemann, S.; et al. Clinical implications of molecular drug resistance testing for Mycobacterium tuberculosis: A TBNET/RESIST-TB consensus statement. Int. J. Tuberc. Lung Dis. 2016, 20, 24–42. [Google Scholar] [CrossRef]
- Avalos, E.; Catanzaro, D.; Catanzaro, A.; Ganiats, T.; Brodine, S.; Alcaraz, J.; Rodwell, T. Frequency and geographic distribution of gyrA and gyrB mutations associated with fluoroquinolone resistance in clinical Mycobacterium tuberculosis isolates: A systematic review. PLoS ONE 2015, 10, e0120470. [Google Scholar] [CrossRef]
- Malik, S.; Willby, M.; Sikes, D.; Tsodikov, O.V.; Posey, J.E. New insights into fluoroquinolone resistance in Mycobacterium tuberculosis: Functional genetic analysis of gyrA and gyrB mutations. PLoS ONE 2012, 7, e39754. [Google Scholar] [CrossRef]
- Mayer, C.; Takiff, H. The Molecular Genetics of Fluoroquinolone Resistance in Mycobacterium tuberculosis. Microbiol. Spectr. 2014, 2, 455–478. [Google Scholar] [CrossRef]
- Kankya, C.; Muwonge, A.; Olet, S.; Munyeme, M.; Biffa, D.; Opuda-Asibo, J.; Skjerve, E.; Oloya, J. Factors associated with pastoral community knowledge and occurrence of mycobacterial infections in human-animal interface areas of Nakasongola and Mubende districts, Uganda. BMC Public Health 2010, 10, 471. [Google Scholar] [CrossRef]
- Cousins, D.V. Mycobacterium bovis infection and control in domestic livestock. Rev. Sci. Technol. 2001, 20, 71–85. [Google Scholar] [CrossRef]
- Cleaveland, S.; Shaw, D.J.; Mfinanga, S.G.; Shirima, G.; Kazwala, R.R.; Eblate, E.; Sharp, M. Mycobacterium bovis in rural Tanzania: Risk factors for infection in human and cattle populations. Tuberculosis 2007, 87, 30–43. [Google Scholar] [CrossRef]
- Cadmus, S.; Palmer, S.; Okker, M.; Dale, J.; Gover, K.; Smith, N.; Jahans, K.; Hewinson, R.G.; Gordon, S.V. Molecular analysis of human and bovine tubercle bacilli from a local setting in Nigeria. J. Clin. Microbiol. 2006, 44, 29–34. [Google Scholar] [CrossRef]
- Brudey, K.; Driscoll, J.R.; Rigouts, L.; Prodinger, W.M.; Gori, A.; Al-Hajoj, S.A.; Allix, C.; Aristimuno, L.; Arora, J.; Baumanis, V.; et al. Mycobacterium tuberculosis complex genetic diversity: Mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006, 6, 23. [Google Scholar] [CrossRef]
- Ayele, W.Y.; Neill, S.D.; Zinsstag, J.; Weiss, M.G.; Pavlik, I. Bovine tuberculosis: An old disease but a new threat to Africa. Int. J. Tuberc. Lung Dis. 2004, 8, 924–937. [Google Scholar]
- Etter, E.; Donado, P.; Jori, F.; Caron, A.; Goutard, F.; Roger, F. Risk analysis and bovine tuberculosis, a re-emerging zoonosis. Ann. N. Y. Acad. Sci. 2006, 1081, 61–73. [Google Scholar] [CrossRef]
- Alemayehu, R.; Girmay, M.; Gobena, A. Bovine tuberculosis is more prevalent in cattle owned by farmers with active tuberculosis in central Ethiopia. Vet. J. 2008, 178, 119–125. [Google Scholar] [CrossRef]
- Aylate, A.; Shah, S.N.; Aleme, H.; Gizaw, T.T. Bovine tuberculosis: Prevalence and diagnostic efficacy of routine meat inspection procedure in Woldiya municipality abattoir north Wollo zone, Ethiopia. Trop. Anim. Health Prod. 2013, 45, 855–864. [Google Scholar] [CrossRef]
