A Novel Bioinformatics Strategy to Analyze Microbial Big Sequence Data for Efficient Knowledge Discovery: Batch-Learning Self-Organizing Map (BLSOM)

Abstract
:1. Introduction
2. An Alignment-Free Clustering Method “BLSOM” Developed for Genome Informatics
3. Basic Characteristics of BLSOM Separation
 ), stickleback (
), stickleback (  ), Takifugu rubripes (
), Takifugu rubripes (  ), Tetraodon nigroviridis (
), Tetraodon nigroviridis (  ), zebrafish (
), zebrafish (  ), chicken (
), chicken (  ), human (
), human (  ), mouse (
), mouse (  ), lizard (
), lizard (  ) and Xenopus (
) and Xenopus (  ). Lattice points containing no sequences after BLSOM calculation are indicated in white blank; (B) G+C%. The G+C% obtained for each lattice point on DegPenta is divided into 21 groups containing an equal number of lattice points, and the highest and lowest groups are shown in dark wine red and dark green, respectively, as presented under this panel [13]; and (C) Examples of pentanucleotides diagnostic for species- and phylotype-dependent separation. Occurrence of pentanucleotides for each lattice point has been calculated and normalized with the occurrence expected from the mononucleotide composition for each lattice point [11]. This observed/expected ratio is indicated in color, presented under the panel C.
), stickleback ( ), Takifugu rubripes ( ), Tetraodon nigroviridis ( ), zebrafish ( ), chicken ( ), human ( ), mouse ( ), lizard ( ) and Xenopus ( ). Lattice points containing no sequences after BLSOM calculation are indicated in white blank; (B) G+C%. The G+C% obtained for each lattice point on DegPenta is divided into 21 groups containing an equal number of lattice points, and the highest and lowest groups are shown in dark wine red and dark green, respectively, as presented under this panel [13]; and (C) Examples of pentanucleotides diagnostic for species- and phylotype-dependent separation. Occurrence of pentanucleotides for each lattice point has been calculated and normalized with the occurrence expected from the mononucleotide composition for each lattice point [11]. This observed/expected ratio is indicated in color, presented under the panel C.
). Lattice points containing no sequences after BLSOM calculation are indicated in white blank; (B) G+C%. The G+C% obtained for each lattice point on DegPenta is divided into 21 groups containing an equal number of lattice points, and the highest and lowest groups are shown in dark wine red and dark green, respectively, as presented under this panel [13]; and (C) Examples of pentanucleotides diagnostic for species- and phylotype-dependent separation. Occurrence of pentanucleotides for each lattice point has been calculated and normalized with the occurrence expected from the mononucleotide composition for each lattice point [11]. This observed/expected ratio is indicated in color, presented under the panel C.
), stickleback ( ), Takifugu rubripes ( ), Tetraodon nigroviridis ( ), zebrafish ( ), chicken ( ), human ( ), mouse ( ), lizard ( ) and Xenopus ( ). Lattice points containing no sequences after BLSOM calculation are indicated in white blank; (B) G+C%. The G+C% obtained for each lattice point on DegPenta is divided into 21 groups containing an equal number of lattice points, and the highest and lowest groups are shown in dark wine red and dark green, respectively, as presented under this panel [13]; and (C) Examples of pentanucleotides diagnostic for species- and phylotype-dependent separation. Occurrence of pentanucleotides for each lattice point has been calculated and normalized with the occurrence expected from the mononucleotide composition for each lattice point [11]. This observed/expected ratio is indicated in color, presented under the panel C.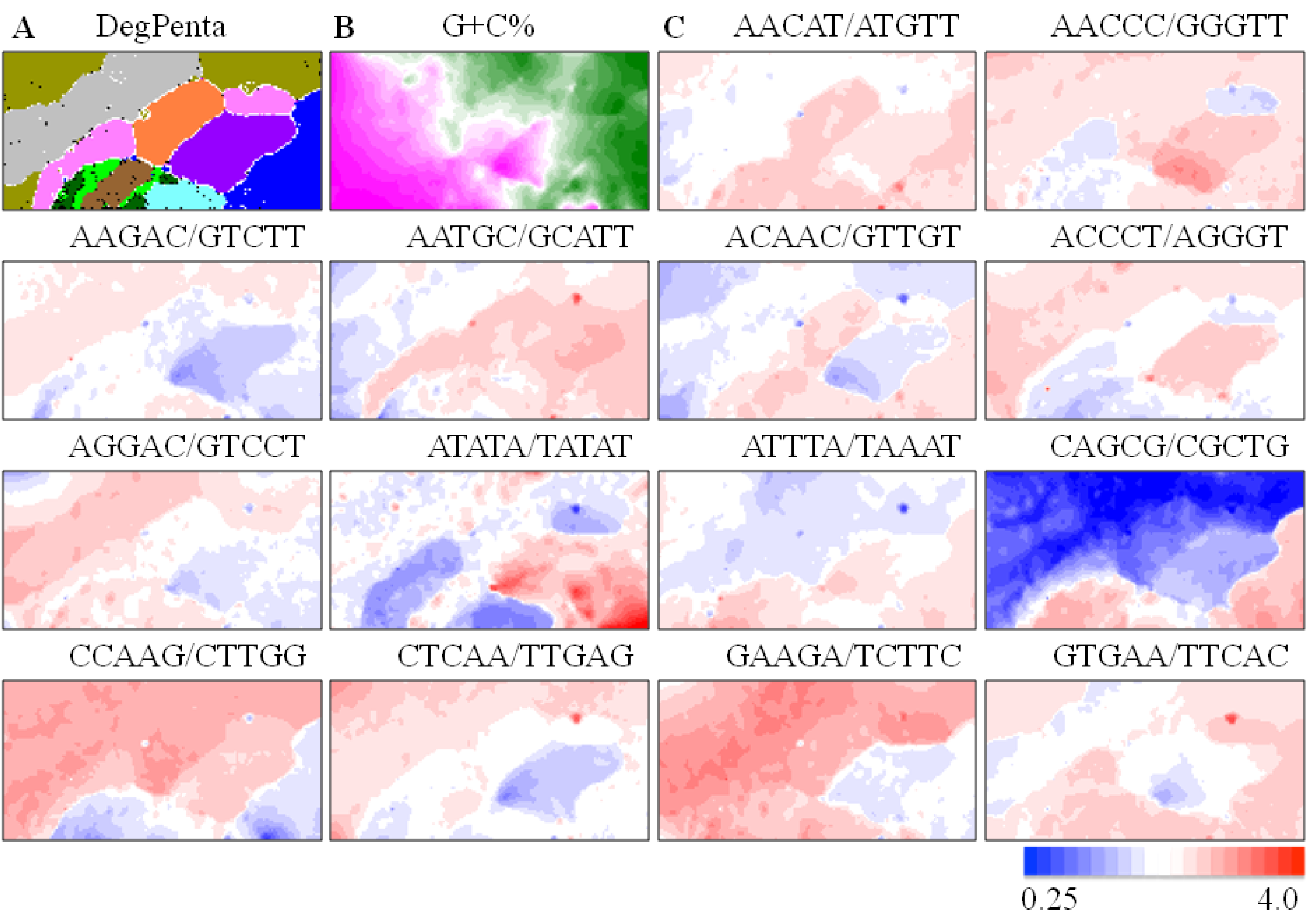
4. Application of BLSOM to Metagenome Studies
4.1. Application to Phylogenetic Classification of Millions of Metagenomic Sequences
 ) or (
) or (  ), respectively; those containing sequences from a single known phylotype are colored as described in the legend; and those including sequences from more than one phylotype or including both environmental sequences and species-known sequences are shown in black; for details, see Abe et al. [10].
