
Characterization of Variant RNAs Encapsidated during Bromovirus Infection by High-Throughput Sequencing
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Propagation, Purification, and RNA Extraction
2.2. High-Throughput Sequencing (HTS)
2.3. Sequence Analysis with Bioinformatics Tools
3. Results
3.1. High Numbers of HTS Reads Do Not Map to the Canonical Viral Genomes
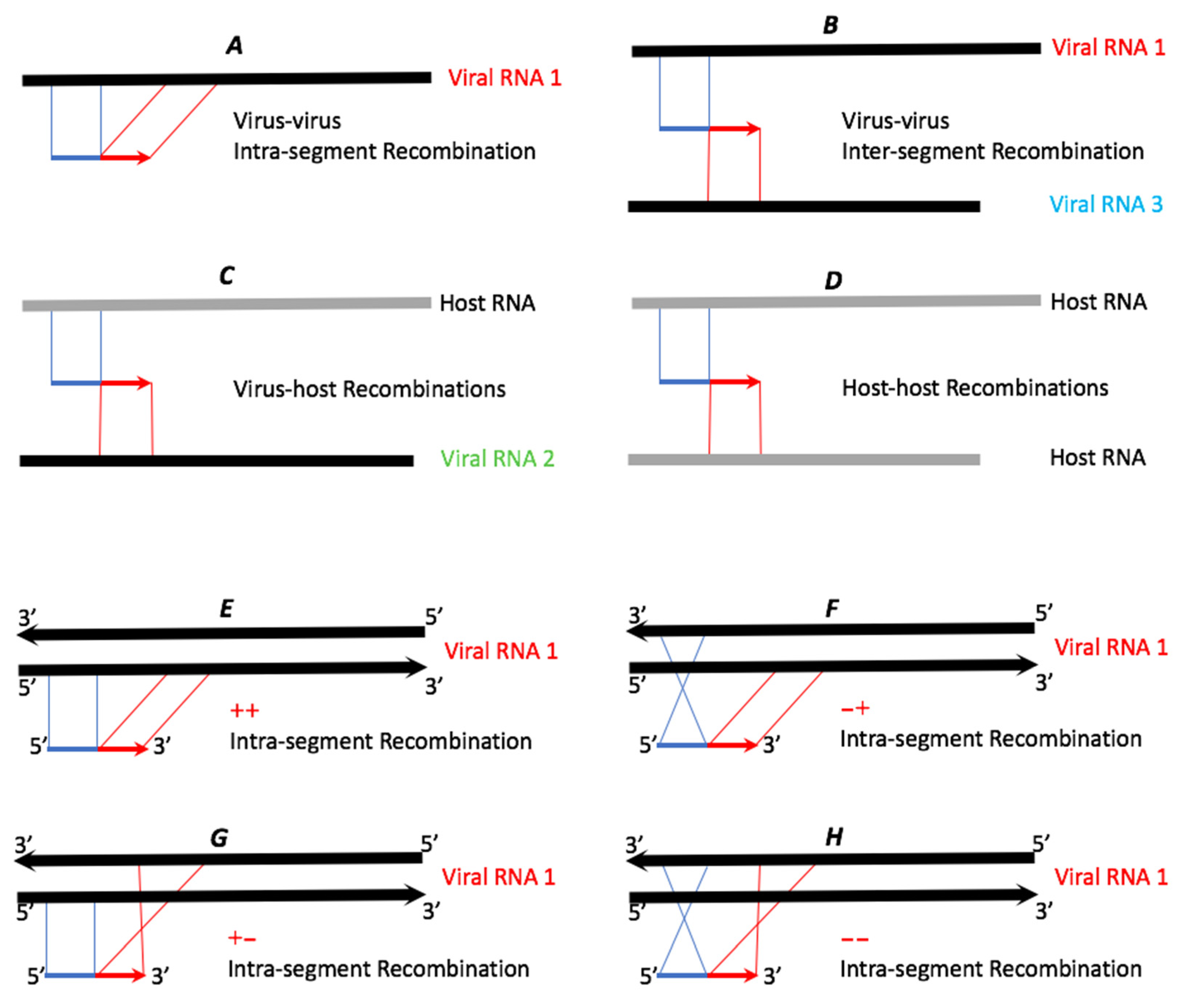
3.2. ViReMa Identified Six Classes of RNA Variants among the Bowtie-Unmapped Reads
- Recombination crosses that include reads split into at least two fragments in the reference RNAs (illustrated in Figure 2). The crossovers that occur in the same RNA segment will appear as insertions or deletions (see below). Out of 31,429,207 BMV reads and 35,192,299 CCMV reads, 1.40% and 1.83% were reported as such recombinant reads.