- Ameni, G.; Vordermeier, M.; Firdessa, R.; Aseffa, A.; Hewinson, G.; Gordon, S.V.; Berg, S. Mycobacterium tuberculosis infection in grazing cattle in central Ethiopia. Vet. J. 2011, 188, 359–361. [Google Scholar] [CrossRef] [Green Version]
- Ameni, G.; Tadesse, K.; Hailu, E.; Deresse, Y.; Medhin, G.; Aseffa, A.; Hewinson, G.; Vordermeier, M.; Berg, S. Transmission of Mycobacterium tuberculosis between farmers and cattle in central Ethiopia. PLoS ONE 2013, 8, e76891. [Google Scholar] [CrossRef]
- Ameni, G.; Erkihun, A. Bovine tuberculosis on small-scale dairy farms in Adama Town, central Ethiopia, and farmer awareness of the disease. Rev. Sci. Technol. 2007, 26, 711–719. [Google Scholar] [CrossRef]
- Ameni, G.; Aseffa, A.; Engers, H.; Young, D.; Hewinson, G.; Vordermeier, M. Cattle husbandry in Ethiopia is a predominant factor affecting the pathology of bovine tuberculosis and gamma interferon responses to mycobacterial antigens. Clin. Vaccine Immunol. 2006, 13, 1030–1036. [Google Scholar] [CrossRef]
- Roug, A.; Perez, A.; Mazet, J.A.; Clifford, D.L.; VanWormer, E.; Paul, G.; Kazwala, R.R.; Smith, W.A. Comparison of intervention methods for reducing human exposure to Mycobacterium bovis through milk in pastoralist households of Tanzania. Prev. Vet. Med. 2014, 115, 157–165. [Google Scholar] [CrossRef]
- Berg, S.; Firdessa, R.; Habtamu, M.; Gadisa, E.; Mengistu, A.; Yamuah, L.; Ameni, G.; Vordermeier, M.; Robertson, B.D.; Smith, N.H.; et al. The burden of mycobacterial disease in ethiopian cattle: Implications for public health. PLoS ONE 2009, 4, e5068. [Google Scholar] [CrossRef]
- Ramirez-Busby, S.M.; Valafar, F. Systematic review of mutations in pyrazinamidase associated with pyrazinamide resistance in Mycobacterium tuberculosis clinical isolates. Antimicrob. Agents Chemother. 2015, 59, 5267–5277. [Google Scholar] [CrossRef]
- Cirillo, D.M.; Cabibbe, A.M.; De Filippo, M.R.; Trovato, A.; Simonetti, T.; Rossolini, G.M.; Tortoli, E. Use of WGS in Mycobacterium tuberculosis routine diagnosis. Int. J. Mycobacteriol. 2016, 5, S252–S253. [Google Scholar] [CrossRef]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591. [Google Scholar] [CrossRef]
- Le Chevalier, F.; Cascioferro, A.; Majlessi, L.; Herrmann, J.L.; Brosch, R. Mycobacterium tuberculosis evolutionary pathogenesis and its putative impact on drug development. Future Microbiol. 2014, 9, 969–985. [Google Scholar] [CrossRef] [Green Version]
- Moran, N.A. Microbial minimalism: Genome reduction in bacterial pathogens. Cell 2002, 108, 583–586. [Google Scholar] [CrossRef]
- Votintseva, A.A.; Bradley, P.; Pankhurst, L.; Del Ojo Elias, C.; Loose, M.; Nilgiriwala, K.; Chatterjee, A.; Smith, E.G.; Sanderson, N.; Walker, T.M.; et al. Same-day diagnostic and surveillance data for tuberculosis via whole genome sequencing of direct respiratory samples. J. Clin. Microbiol. 2017, 55, 1285–1298. [Google Scholar] [CrossRef]
- Joshi, D.; Harris, N.B.; Waters, R.; Thacker, T.; Mathema, B.; Krieswirth, B.; Sreevatsan, S. Single nucleotide polymorphisms in the Mycobacterium bovis genome resolve phylogenetic relationships. J. Clin. Microbiol. 2012, 50, 3853–3861. [Google Scholar] [CrossRef]