), respectively; those containing sequences from a single known phylotype are colored as described in the legend; and those including sequences from more than one phylotype or including both environmental sequences and species-known sequences are shown in black; for details, see Abe et al. [10]. ) bar in the “Sargasso Seq. unclassified” panel (Figure 2C); 79% of the Sargasso sequences belonged to this phylotype-unclassified category. These Sargasso sequences should correspond to sequences derived from genomes that had been poorly-characterized at that time. The remaining 21% of the Sargasso sequences are associated with species-known sequences (black lattices in Figure 2A); the number of Sargasso sequences classified into lattice points containing species-known sequences is indicated by the height of the colored bar in the “3D, Sargasso Seq. classified” panel (Figure 2D), which represents the phylotype of the associated species-known sequences; and 92 genera, whose sequences are associated with the Sargasso sequences, have been reported together with the numbers of the associated Sargasso sequences [10].) or ( ); those including environmental and species-known sequences or those from more than one known phylotype are indicated in black, and those containing species-known sequences from a single phylotype are indicated in color as follows: α-proteobacteria ( ), β-proteobacteria (
) bar in the “Sargasso Seq. unclassified” panel (Figure 2C); 79% of the Sargasso sequences belonged to this phylotype-unclassified category. These Sargasso sequences should correspond to sequences derived from genomes that had been poorly-characterized at that time. The remaining 21% of the Sargasso sequences are associated with species-known sequences (black lattices in Figure 2A); the number of Sargasso sequences classified into lattice points containing species-known sequences is indicated by the height of the colored bar in the “3D, Sargasso Seq. classified” panel (Figure 2D), which represents the phylotype of the associated species-known sequences; and 92 genera, whose sequences are associated with the Sargasso sequences, have been reported together with the numbers of the associated Sargasso sequences [10].) or ( ); those including environmental and species-known sequences or those from more than one known phylotype are indicated in black, and those containing species-known sequences from a single phylotype are indicated in color as follows: α-proteobacteria ( ), β-proteobacteria (  ), γ-proteobacteria (
), γ-proteobacteria (  ), δ-proteobacteria (
), δ-proteobacteria (  ), ε-proteobacteria (
), ε-proteobacteria (  ), Actinobacteria ( ), Aquificae (
), Actinobacteria ( ), Aquificae (  ), Bacteroidetes (
), Bacteroidetes (  ), Chlamydiae (
), Chlamydiae (  ), Chlorobi (
), Chlorobi (  ), Chloroflexi ( ), Crenarchaeota ( ), Cyanobacteria (
), Chloroflexi ( ), Crenarchaeota ( ), Cyanobacteria (  ), Deinococcus-Thermus (
), Deinococcus-Thermus (  ), Dictyoglomi (
), Dictyoglomi (  ), Euryarchaeota ( ), Fibrobacteres (
), Euryarchaeota ( ), Fibrobacteres (  ), Firmicutes (
), Firmicutes (  ), Fusobacteria ( ), Nitrospirae (
), Fusobacteria ( ), Nitrospirae (  ), Planctomycetes (
), Planctomycetes (  ), Spirochaetales (
), Spirochaetales (  ), Thermodesulfobacteriales (
), Thermodesulfobacteriales (  ), Thermotogales ( ) and Verrucomicrobiae (
), Thermotogales ( ) and Verrucomicrobiae (  ); (B) “3D, Biofilm Seq.”. The square root of the number of biofilm sequences classified into each lattice point is shown by the height of the bar distinctively colored to show the dominant species reported by Tyson et al. [31]: Ferroplasma ( ), Leptospirillum ( ) and Thermoplasmatales ( ); (C) “3D, Sargasso Seq. unclassified”. The number of Sargasso sequences located at the lattice points containing no species-known sequences is shown by the height of the bar indicated in color ( ), as described in B; and (D) “3D, Sargasso Seq. classified”. The number of Sargasso sequences classified into the lattice points containing species-known sequences from a single phylotype is indicated by the height of the bar distinctively colored to show the phylotype, as described in A. For details, see Abe et al. [10].
) or ( ); those including environmental and species-known sequences or those from more than one known phylotype are indicated in black, and those containing species-known sequences from a single phylotype are indicated in color as follows: α-proteobacteria ( ), β-proteobacteria ( ), γ-proteobacteria ( ), δ-proteobacteria ( ), ε-proteobacteria ( ), Actinobacteria ( ), Aquificae ( ), Bacteroidetes ( ), Chlamydiae ( ), Chlorobi ( ), Chloroflexi ( ), Crenarchaeota ( ), Cyanobacteria ( ), Deinococcus-Thermus ( ), Dictyoglomi ( ), Euryarchaeota ( ), Fibrobacteres ( ), Firmicutes ( ), Fusobacteria ( ), Nitrospirae ( ), Planctomycetes ( ), Spirochaetales ( ), Thermodesulfobacteriales ( ), Thermotogales ( ) and Verrucomicrobiae ( ); (B) “3D, Biofilm Seq.”. The square root of the number of biofilm sequences classified into each lattice point is shown by the height of the bar distinctively colored to show the dominant species reported by Tyson et al. [31]: Ferroplasma ( ), Leptospirillum ( ) and Thermoplasmatales ( ); (C) “3D, Sargasso Seq. unclassified”. The number of Sargasso sequences located at the lattice points containing no species-known sequences is shown by the height of the bar indicated in color ( ), as described in B; and (D) “3D, Sargasso Seq. classified”. The number of Sargasso sequences classified into the lattice points containing species-known sequences from a single phylotype is indicated by the height of the bar distinctively colored to show the phylotype, as described in A. For details, see Abe et al. [10].
); (B) “3D, Biofilm Seq.”. The square root of the number of biofilm sequences classified into each lattice point is shown by the height of the bar distinctively colored to show the dominant species reported by Tyson et al. [31]: Ferroplasma ( ), Leptospirillum ( ) and Thermoplasmatales ( ); (C) “3D, Sargasso Seq. unclassified”. The number of Sargasso sequences located at the lattice points containing no species-known sequences is shown by the height of the bar indicated in color ( ), as described in B; and (D) “3D, Sargasso Seq. classified”. The number of Sargasso sequences classified into the lattice points containing species-known sequences from a single phylotype is indicated by the height of the bar distinctively colored to show the phylotype, as described in A. For details, see Abe et al. [10].
