
VEEV TC-83 Triggers Dysregulation of the Tryptophan–Kynurenine Pathway in the Central Nervous System That Correlates with Cognitive Impairment in Tg2576 Mice
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, and Biosafety
2.2. Animal Experiments
2.3. Active Avoidance
2.4. Tissue Preparation
2.5. RNA Isolation and qRT-PCR
2.6. Western Blot Analysis
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Enzyme Activity Assay
2.9. Statistical Analysis
3. Results
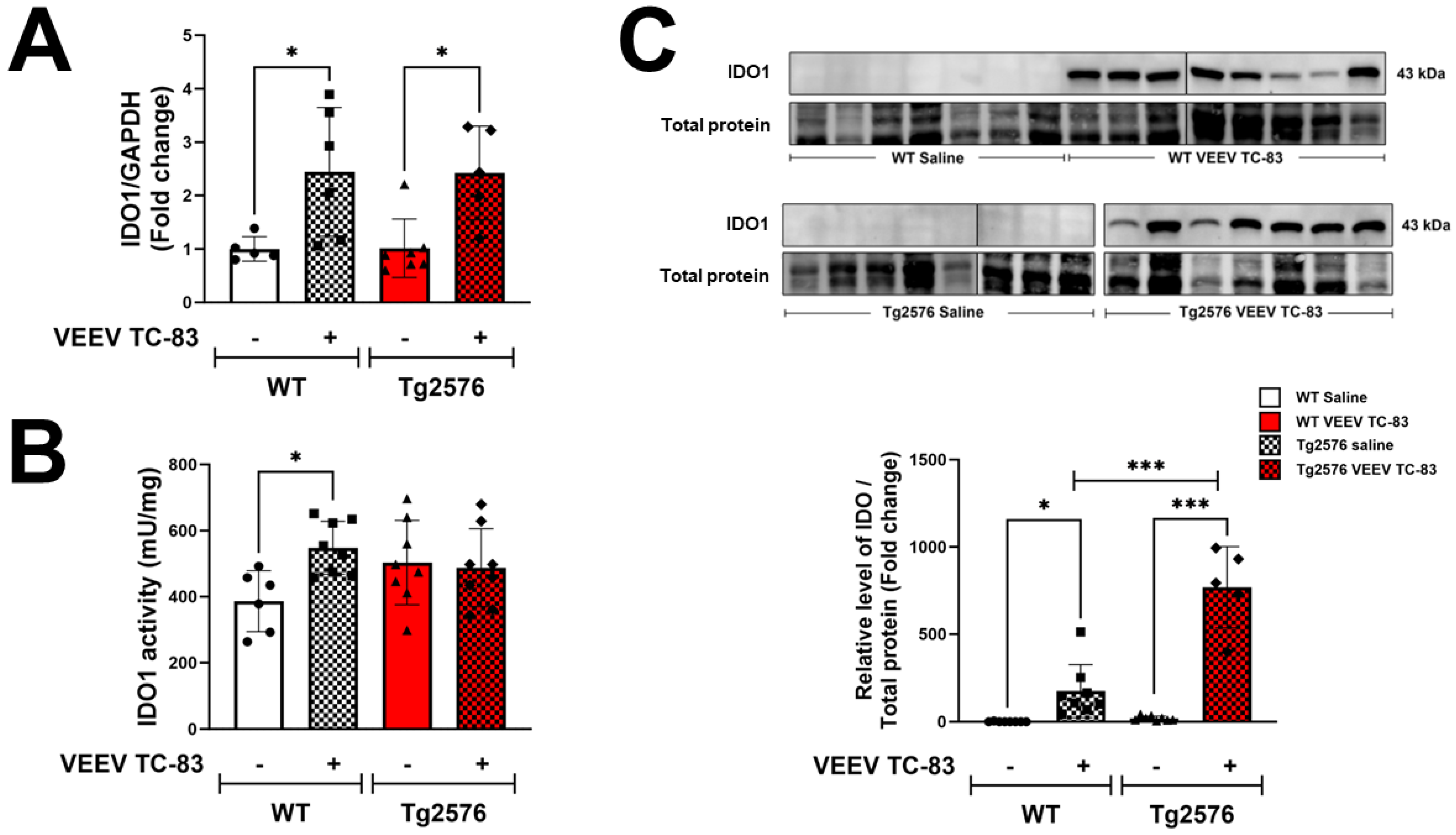
3.1. Increased IDO Levels in CNS Are Associated with VEEV TC-83 Infection
3.2. IDO Levels Correlate to Inflammatory Mediators and Active Avoidance Behavior
3.3. Tryptophan Catabolism and KP Activation Are Upregulated during Infection
3.4. VEEV TC-83 Impacts QUIN and NAD+ Levels in Tg2576 Mice Leading to Behavioral Changes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef] [PubMed]
- Westaway, E.G.; Brinton, M.A.; Gaidamovich, S.; Horzinek, M.C.; Igarashi, A.; Kaariainen, L.; Lvov, D.K.; Porterfield, J.S.; Russell, P.K.; Trent, D.W. Togaviridae. Intervirology 1985, 24, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, P.V.; Estrada-Franco, J.G.; Navarro-Lopez, R.; Ferro, C.; Haddow, A.D.; Weaver, S.C. Endemic Venezuelan equine encephalitis in the Americas: Hidden under the dengue umbrella. Future Virol. 2011, 6, 721–740. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ronca, S.E.; Dineley, K.T.; Paessler, S. Neurological Sequelae Resulting from Encephalitic Alphavirus Infection. Front. Microbiol. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Weaver, S.C.; Ferro, C.; Barrera, R.; Boshell, J.; Navarro, J.C. Venezuelan equine encephalitis. Annu. Rev. Entomol. 2004, 49, 141–174. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.M.; Martin, D.H. Venezuelan equine encephalitis. Adv. Vet. Sci. Comp. Med. 1974, 18, 79–116. [Google Scholar] [PubMed]
- Williams, E.P.; Xue, Y.; Lee, J.; Fitzpatrick, E.A.; Kong, Y.; Reichard, W.; Writt, H.; Jonsson, C.B. Deep spatial profiling of Venezuelan equine encephalitis virus reveals increased genetic diversity amidst neuroinflammation and cell death during brain infection. J. Virol. 2023, 97, e0082723. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schoneboom, B.A.; Fultz, M.J.; Miller, T.H.; McKinney, L.C.; Grieder, F.B. Astrocytes as targets for Venezuelan equine encephalitis virus infection. J. Neurovirol. 1999, 5, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Blackhurst, B.M.; Funk, K.E. Viral pathogens increase risk of neurodegenerative disease. Nat. Rev. Neurol. 2023, 19, 259–260. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 2023, 111, 1086–1093.e2. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Romanescu, C.; Schreiner, T.G.; Mukovozov, I. The Role of Human Herpesvirus 6 Infection in Alzheimer’s Disease Pathogenicity—A Theoretical Mosaic. J. Clin. Med. 2022, 11, 3061. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Feng, S.; Liu, Y.; Zhou, Y.; Shu, Z.; Cheng, Z.; Brenner, C.; Feng, P. Mechanistic insights into the role of herpes simplex virus 1 in Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1245904. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mody, P.H.; Marvin, K.N.; Hynds, D.L.; Hanson, L.K. Cytomegalovirus infection induces Alzheimer’s disease-associated alterations in tau. J. Neurovirol. 2023, 29, 400–415. [Google Scholar] [CrossRef] [PubMed]