- Garcia-Betancur, J.C.; Menendez, M.C.; Del Portillo, P.; Garcia, M.J. Alignment of multiple complete genomes suggests that gene rearrangements may contribute towards the speciation of Mycobacteria. Infect. Genet. Evol. 2012, 12, 819–826. [Google Scholar] [CrossRef]
- Salipante, S.J.; SenGupta, D.J.; Cummings, L.A.; Land, T.A.; Hoogestraat, D.R.; Cookson, B.T. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J. Clin. Microbiol. 2015, 53, 1072–1079. [Google Scholar] [CrossRef]
- Takiff, H.E.; Feo, O. Clinical value of whole-genome sequencing of Mycobacterium tuberculosis. Lancet Infect. Dis. 2015, 15, 1077–1090. [Google Scholar] [CrossRef]
- Jagielski, T.; Minias, A.; Van Ingen, J.; Rastogi, N.; Brzostek, A.; Zaczek, A.; Dziadek, J. Methodological and Clinical Aspects of the Molecular Epidemiology of Mycobacterium tuberculosis and Other Mycobacteria. Clin. Microbiol. Rev. 2016, 29, 239–290. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Hu, Y.; Wang, Q.; Li, H.M.; Gao, G.F.; Liu, C.H.; Zhu, B. Comparative genomic analysis of Mycobacterium tuberculosis clinical isolates. BMC Genom. 2014, 15, 469. [Google Scholar] [CrossRef]
- Whitfield, M.G.; Soeters, H.M.; Warren, R.M.; York, T.; Sampson, S.L.; Streicher, E.M.; Van Helden, P.D.; Van Rie, A. A Global Perspective on Pyrazinamide Resistance: Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0133869. [Google Scholar] [CrossRef]
- Scorpio, A.; Zhang, Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef]
- Maslov, D.A.; Zaichikova, M.V.; Chernousova, L.N.; Shur, K.V.; Bekker, O.B.; Smirnova, T.G.; Larionova, E.E.; Andreevskaya, S.N.; Zhang, Y.; Danilenko, V.N. Resistance to pyrazinamide in Russian Mycobacterium tuberculosis isolates: pncA sequencing versus Bactec MGIT 960. Tuberculosis 2015, 95, 608–612. [Google Scholar] [CrossRef]
- Vilcheze, C.; Wang, F.; Arai, M.; Hazbon, M.H.; Colangeli, R.; Kremer, L.; Weisbrod, T.R.; Alland, D.; Sacchettini, J.C.; Jacobs, W.R., Jr. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat. Med. 2006, 12, 1027–1029. [Google Scholar] [CrossRef]
- Aye, K.S.; Nakajima, C.; Yamaguchi, T.; Win, M.M.; Shwe, M.M.; Win, A.A.; Lwin, T.; Nyunt, W.W.; Ti, T.; Suzuki, Y. Genotypic characterization of multi-drug-resistant Mycobacterium tuberculosis isolates in Myanmar. J. Infect. Chemother. 2016, 22, 174–179. [Google Scholar] [CrossRef]
- Khosravi, A.D.; Goodarzi, H.; Alavi, S.M. Detection of genomic mutations in katG, inhA and rpoB genes of Mycobacterium tuberculosis isolates using polymerase chain reaction and multiplex allele-specific polymerase chain reaction. Braz. J. Infect. Dis. 2012, 16, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Vordermeier, H.M.; Perez De Val, B.; Buddle, B.M.; Villarreal-Ramos, B.; Jones, G.J.; Hewinson, R.G.; Domingo, M. Vaccination of domestic animals against tuberculosis: Review of progress and contributions to the field of the TBSTEP project. Res. Vet. Sci. 2014, 97, S53–S60. [Google Scholar] [CrossRef]
- Tschopp, R.; Aseffa, A.; Schelling, E.; Berg, S.; Hailu, E.; Gadisa, E.; Habtamu, M.; Argaw, K.; Zinsstag, J. Bovine tuberculosis at the wildlife-livestock-human interface in Hamer Woreda, South Omo, Southern Ethiopia. PLoS ONE 2010, 5, e12205. [Google Scholar] [CrossRef]