) or ( ); those including environmental and species-known sequences or those from more than one known phylotype are indicated in black, and those containing species-known sequences from a single phylotype are indicated in color as follows: α-proteobacteria ( ), β-proteobacteria ( ), γ-proteobacteria ( ), δ-proteobacteria ( ), ε-proteobacteria ( ), Actinobacteria ( ), Aquificae ( ), Bacteroidetes ( ), Chlamydiae ( ), Chlorobi ( ), Chloroflexi ( ), Crenarchaeota ( ), Cyanobacteria ( ), Deinococcus-Thermus ( ), Dictyoglomi ( ), Euryarchaeota ( ), Fibrobacteres ( ), Firmicutes ( ), Fusobacteria ( ), Nitrospirae ( ), Planctomycetes ( ), Spirochaetales ( ), Thermodesulfobacteriales ( ), Thermotogales ( ) and Verrucomicrobiae ( ); (B) “3D, Biofilm Seq.”. The square root of the number of biofilm sequences classified into each lattice point is shown by the height of the bar distinctively colored to show the dominant species reported by Tyson et al. [31]: Ferroplasma ( ), Leptospirillum ( ) and Thermoplasmatales ( ); (C) “3D, Sargasso Seq. unclassified”. The number of Sargasso sequences located at the lattice points containing no species-known sequences is shown by the height of the bar indicated in color ( ), as described in B; and (D) “3D, Sargasso Seq. classified”. The number of Sargasso sequences classified into the lattice points containing species-known sequences from a single phylotype is indicated by the height of the bar distinctively colored to show the phylotype, as described in A. For details, see Abe et al. [10].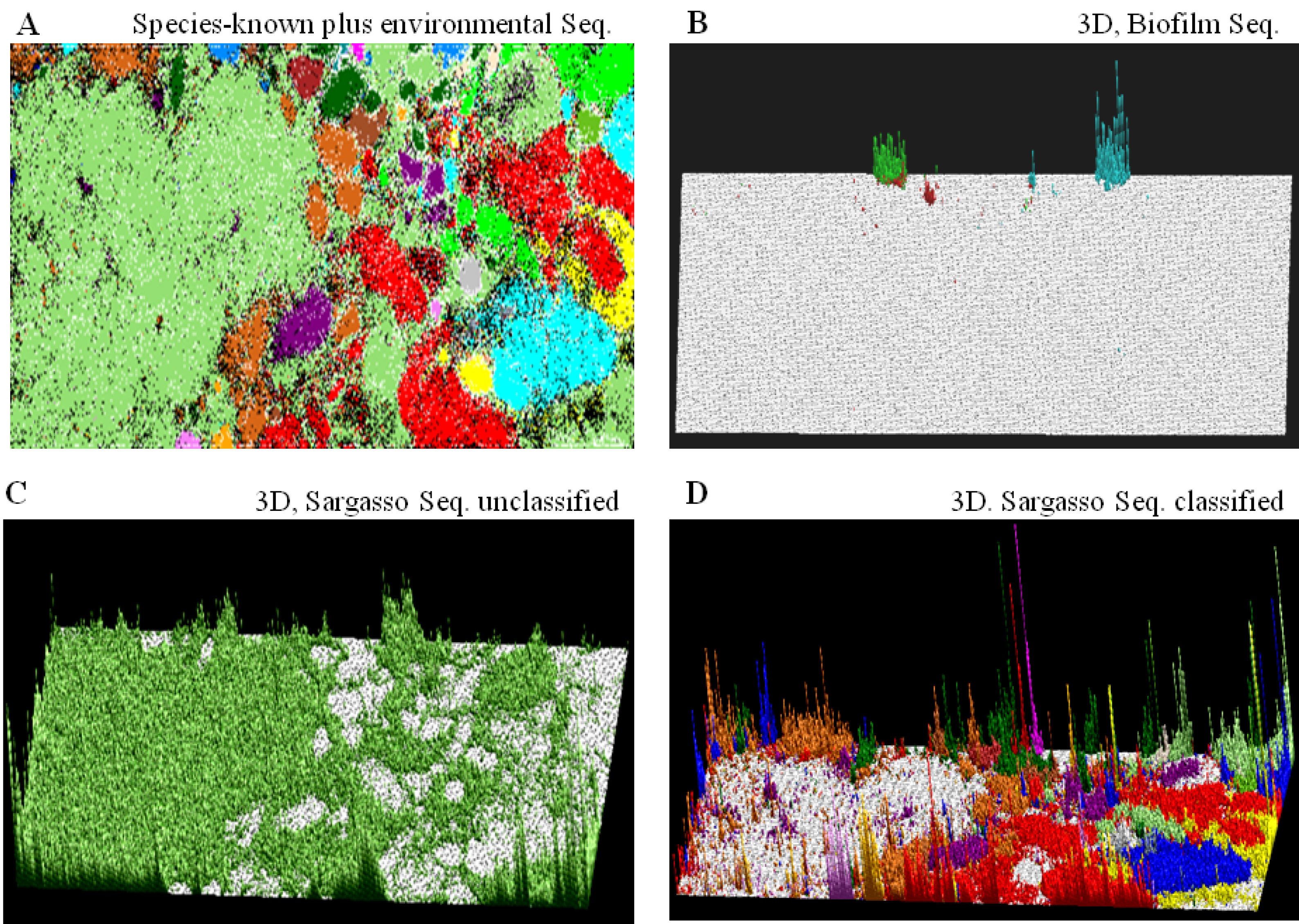
4.2. A General Strategy of Phylogenetic Assignments of Metagenomic Sequences

4.3. An Application to a Microbiome Analysis of Ticks and Other Applications
5. Oligonucleotide-BLSOM Applied to Studies of the Influenza Virus Genomes
5.1. Host-Dependent Clustering of Influenza Virus Genome Sequences
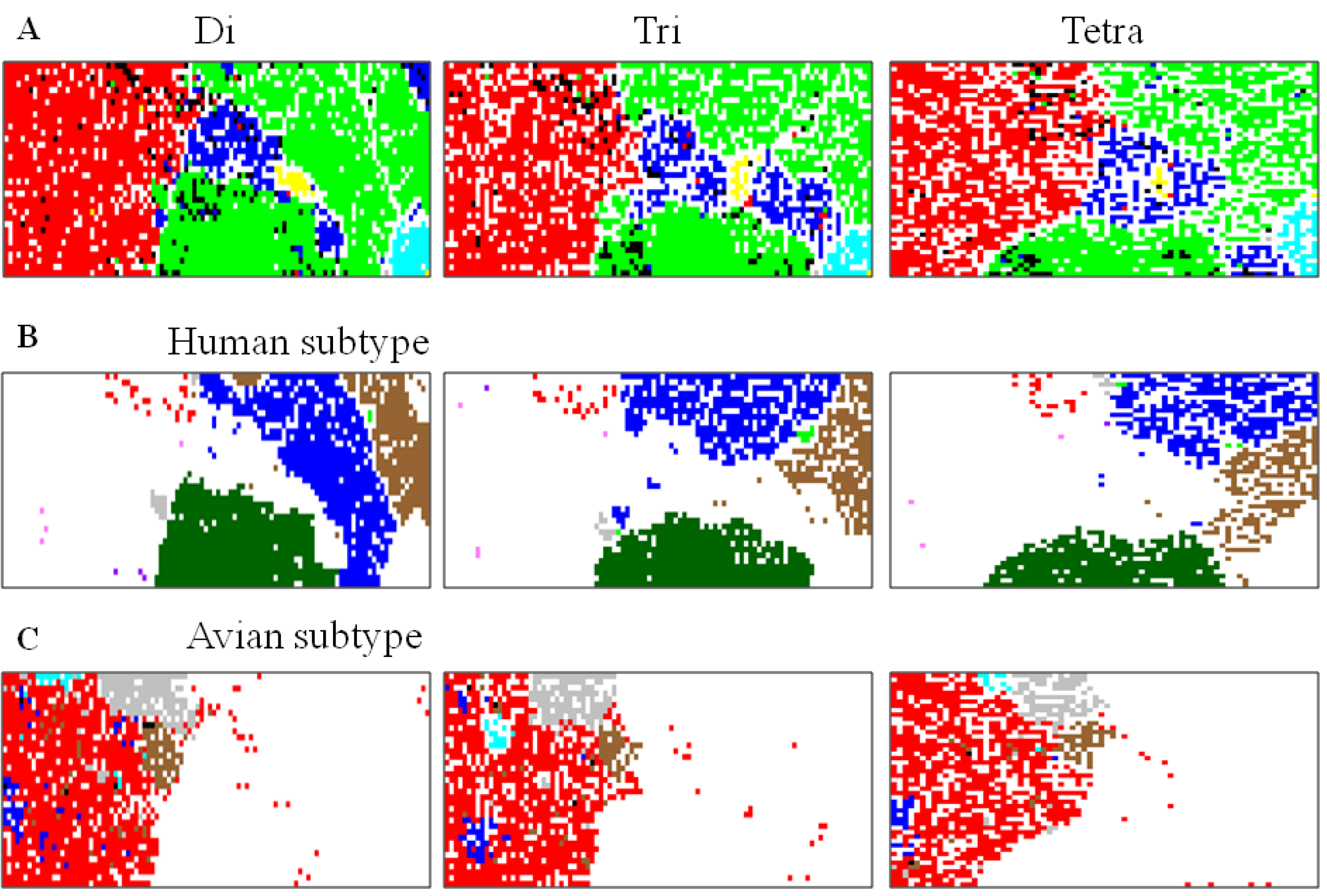
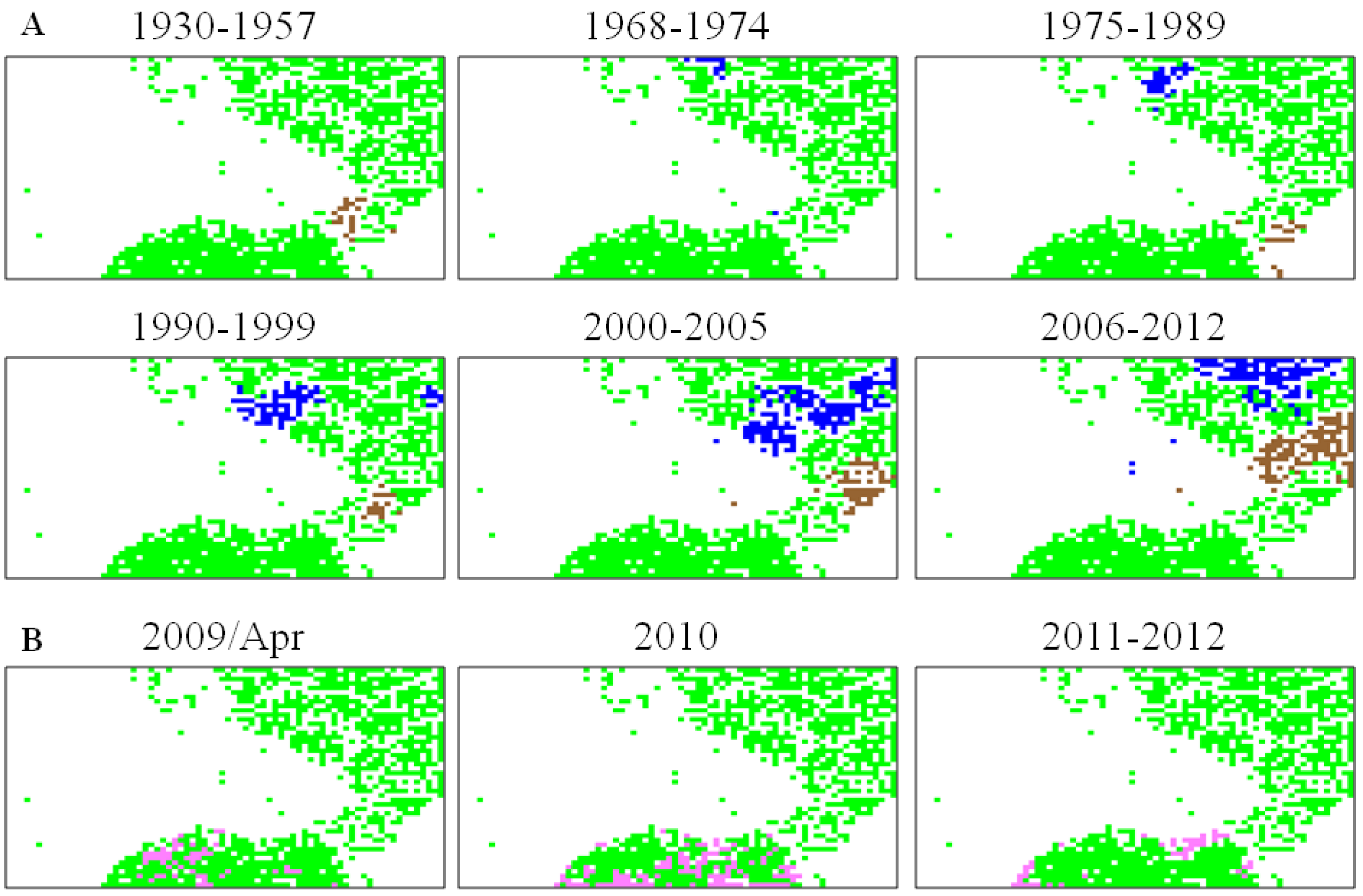
5.2. Retrospective Time Series Changes Visualized for Human Viruses
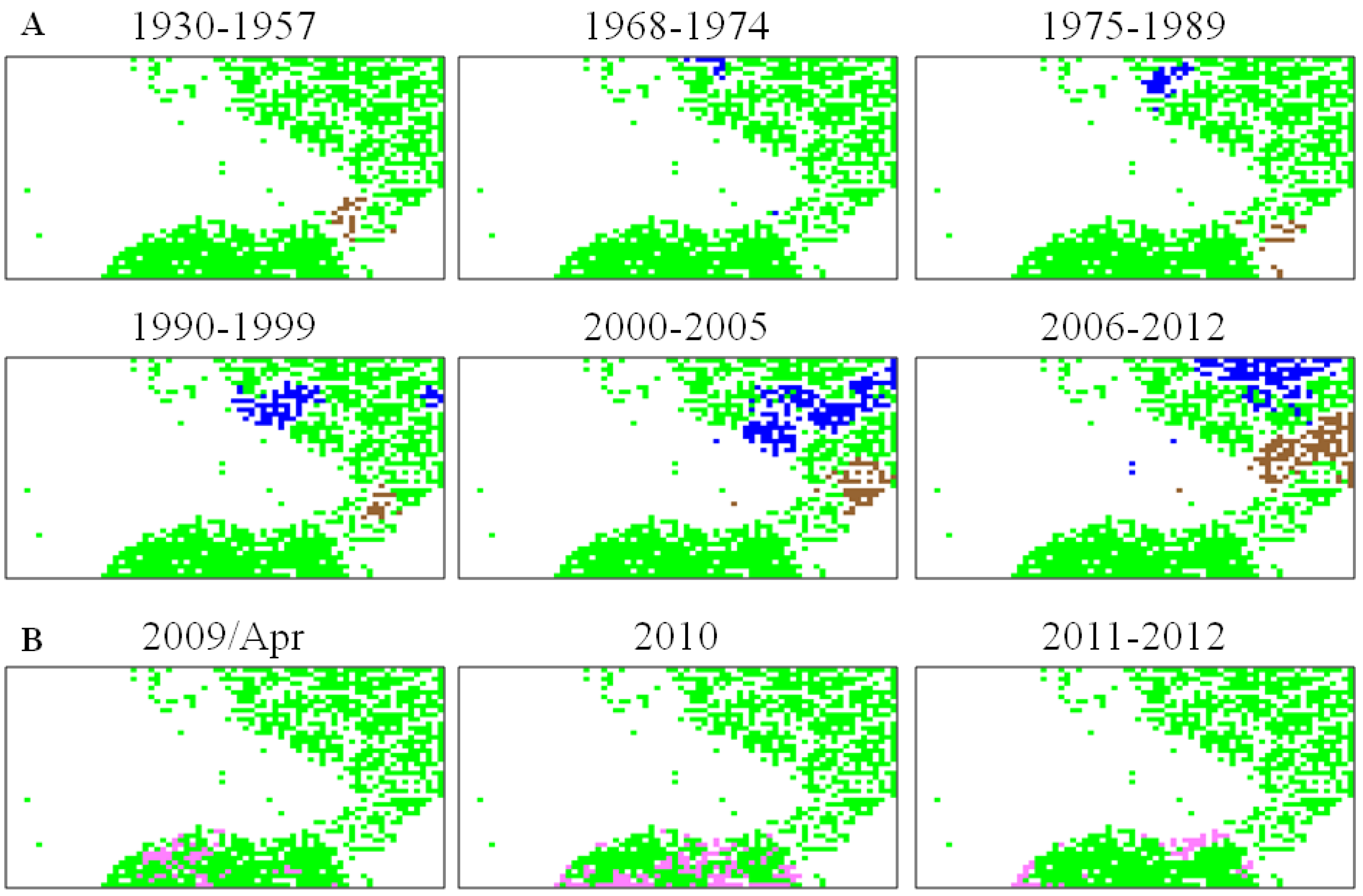
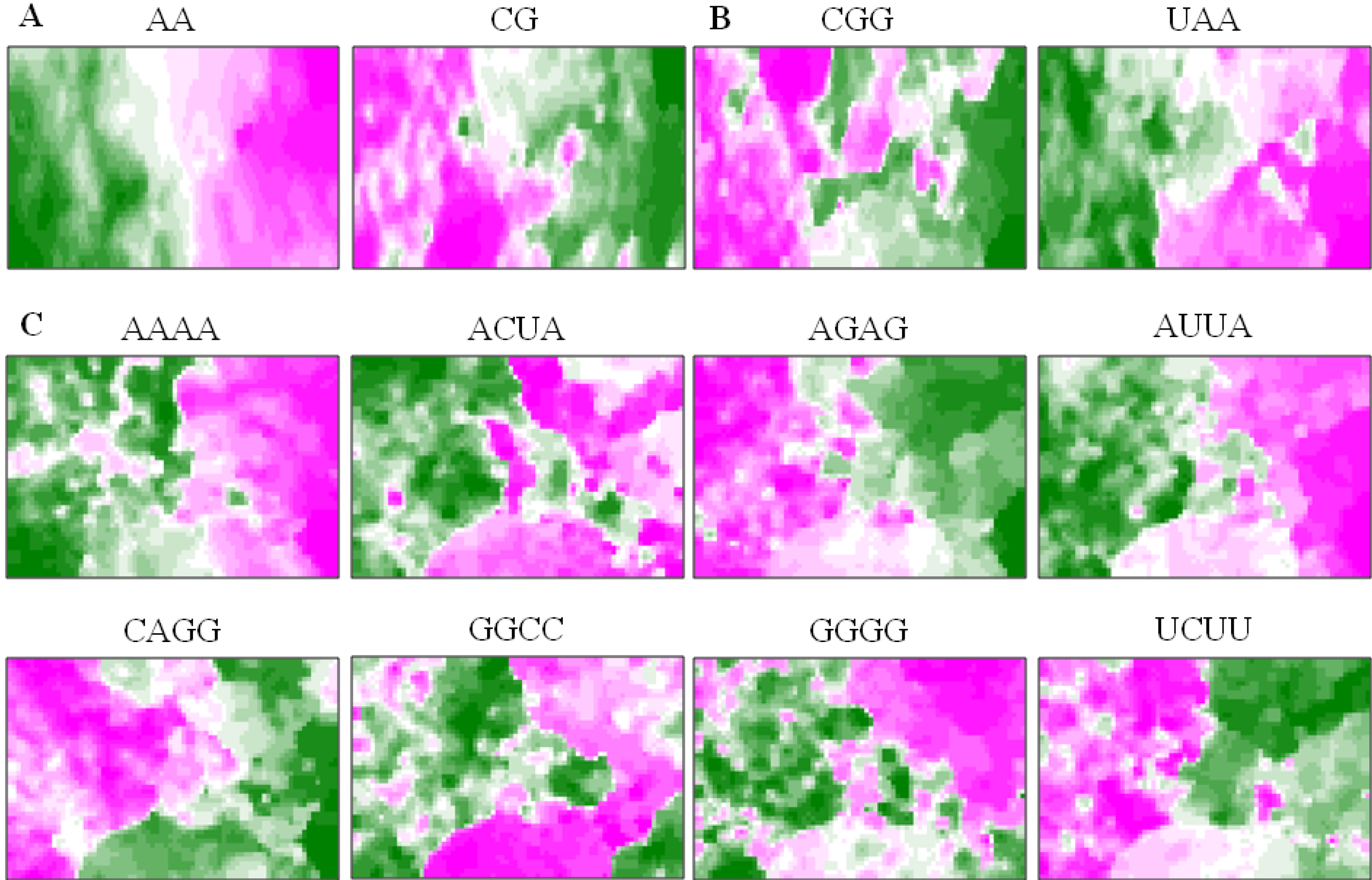
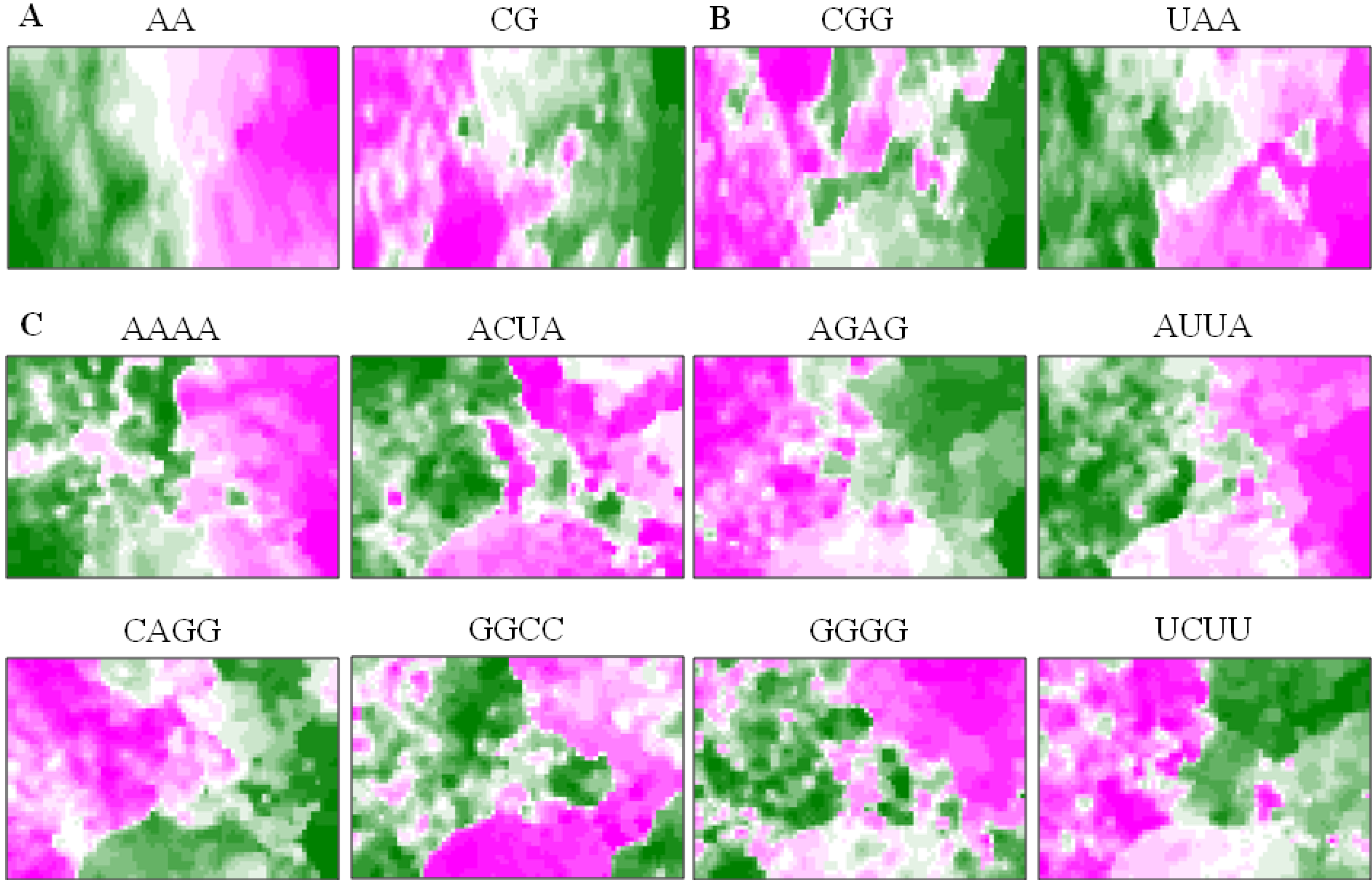
5.3. Diagnostic Oligonucleotides Responsible for Host-Dependent Separation
5.4. A Strategy for Finding Potentially Hazardous Strains

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Diagnostic Tetranucleotides |
|---|---|
| High | AAGU, ACUA, ACUU, AUGA, AUUA, |
| AUUU, CUAA, CUUU, GCCG, GGCC, | |
| UAAG, UAUC, UCAU, UUAA, UUAU | |
| Low | ACCG, ACGC, AUCU, CUCA, CUGA, |