- Dal Canto, M.C.; Rabinowitz, S.G. Experimental models of virus-induced demyelination of the central nervous system. Ann. Neurol. 1982, 11, 109–127. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dal Canto, M.C.; Rabinowitz, S.G. Central nervous system demyelination in Venezuelan equine encephalomyelitis infection. J. Neurol. Sci. 1981, 49, 397–418. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, M.D.; Varney, B.; Sundaram, G.; Lennon, M.J.; Lim, C.K.; Jacobs, K.; Guillemin, G.J.; Brew, B.J. Recent evidence for an expanded role of the kynurenine pathway of tryptophan metabolism in neurological diseases. Neuropharmacology 2017, 112 Pt B, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Mithaiwala, M.N.; Santana-Coelho, D.; Porter, G.A.; O’Connor, J.C. Neuroinflammation and the Kynurenine Pathway in CNS Disease: Molecular Mechanisms and Therapeutic Implications. Cells 2021, 10, 1548. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, Y.S.; Ogbechi, J.; Clanchy, F.I.; Williams, R.O.; Stone, T.W. IDO and Kynurenine Metabolites in Peripheral and CNS Disorders. Front. Immunol. 2020, 11, 388. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huengsberg, M.; Winer, J.B.; Gompels, M.; Round, R.; Ross, J.; Shahmanesh, M. Serum kynurenine-to-tryptophan ratio increases with progressive disease in HIV-infected patients. Clin. Chem. 1998, 44, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Larrea, L.; Peyron, R. Motor cortex stimulation for neuropathic pain: From phenomenology to mechanisms. Neuroimage 2007, 37 (Suppl. S1), S71–S79. [Google Scholar] [CrossRef] [PubMed]
- Atlas, A.; Franzen-Rohl, E.; Soderlund, J.; Jonsson, E.G.; Samuelsson, M.; Schwieler, L.; Skoldenberg, B.; Engberg, G. Sustained elevation of kynurenic Acid in the cerebrospinal fluid of patients with herpes simplex virus type 1 encephalitis. Int. J. Tryptophan Res. 2013, 6, 89–96. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Baran, H.; Hainfellner, J.A.; Kepplinger, B. Kynurenic Acid Metabolism in Various Types of Brain Pathology in HIV-1 Infected Patients. Int. J. Tryptophan Res. 2012, 5, 49–64. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, Y.; Zhao, M.; Chen, X.; Zhang, R.; Le, A.; Hong, M.; Zhang, Y.; Jia, L.; Zang, W.; Jiang, C.; et al. Tryptophan Metabolism in Central Nervous System Diseases: Pathophysiology and Potential Therapeutic Strategies. Aging Dis. 2023, 14, 858–878. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gonzalez Esquivel, D.; Ramirez-Ortega, D.; Pineda, B.; Castro, N.; Rios, C.; Perez de la Cruz, V. Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacology 2017, 112 Pt B, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sanchez, M.; Jimenez, J.; Narvaez, A.; Antequera, D.; Llamas-Velasco, S.; Martin, A.H.; Arjona, J.A.M.; Munain, A.L.; Bisa, A.L.; Marco, M.P.; et al. Kynurenic Acid Levels are Increased in the CSF of Alzheimer’s Disease Patients. Biomolecules 2020, 10, 571. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shen, X.N.; Li, J.Q.; Wang, H.F.; Li, H.Q.; Huang, Y.Y.; Yang, Y.X.; Tan, L.; Dong, Q.; Yu, J.T.; Alzheimer’s Disease Neuroimaging, I. Plasma amyloid, tau, and neurodegeneration biomarker profiles predict Alzheimer’s disease pathology and clinical progression in older adults without dementia. Alzheimers Dement. 2020, 12, e12104. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thomas, T.; Stefanoni, D.; Reisz, J.A.; Nemkov, T.; Bertolone, L.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hansen, K.C.; Hod, E.A.; et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight 2020, 5, e140327. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fraser, D.D.; Slessarev, M.; Martin, C.M.; Daley, M.; Patel, M.A.; Miller, M.R.; Patterson, E.K.; O’Gorman, D.B.; Gill, S.E.; Wishart, D.S.; et al. Metabolomics Profiling of Critically Ill Coronavirus Disease 2019 Patients: Identification of Diagnostic and Prognostic Biomarkers. Crit. Care Explor. 2020, 2, e0272. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001, 21, 372–381. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dineley, K.T.; Xia, X.; Bui, D.; Sweatt, J.D.; Zheng, H. Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J. Biol. Chem. 2002, 277, 22768–22780. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.M.; Kayed, R.; Zheng, H.; Sweatt, J.D.; Dineley, K.T. Loss of alpha7 nicotinic receptors enhances beta-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2010, 30, 2442–2453. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ashe, K.H.; Zahs, K.R. Probing the biology of Alzheimer’s disease in mice. Neuron 2010, 66, 631–645. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Taylor, M.W.; Feng, G.S. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991, 5, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Nomiyama, S.; Hirata, F.; Hayaishi, O. Indoleamine 2,3-dioxygenase. Purification and some properties. J. Biol. Chem. 1978, 253, 4700–4706. [Google Scholar] [CrossRef] [PubMed]
- Mbongue, J.C.; Nicholas, D.A.; Torrez, T.W.; Kim, N.S.; Firek, A.F.; Langridge, W.H. The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines 2015, 3, 703–729. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fongsaran, C.; Jirakanwisal, K.; Peng, B.-H.; Fracassi, A.; Taglialatela, G.; Dineley, K.T.; Paessler, S.; Cisneros, I.E. Arbovirus infection increases the risk for the development of neurodegenerative disease pathology in the murine model. Brain Behav. Immun. Health 2024, 38, 100780. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Zwolinska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Campbell, B.M.; Charych, E.; Lee, A.W.; Moller, T. Kynurenines in CNS disease: Regulation by inflammatory cytokines. Front. Neurosci. 2014, 8, 12. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alves de Souza, T.M.; Fernandes-Santos, C.; Araujo da Paixao de Oliveira, J.; Tome, L.C.T.; Fiestas-Solorzano, V.E.; Nunes, P.C.G.; Guimaraes, G.M.C.; Sanchez-Arcila, J.C.; Paiva, I.A.; de Souza, L.J.; et al. Increased Indoleamine 2,3-Dioxygenase 1 (IDO-1) Activity and Inflammatory Responses during Chikungunya Virus Infection. Pathogens 2022, 11, 444. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2005, 49, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Ostapiuk, A.; Urbanska, E.M. Kynurenic acid in neurodegenerative disorders-unique neuroprotection or double-edged sword? CNS Neurosci. Ther. 2022, 28, 19–35. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schaeffer, E.L.; Figueiro, M.; Gattaz, W.F. Insights into Alzheimer disease pathogenesis from studies in transgenic animal models. Clinics 2011, 66 (Suppl. S1), 45–54. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Quak, J.; Doornbos, B.; Roest, A.M.; Duivis, H.E.; Vogelzangs, N.; Nolen, W.A.; Penninx, B.W.; Kema, I.P.; de Jonge, P. Does tryptophan degradation along the kynurenine pathway mediate the association between pro-inflammatory immune activity and depressive symptoms? Psychoneuroendocrinology 2014, 45, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Dantzer, R.; Kelley, K.W.; Lawson, M.A.; Woolwine, B.J.; Vogt, G.; Spivey, J.R.; Saito, K.; Miller, A.H. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: Relationship to CNS immune responses and depression. Mol. Psychiatry 2010, 15, 393–403. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schrocksnadel, K.; Wirleitner, B.; Winkler, C.; Fuchs, D. Monitoring tryptophan metabolism in chronic immune activation. Clin. Chim. Acta 2006, 364, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Akimoto, H.; Kagawa, S.; Guillemin, G.J.; Takikawa, O. Proinflammatory cytokine interferon-gamma increases induction of indoleamine 2,3-dioxygenase in monocytic cells primed with amyloid beta peptide 1-42: Implications for the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 110, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Castro-Portuguez, R.; Sutphin, G.L. Kynurenine pathway, NAD(+) synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 2020, 132, 110841. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitron, R.; Ugalde Muniz, P.; Pineda, B.; Pedraza-Chaverri, J.; Rios, C.; Perez-de la Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxid. Med. Cell Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ryu, J.K.; Kim, S.U.; McLarnon, J.G. Blockade of quinolinic acid-induced neurotoxicity by pyruvate is associated with inhibition of glial activation in a model of Huntington’s disease. Exp. Neurol. 2004, 187, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Perez-Severiano, F.; Escalante, B.; Rios, C. Nitric oxide synthase inhibition prevents acute quinolinate-induced striatal neurotoxicity. Neurochem. Res. 1998, 23, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Fernandez-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T.; et al. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2017, 155, 76–95. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Meininger, V.; Brew, B.J. Implications for the kynurenine pathway and quinolinic acid in amyotrophic lateral sclerosis. Neurodegener. Dis. 2005, 2, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Vandresen-Filho, S.; Severino, P.C.; Constantino, L.C.; Martins, W.C.; Molz, S.; Dal-Cim, T.; Bertoldo, D.B.; Silva, F.R.; Tasca, C.I. N-methyl-D-aspartate preconditioning prevents quinolinic acid-induced deregulation of glutamate and calcium homeostasis in mice hippocampus. Neurotox Res. 2015, 27, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Ting, K.; Cullen, K.M.; Braidy, N.; Brew, B.J.; Guillemin, G.J. The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS ONE 2009, 4, e6344. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Grant, R.S. Indoleamine 2,3-Dioxygenase Activity Increases NAD+ Production in IFN-gamma-Stimulated Human Primary Mononuclear Cells. Int. J. Tryptophan Res. 2018, 11, 1178646917751636. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moffett, J.R.; Arun, P.; Puthillathu, N.; Vengilote, R.; Ives, J.A.; Badawy, A.A.; Namboodiri, A.M. Quinolinate as a Marker for Kynurenine Metabolite Formation and the Unresolved Question of NAD(+) Synthesis During Inflammation and Infection. Front. Immunol. 2020, 11, 31. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Perez-De La Cruz, V.; Carrillo-Mora, P.; Santamaria, A. Quinolinic Acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int. J. Tryptophan Res. 2012, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fongsaran, C.; Dineley, K.T.; Paessler, S.; Cisneros, I.E. VEEV TC-83 Triggers Dysregulation of the Tryptophan–Kynurenine Pathway in the Central Nervous System That Correlates with Cognitive Impairment in Tg2576 Mice. Pathogens 2024, 13, 397. https://doi.org/10.3390/pathogens13050397
Fongsaran C, Dineley KT, Paessler S, Cisneros IE. VEEV TC-83 Triggers Dysregulation of the Tryptophan–Kynurenine Pathway in the Central Nervous System That Correlates with Cognitive Impairment in Tg2576 Mice. Pathogens. 2024; 13(5):397. https://doi.org/10.3390/pathogens13050397
Chicago/Turabian StyleFongsaran, Chanida, Kelly T. Dineley, Slobodan Paessler, and Irma E. Cisneros. 2024. "VEEV TC-83 Triggers Dysregulation of the Tryptophan–Kynurenine Pathway in the Central Nervous System That Correlates with Cognitive Impairment in Tg2576 Mice" Pathogens 13, no. 5: 397. https://doi.org/10.3390/pathogens13050397