- Srivastava, K.; Chauhan, D.S.; Gupta, P.; Singh, H.B.; Sharma, V.D.; Yadav, V.S.; Sreekumaran; Thakral, S.S.; Dharamdheeran, J.S.; Nigam, P.; et al. Isolation of Mycobacterium bovis and M-tuberculosis from cattle of some farms in north India—Possible relevance in human health. Indian J. Med. Res. 2008, 128, 26–31. [Google Scholar]
- Ehrt, S.; Rhee, K.; Schnappinger, D. Mycobacterial genes essential for the pathogen’s survival in the host. Immunol. Rev. 2015, 264, 319–326. [Google Scholar] [CrossRef]
- Pankhurst, L.J.; Del Ojo Elias, C.; Votintseva, A.A.; Walker, T.M.; Cole, K.; Davies, J.; Fermont, J.M.; Gascoyne-Binzi, D.M.; Kohl, T.A.; Kong, C.; et al. Rapid, comprehensive, and affordable mycobacterial diagnosis with whole-genome sequencing: A prospective study. Lancet Respir. Med. 2016, 4, 49–58. [Google Scholar] [CrossRef]
- Sabat, A.J.; Budimir, A.; Nashev, D.; Sa-Leao, R.; Van Dijl, J.; Laurent, F.; Grundmann, H.; Friedrich, A.W.; ESGEM. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Eurosurveillance 2013, 18, 20380. [Google Scholar] [CrossRef] [Green Version]
- Kjeldsen, M.K.; Bek, D.; Rasmussen, E.M.; Prieme, A.; Thomsen, V.O. Line probe assay for differentiation within Mycobacterium tuberculosis complex. Evaluation on clinical specimens and isolates including Mycobacterium pinnipedii. Scand. J. Infect. Dis. 2009, 41, 635–641. [Google Scholar] [CrossRef]
- Harris, S.R.; Okoro, C.K. Whole-Genome Sequencing for Rapid and Accurate Identification of Bacterial Transmission Pathways. New Approaches Prokaryotic Syst. 2014, 41, 123–152. [Google Scholar] [CrossRef]
- Zakham, F.; Aouane, O.; Ussery, D.; Benjouad, A.; Ennaji, M.M. Computational genomics-proteomics and Phylogeny analysis of twenty one mycobacterial genomes (Tuberculosis and non Tuberculosis strains). Microb. Inf. Exp. 2012, 2, 7. [Google Scholar] [CrossRef]
- Schurch, A.C.; van Soolingen, D. DNA fingerprinting of Mycobacterium tuberculosis: From phage typing to whole-genome sequencing. Infect. Genet. Evol. 2012, 12, 602–609. [Google Scholar] [CrossRef]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Primer Type and Target Locus | Forward Primer | Reverse Primer | Size (bp) |
|---|---|---|---|
| 16SrRNA | 5′ ACG GTG GGT ACT AGG TGT GGG TTT C 3′ | 5′ TCT GCG ATT ACT AGC GAC TCC GAC TTC A 3′ | 543 |
| IS6110 | 5′ TCA GCC GCG TCC ACG CCG CCA 3′ | 5′ CCG ACC GCT CCG ACC GAC GGT 3′ | 786 |
| RD9 (Rv2073c) | 5′ TCG CCG CTG CCA GAT GAG TC3′ | 5′ TTT GGG AGC CGC CGG TGG TGA TGA 3′ | 600 |
| RD4 (Rv1510) | 5′ GTG CGC TCC ACC CAA ATA GTT GC3′ | 5′ TGT CGA CCT GGG GCA CAA ATC AGT C 3′ | 1033 |
| RD12 (Rv3120) | 5′ GTC GGC GAT AGA CCA TGA GTC CGT CTC CAT3′ | 5′ GCG AAA AGT GGG CGG ATG CCA GAA TAG T 3′ | 404 |
| RD1 (Rv3877/8) | 5′ CGA CGG GTC TGA CGG CCA AAC TCA TC3′ | 5′ CTT GCT CGG TGG CCG GTT TTT CAG C 3′ | 999 |
| Regions of Difference (RD) | Suspected MTC | Frequency | Total Tested | |
|---|---|---|---|---|
| Positive + | Negative − | |||
| 16S rRNA | Mycobacteria | 133 | 3 | 136 |
| IS 6110 | Mycobacterium tuberculosis complex | 133 | 3 | 136 |
| RD9 | MTC other than M. africanum and M. bovis | 126 | 7 | 133 |
| RD4 | M. bovis/M. bovis BCG/M. caprae | 5 | 2 | 7 |
| RD12 | M. bovis/M. bovis BCG | 0 | 2 | 2 |
| RD1 | M. bovis | 2 | 0 | 2 |