| GAGC, GAGG, GAUC, GCAG, GCUG, GGAG |
| Point | Subtype | Year | Strain |
|---|---|---|---|
| 18 | H1N1 | 2009 | A/turkey/Ontario/FAV110 |
| 18 | H1N1 | 1988 | A/turkey/NC/19762 |
| 18 | H1N1 | 1988 | A/turkey/NC/17026 |
| 17 | H1N1 | 2009 | A/turkey/Ontario/FAV114-17 |
| 17 | H1N1 | 1992 | A/turkey/IA/21089-3 |
| 17 | H1N1 | 1991 | A/chicken/PA/35154 |
| 15 | H1N1 | 1980 | A/turkey/Kansas/4880 |
| 14 | H4N2 | 2006 | A/Pekin duck/California/P30 |
| 11 | H3N8 | 1987 | A/duck/LA/17G |
| 11 | H3N2 | 2011 | A/turkey/Ontario/FAV-9 |
| 11 | H1N2 | 2001 | A/duck/NC/91347 |
| 10 | H3N2 | 2011 | A/turkey/Ontario/FAV-10 |
| 9 | H6N2 | 2004 | A/chicken/CA/S0403106 |
| 9 | H6N2 | 2002 | A/wild duck/Shantou/867 |
| 9 | H5N2 | 2012 | A/chicken/Taiwan/A1997 |
| 9 | H5N2 | 2002 | A/chicken/Guatemala/194573 |
5.5. BLSOM Analyses of Individual Segments
6. Other Applications of BLSOM and Future Prospects
6.1. Addition of Computer-Generated Random Sequences
6.2. Peptide-BLSOM for Functional Prediction of Poorly-Characterized Protein Candidates
Acknowledgments
Conflicts of Interest
References
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Kanaya, S.; Kinouchi, M.; Abe, T.; Kudo, Y.; Yamada, Y.; Nishi, T.; Mori, H.; Ikemura, T. Analysis of codon usage diversity of bacterial genes with a self-organizing map (SOM): Characterization of horizontally transferred genes with emphasis on the E. coli O157 genome. Gene 2001, 276, 89–99. [Google Scholar] [CrossRef]
- Abe, T.; Kanaya, S.; Kinouchi, M.; Ichiba, Y.; Kozuki, T.; Ikemura, T. Informatics for unveiling hidden genome signatures. Genome Res. 2003, 13, 693–702. [Google Scholar] [CrossRef]
- Abe, T.; Sugawara, H.; Kanaya, S.; Ikemura, T. Sequences from almost all prokaryotic, eukaryotic, and viral genomes available could be classified according to genomes on a large-scale Self-Organizing Map constructed with the Earth Simulator. J. Earth Simulator 2006, 6, 17–23. [Google Scholar]
- Karlin, S.; Campbell, A.M.; Mrazek, J. Comparative DNA analysis across diverse genomes. Annu. Rev. Genet. 1998, 32, 185–225. [Google Scholar] [CrossRef]
- Kohonen, T. Self-organized formation of topologi-cally correct feature maps. Biol. Cybern. 1982, 43, 59–69. [Google Scholar] [CrossRef]
- Kohonen, T.; Oja, E.; Simula, O.; Visa, A.; Kangas, J. Engineering applications of the self-organizing map. Proc. IEEE 1996, 84, 1358–1384. [Google Scholar] [CrossRef]
- Abe, T.; Sugawara, H.; Kinouchi, M.; Kanaya, S.; Ikemura, T. Novel phylogenetic studies of genomic sequence fragments derived from uncultured microbe mixtures in environmental and clinical samples. DNA Res. 2005, 12, 281–290. [Google Scholar]