- Ambiguous recombinants included reads with pads longer than 25 nts at either end, or reads with pads of any size in the middle (12.73% and 9.45% of reads, respectively).
- Nucleotide substitutions in reads carrying larger than two consecutive nucleotide mismatches. We found that 1.25% and 1.21% reads of BMV and CCMV fell into this group. The single nucleotide mismatches (or SNPs) were not reported by ViReMa (see the additional analysis of SNPs below).
- Micro-insertions included reads mapped to a single reference after exclusion of a small number (≤4 nts) of nucleotides in the middle, involving 0.01% and 0.23% reads in the two viruses. Reads with longer than 5 nts insertions were reported as ambiguous recombinants by ViReMa.
- Single mapping reads carried the pads of unmapped regions shorter than 25 nts, as well as reads with single-base mismatches in the middle. Those involved, 80.40% and 82.08% reads for BMV and CCMV, respectively.
- Unmapped reads. Only 4.21% and 5.20% reads fell into this category.
3.3. Inter-Segmental vs. Intra-Segmental Recombinants
3.4. Mapping of Substitutions to the Reference Genomes
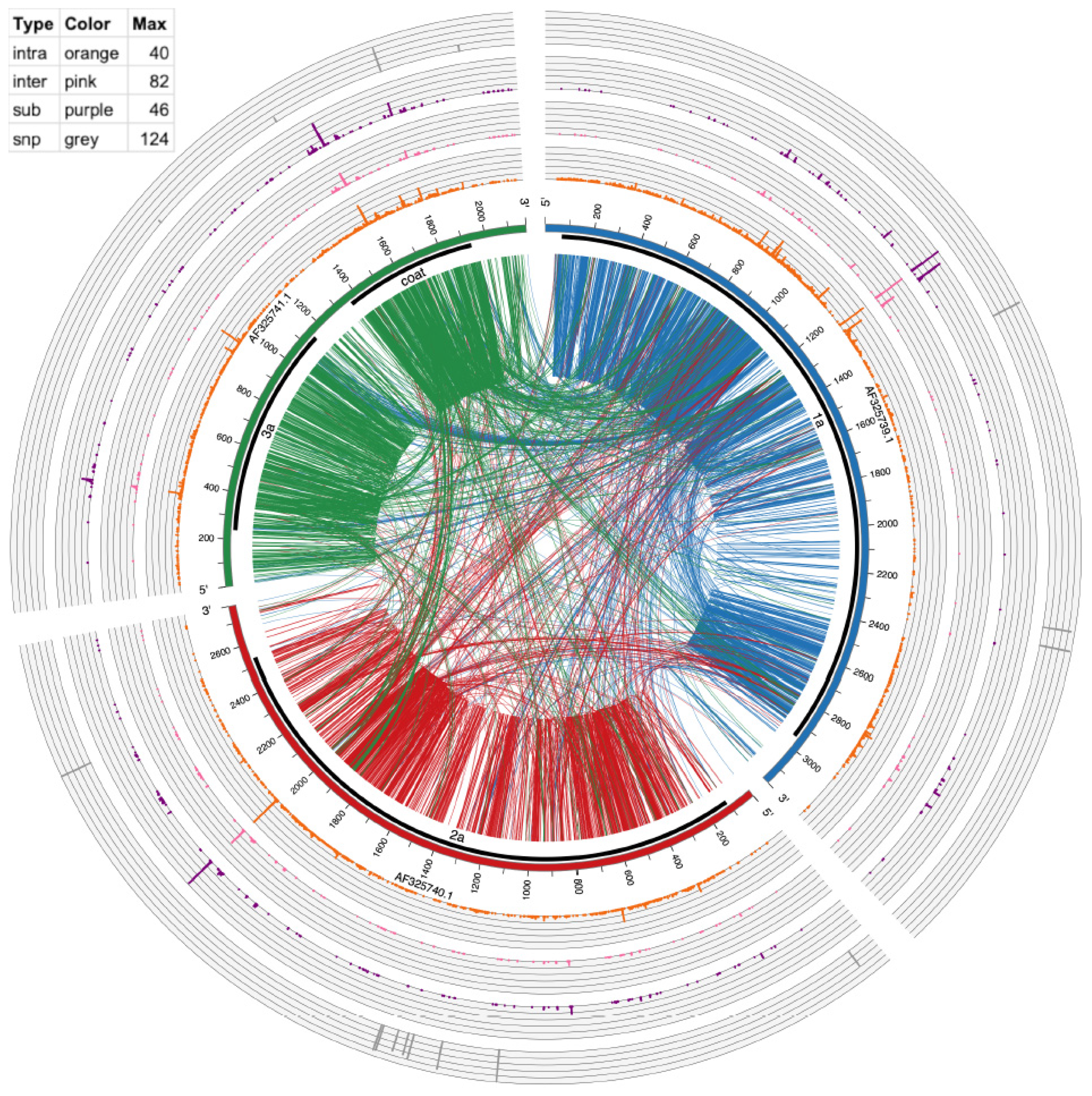
3.5. Visualizing of Recombination Hotspots with Circos Plots
3.6. Recombination Events between Virus and Host RNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 2015, 479, 2–25. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Sztuba-Solińska, J.; Urbanowicz, A.; Figlerowicz, M.; Bujarski, J.J. RNA-RNA recombination in plant virus replication and evolution. Annu. Rev. Phytopathol. 2011, 49, 415–443. [Google Scholar] [CrossRef]
- Bujarski, J.J. Genetic recombination in plant-infecting messenger-sense RNA viruses: Overview and research perspectives. Front. Plant Sci. 2013, 4, 68. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.-Y.; Wang, J.; Shen, Y.; Zhou, B.; Duan, L.; Cheung, C.-L.; Ma, C.; Lycett, S.J.; Leung, C.Y.-H.; Chen, X. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature 2013, 502, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A. Recombination among picornaviruses. Rev. Med. Virol. 2010, 20, 327–337. [Google Scholar] [CrossRef]
- Martin-Valls, G.; Kvisgaard, L.K.; Tello, M.; Darwich, L.; Cortey, M.; Burgara-Estrella, A.; Hernández, J.; Larsen, L.E.; Mateu, E. Analysis of ORF5 and full-length genome sequences of porcine reproductive and respiratory syndrome virus isolates of genotypes 1 and 2 retrieved worldwide provides evidence that recombination is a common phenomenon and may produce mosaic isolates. J. Virol. 2014, 88, 3170–3181. [Google Scholar] [CrossRef]