| Sample Type | Drug Resistance Patterns | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NNNNN | NNNNR | NSSSR | RRRRR | RRRSR | RRSRR | RRSSR | SRRRR | SRRSR | SRSSR | SSSSR | Total | |
| Blood | 2 | 2 | ||||||||||
| Bronchial wash | 1 | 1 | ||||||||||
| Gastric Aspirates | 2 | 2 | ||||||||||
| Isolate | 2 | 1 | 2 | 2 | 12 | 19 | ||||||
| Pleural fluid | 1 | 1 | 1 | 3 | ||||||||
| Sputum | 2 | 44 | 5 | 6 | 1 | 36 | 3 | 2 | 10 | 109 | ||
| Total | 2 | 2 | 1 | 44 | 6 | 7 | 1 | 40 | 3 | 4 | 26 | 136 |
| Bioproject ID | Biosample ID | Genome/Genbank Accession | Sample Name | Sample Source | Sample Type | Interval |
|---|---|---|---|---|---|---|
| PRJNA233395 | SAMN02567763 | JKAK00000000 | Mycobacterium tuberculosis D 4155 | Pleural biopsy | Human | Baseline |
| PRJNA233396 | SAMN02567764 | JKAJ00000000 | Mycobacterium tuberculosis Kc 32216 | Isolate | Human | Baseline |
| PRJNA233392 | SAMN02567760 | JKAN00000000 | Mycobacterium tuberculosis Wt 21231 | Sputum | Human | Baseline |
| PRJNA233393 | SAMN02567761 | JKAM00000000 | Mycobacterium bovis Bz_ 31150 | Bronchial wash | Chimpanzee | Unknown |
| PRJNA233394 | SAMN02567762 | JKAL00000000 | Mycobacterium bovis B2_7505 | Sputum | Human | Unknown |
| PRJNA233397 | SAMN02567765 | JKAI00000000 | Mycobacterium tuberculosis Wt 21419 | Sputum | Human | Baseline |
| PRJNA233398 | SAMN02567766 | JKAH00000000 | Mycobacterium bovis Mr 4387 | Sputum | Human | Unknown |
| PRJNA233399 | SAMN02567767 | JKAG00000000 | Mycobacterium tuberculosis Kc 9614 | Sputum | Human | 3 mo. after start of treatment |
| Isolate | Host | Country |
|---|---|---|
| Af2122_97 M. bovis reference strain | Cattle | United Kingdom |
| M. bovis B2_7505 | Human | Uganda |
| M. bovis BZ_31150 | Chimpanzee | Uganda |
| M. tuberculosis WT 21231 | Human | Uganda |
| M. tuberculosis WT 21419 | Human | Uganda |
| M. tuberculosis KC_32216 | Human | Uganda |
| M. tuberculosis KC 9614 | Human | Uganda |
| M. tuberculosis D 4155 | Human | Uganda |
| M. tuberculosis MR 438xz | Human | Uganda |
| MBO_4303- Mycobacterium bovis strain 04-303 | Wild boar living on free-range | Argentina |
| MBAN5v1 = Mycobacterium bovis Strain AN5 | Used for Production of Purified Protein Derivative—Brazil | Brazil |
| ASM_93432v1 = Mycobacterium bovis strain SP38 | Isolated from the lungs of a bovine | Sao Paolo, Brazil |
| NE_Elk | Elk, Cervus canadensis | Minnesota, USA |
| MBO_Consentino | Elk, Cervus canadensis | Minnesota, USA |
| MBO_Elk_94_0704-Mycobacterium bovis | Elk, Cervus canadensis | (USDA/APHIS/VS/NVSL) |
| M. tuberculosis KZN strain (Kwazulu Natal Isolate) | Human | South Africa |
| M. tuberculosis CDC 1551 | Human | USA |
| Drug | Gene | Gene Product | Mutations |
|---|---|---|---|
| INH | KatG inhA | Catalase/peroxidase NADH-dependent enoyl-(ACP) reductase | S315T, R463L, A243P, V73A, W191G, Q525P S94A |
| RMP | rpoB | DNA directed RNA polymerase subunit beta | D435Y, D435V, S450L, M390T |
| PZA | pncA | Pyrazinamidase/ nicotinamidase pncA | H57D, P54Q, A134V, V163A, V131F |
| STR | rpsL rrs | rpsL 30S ribosomal protein S12 16S ribosomal RNA | None seenR309C, R338G, R173C |
| EMB | embA embC | Indolylacetylinositol arabinosyltransferase A Indolylacetylinositol arabinosyltransferase C | G884D, T608N T270I, V52X, M800V |