- Abe, T.; Sugawara, H.; Kinouchi, M.; Kanaya, S.; Ikemura, T. A large-scale Self-Organizing Map (SOM) unveils sequence characteristics of a wide range of eukaryote genomes. Gene 2006, 365, 27–34. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Wada, K.; Wada, Y.; Abe, T.; Ikemura, T. Notable clustering of transcription-factor-binding motifs in human pericentric regions and its biological significance. Chromosome Res. 2013, 21, 461–474. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Abe, T.; Wada, K.; Itoh, M.; Ikemura, T. Prediction of directional changes of influenza a virus genome sequences with emphasis on pandemic H1N1/09 as a model case. DNA Res. 2011, 18, 125–136. [Google Scholar] [CrossRef]
- Bernardi, G.; Olofsson, B.; Filipski, J.; Zerial, M.; Salinas, J.; Cuny, G.; Meunier-Rotival, M.; Rodier, F. The mosaic genome of warm-blooded vertebrates. Science 1985, 228, 953–958. [Google Scholar]
- Ikemura, T. Codon usage and transfer RNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar]
- Ikemura, T.; Aota, S. Global variation in G + C content along vertebrate genome DNA: Possible correlation with chromosome band structures. J. Mol. Biol. 1988, 203, 1–13. [Google Scholar] [CrossRef]
- Ikemura, T.; Wada, K. Evident diversity of codon usage patterns of human genes with respect to chromosome banding patterns and chromosome numbers; relation between nucleotide sequence data and cytogenetic data. Nucleic Acids Res. 1991, 19, 4333–4339. [Google Scholar] [CrossRef]
- Bernardi, G. Structural and Evolutionary Genomics: Natural Selection in Genome Evolution; Elsevier Science: New York, NY, USA, 2004. [Google Scholar]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar]
- DeLong, E.F. Microbial population genomics and ecology. Curr. Opin. Microbiol. 2002, 5, 520–524. [Google Scholar] [CrossRef]
- Lorenz, P.; Liebeton, K.; Niehaus, F.; Eck, J. Screening for novel enzymes for biocatalytic processes: Accessing the metagenome as a resource of novel functional sequence space. Curr. Opin. Biotechnol. 2002, 13, 572–577. [Google Scholar] [CrossRef]
- Hugenholtz, P.; Pace, N.R. Identifying microbial diversity in the natural environment: A molecular phylogenetic approach. Trends Biotechnol. 1996, 14, 190–197. [Google Scholar] [CrossRef]
- Rondon, M.R.; August, P.R.; Bettermann, A.D.; Brady, S.F.; Grossman, T.H.; Liles, M.R.; Loiacono, K.A.; Lynch, B.A.; MacNeil, I.A.; Minor, C.; et al. Cloning the soil metagenome: A strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl. Environ. Microbiol. 2000, 66, 2541–2547. [Google Scholar] [CrossRef]
- Schloss, P.D.; Handelsman, J. Biotechnological prospects from metagenomics. Curr. Opin. Biotechnol. 2003, 14, 303–310. [Google Scholar] [CrossRef]
- DeLong, E.F.; Preston, C.M.; Mincer, T.; Rich, V.; Hallam, S.J.; Frigaard, N.U.; Martinez, A.; Sullivan, M.B.; Edwards, R.; Brito, B.R.; et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 2006, 311, 496–503. [Google Scholar]
- Frias-Lopez, J.; Shi, Y.; Tyson, G.W.; Coleman, M.L.; Schuster, S.C.; Chisholm, S.W.; Delong, E.F. Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. USA 2008, 105, 3805–3810. [Google Scholar]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, K.; Itoh, T.; Kuwahara, T.; Oshima, K.; Toh, H.; Toyoda, A.; Takami, H.; Morita, H.; Sharma, V.K.; Srivastava, T.P.; et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007, 14, 169–181. [Google Scholar] [CrossRef]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef]
- Tyson, G.W.; Chapman, J.; Hugenholtz, P.; Allen, E.E.; Ram, R.J.; Richardson, P.M.; Solovyev, V.V.; Rubin, E.M.; Rokhsar, D.S.; Banfield, J.F. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004, 428, 37–43. [Google Scholar] [CrossRef]
- Dick, G.J.; Andersson, A.F.; Baker, B.J.; Simmons, S.L.; Thomas, B.C.; Yelton, A.P.; Banfield, J.F. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 2009, 10. [Google Scholar] [CrossRef]
- Ultsch, A.; Moerchen, F. ESOM-Maps: Tools for Clustering, Visualization, and Classification with Emergent SOM; Technical Report No. 46; University of Marburg: Marburg, Germany, 2005. [Google Scholar]
- Nakao, R.; Abe, T.; Nijhof, A.M.; Yamamoto, S.; Jongejan, F.; Ikemura, T.; Sugimoto, C. A novel approach, based on BLSOMs (Batch Learning Self-Organizing Maps), to the microbiome analysis of ticks. ISME J. 2013, 7, 1003–1015. [Google Scholar] [CrossRef]
- Hayashi, H.; Abe, T.; Sakamoto, M.; Ohara, H.; Ikemura, T.; Sakka, K.; Benno, Y. Direct cloning of genes encoding novel xylanases from human gut. Can. J. Microbiol. 2005, 51, 251–259. [Google Scholar] [CrossRef]
- Uchiyama, T.; Abe, T.; Ikemura, T.; Watanabe, K. Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat. Biotechnol. 2005, 23, 88–93. [Google Scholar]
- Kosaka, T.; Kato, S.; Shimoyama, T.; Ishii, S.; Abe, T.; Watanabe, K. The genome of Pelotomaculum thermopropionicum reveals niche-associated evolution in anaerobic microbiota. Genome Res. 2008, 18, 442–448. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Swine influenza A (H1N1) infection in two children—South California, March–April 2009. Morb. Mortal. Wkly. Rep. 2009, 58, 400–402. [Google Scholar]