| OFX | gyrA | DNA gyrase subunit A | E21Q, S95T, G668D, D639A, R418W, D199G, T80A |
| Mutations in Target Gene (Corresponding Drugs) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Isolate | Antimycobacterial Agent Resistance Profile | PncA- Rv2043c (PZA) | KatG—Rv1908c (INH) | RpoB- Rv0667 (RMP) | EmbA- Rv3794 | EmbC Rv3793 | InhA -Rv1484 (INH) | GyrA- Rv0006 | 16s rRNA-rrs Rvnr01 |
| M. bovis Af2122/97 | PZA | H57D | R463L | - | - | T270I | - | E21Q; S95T D639A; G668D | - |
| M. tuberculosis CDC_1551 | n/a | - | - | - | - | V52X | - | E21Q; S95T G668D | - |
| M. bovis B2_7505 | PZA, INH, RMP, EMB, STR | H57D | S315T R463L | D435Y | - | - | S94A | E21Q; S95T R418W; G668D | R309C |
| M. bovis Bz_31150 | PZA | H57D | R463L | - | - | T270I | - | E21Q; S95T D199G; G668D | - |
| MBO Ravenel | n/a | - | R463L A243P | - | - | M800V | - | D639A G668D | - |
| MBO Elk | n/a | H57D | R463L | - | - | T270I | - | E21Q; S95T D639A; G668D | - |
| MBO Consentino | n/a | H57D | R463L V73A | - | - | T270I | - | E21Q; S95T D639A; G668D | - |
| Mtb MR 4387 | STM, RMP, INH, EMB, PZA | A134V | R463L | D435V | G884D | - | - | E21Q; S95T G668D | R173C R338G |
| Mtb D4155 | PZA | - | R463L | - | G884D | - | - | E21Q; S95T G668D | R338G |
| Mtb Kc 32216 | PZA | _ | - | - | - | - | - | E21Q; T80A S95T; G668D | - |
| Mtb Kc 9614 | RMP, INH, EMB, PZA | Y(-4)C | S315T | S450L | T608N | - | - | E21Q; T80A S95T; G668D | - |
| WT 21419 | RMP, INH, EMB, PZA | V163A | W191G | S450L | T608N | - | - | E21Q; T80A S95T; G668D | - |
| WT 21231 | STM, RMP, INH, EMB, PZA | V131F | Q525P | M390T; S450L | - | - | - | E21Q | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wanzala, S.I.; Nakavuma, J.; Travis, D.; Kia, P.; Ogwang, S.; Waters, W.R.; Thacker, T.; Johnson, T.; Hadi, S.A.; Sreevatsan, S. Retrospective Analysis of Archived Pyrazinamide Resistant Mycobacterium tuberculosis Complex Isolates from Uganda—Evidence of Interspecies Transmission. Microorganisms 2019, 7, 221. https://doi.org/10.3390/microorganisms7080221
Wanzala SI, Nakavuma J, Travis D, Kia P, Ogwang S, Waters WR, Thacker T, Johnson T, Hadi SA, Sreevatsan S. Retrospective Analysis of Archived Pyrazinamide Resistant Mycobacterium tuberculosis Complex Isolates from Uganda—Evidence of Interspecies Transmission. Microorganisms. 2019; 7(8):221. https://doi.org/10.3390/microorganisms7080221
Chicago/Turabian StyleWanzala, Sylvia I., Jesca Nakavuma, Dominic Travis, Praiscillia Kia, Sam Ogwang, Wade Ray Waters, Tyler Thacker, Timothy Johnson, Syeda Anum Hadi, and Srinand Sreevatsan. 2019. "Retrospective Analysis of Archived Pyrazinamide Resistant Mycobacterium tuberculosis Complex Isolates from Uganda—Evidence of Interspecies Transmission" Microorganisms 7, no. 8: 221. https://doi.org/10.3390/microorganisms7080221
APA StyleWanzala, S. I., Nakavuma, J., Travis, D., Kia, P., Ogwang, S., Waters, W. R., Thacker, T., Johnson, T., Hadi, S. A., & Sreevatsan, S. (2019). Retrospective Analysis of Archived Pyrazinamide Resistant Mycobacterium tuberculosis Complex Isolates from Uganda—Evidence of Interspecies Transmission. Microorganisms, 7(8), 221. https://doi.org/10.3390/microorganisms7080221