- Smith, G.J.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and genetic characteristics of swine-origin 2009 A (H1N1) influenza viruses circulating in humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef]
- Bush, R.M.; Bende, C.A.; Subbarao, K.; Cox, N.J.; Fitch, W.M. Predicting the evolution of human influenza A. Science 1999, 286, 1921–1925. [Google Scholar] [CrossRef]
- Suzuki, Y.; Gojobori, T. A method for detecting positive selection at single amino acid sites. Mol. Biol. Evol. 1999, 16, 1315–1328. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Abe, T.; Wada, Y.; Wada, K.; Ikemura, T. Novel bioinformatics strategies for prediction of directional sequence changes in influenza virus genomes and for surveillance of potentially hazardous strains. BMC Infect. Dis. 2013, 13, 386–394. [Google Scholar] [CrossRef]
- Bao, Y.; Bolotov, P.; Dernovoy, D.; Kiryutin, B.; Zaslavsky, L.; Tatusova, T.; Ostell, J.; Lipman, D. The influenza virus resource at the National Center for Biotechnology Information. J. Virol. 2008, 82, 596–601. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Ikemura, T.; Wada, K; Wada, Y.; Abe, T. Novel Bioinformatics Method to Analyze More than 10,000 Influenza Virus Strains Easily at Once Batch-Learning Self Organizing Map (BLSOM). In Advance in Viral Genome Research; Borrelli, J.A., Giannini, Y.D., Eds.; Nova Science Publishers: New York, NY, USA, 2013; pp. 95–112. [Google Scholar]
- Garcia-Sastre, A. Inhibition of interferon-mediated antiviral responses by influenza A viruses and other negative-strand RNA viruses. Virology 2001, 279, 375–384. [Google Scholar] [CrossRef]
- Voinnet, O. Induction and suppression of RNA silencing: Insights from viral infections. Nat. Rev. Genet. 2005, 6, 206–220. [Google Scholar] [CrossRef]
- Nelson, M.I.; Holmes, E.C. The evolution of epidemic influenza. Nat. Rev. Genet. 2007, 8, 196–205. [Google Scholar] [CrossRef]
- Alexey, A.; Moelling, K. Dicer is involved in protection against influenza A virus infection. J. Gen. Virol. 2007, 88, 2627–2635. [Google Scholar] [CrossRef]
- Liu, D.; Shi, W.; Shi, Y.; Wang, D.; Xiao, H.; Li, W.; Bi, Y.; Wu, Y.; Li, X.; Yan, J.; et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: Phylogenetic, structural, and coalescent analyses. Lancet 2013, 381, 1926–1932. [Google Scholar] [CrossRef]
- Rabadan, R.; Levine, A.J.; Robins, H. Comparison of avian and human influenza A viruses reveals a mutational bias on the viral genomes. J. Virol. 2006, 80, 11887–11891. [Google Scholar] [CrossRef]
- Berhane, Y.; Ojkic, D.; Neufeld, J.; Leith, M.; Hisanaga, T.; Kehler, H.; Ferencz, A.; Wojcinski, H.; Cottam-Birt, C.; Suderman, M.; et al. Molecular characterization of pandemic H1N1 influenza viruses isolated from turkeys and pathogenicity of a human pH1N1 isolate in turkeys. Avian Dis. 2010, 54, 1275–1285. [Google Scholar] [CrossRef]
- Abe, T.; Wada, K.; Iwasaki, Y.; Ikemura, T. Novel bioinformatics for inter- and intraspecies comparison of genome signatures in plant genomes. Plant Biotechnol. 2009, 26, 469–477. [Google Scholar] [CrossRef]
- Uehara, H.; Iwasaki, Y.; Wada, C.; Ikemura, T.; Abe, T. A novel bioinformatics strategy for searching industrially useful genome resources from metagenomic sequence libraries. Genes Genet. Sys. 2011, 86, 53–66. [Google Scholar] [CrossRef]
- Abe, T.; Kanaya, S.; Uehara, H.; Ikemura, T. A novel bioinformatics strategy for function prediction of poorly-characterized protein genes obtained from metagenome analyses. DNA Res. 2009, 16, 287–298. [Google Scholar] [CrossRef]
- Ikeda, S.; Abe, T.; Nakamura, Y.; Kibinge, N.; Hirai Morita, A.; Nakatani, A.; Ono, N.; Ikemura, T.; Nakamura, K.; Altaf-Ul-Amin, M.; et al. Systematization of the protein sequence diversity in enzymes related to secondary metabolic pathways in plants, in the context of big data biology inspired by the KNApSAcK Motorcycle database. Plant Cell Physiol. 2013, 54, 711–727. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Iwasaki, Y.; Abe, T.; Wada, K.; Wada, Y.; Ikemura, T. A Novel Bioinformatics Strategy to Analyze Microbial Big Sequence Data for Efficient Knowledge Discovery: Batch-Learning Self-Organizing Map (BLSOM). Microorganisms 2013, 1, 137-157. https://doi.org/10.3390/microorganisms1010137
Iwasaki Y, Abe T, Wada K, Wada Y, Ikemura T. A Novel Bioinformatics Strategy to Analyze Microbial Big Sequence Data for Efficient Knowledge Discovery: Batch-Learning Self-Organizing Map (BLSOM). Microorganisms. 2013; 1(1):137-157. https://doi.org/10.3390/microorganisms1010137
Chicago/Turabian StyleIwasaki, Yuki, Takashi Abe, Kennosuke Wada, Yoshiko Wada, and Toshimichi Ikemura. 2013. "A Novel Bioinformatics Strategy to Analyze Microbial Big Sequence Data for Efficient Knowledge Discovery: Batch-Learning Self-Organizing Map (BLSOM)" Microorganisms 1, no. 1: 137-157. https://doi.org/10.3390/microorganisms1010137