- Villabona-Arenas, C.J.; Zanotto, P.M.d.A. Worldwide spread of dengue virus type 1. PLoS ONE 2013, 8, e62649. [Google Scholar] [CrossRef]
- Weiss, B.G.; Schlesinger, S. Recombination between sindbis virus RNAs. J. Virol. 1991, 65, 4017–4025. [Google Scholar] [CrossRef]
- Pagán, I.; Holmes, E.C. Long-term evolution of the Luteoviridae: Time scale and mode of virus speciation. J. Virol. 2010, 84, 6177–6187. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ahlquist, P. Filling a GAP(DH) in asymmetric viral RNA synthesis. Cell Host Microbe 2008, 3, 124–125. [Google Scholar] [CrossRef]
- Palasingam, K.; Shaklee, P.N. Reversion of Q beta RNA phage mutants by homologous RNA recombination. J. Virol. 1992, 66, 2435–2442. [Google Scholar] [CrossRef] [PubMed]
- Han, G.-Z.; Worobey, M. Homologous recombination in negative sense RNA viruses. Viruses 2011, 3, 1358–1373. [Google Scholar] [CrossRef] [PubMed]
- Delviks-Frankenberry, K.; Galli, A.; Nikolaitchik, O.; Mens, H.; Pathak, V.K.; Hu, W.S. Mechanisms and factors that influence high frequency retroviral recombination. Viruses 2011, 3, 1650–1680. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.L. Genome packaging by spherical plant RNA viruses. Annu. Rev. Phytopathol. 2006, 44, 61–87. [Google Scholar] [CrossRef]
- Kolondam, B.; Rao, P.; Sztuba-Solinska, J.; Weber, P.H.; Dzianott, A.; Johns, M.A.; Bujarski, J.J. Co-infection with two strains of Brome mosaic bromovirus reveals common RNA recombination sites in different hosts. Virus Evol. 2015, 1, vev021. [Google Scholar] [CrossRef]
- Figlerowicz, M.; Nagy, P.D.; Bujarski, J.J. A mutation in the putative RNA polymerase gene inhibits nonhomologous, but not homologous, genetic recombination in an RNA virus. Proc. Natl. Acad. Sci. USA 1997, 94, 2073–2078. [Google Scholar] [CrossRef]
- Figlerowicz, M.; Nagy, P.D.; Tang, N.; Kao, C.C.; Bujarski, J.J. Mutations in the N terminus of the brome mosaic virus polymerase affect genetic RNA-RNA recombination. J. Virol. 1998, 72, 9192–9200. [Google Scholar] [CrossRef]
- Allison, R.; Thompson, C.; Ahlquist, P. Regeneration of a functional RNA virus genome by recombination between deletion mutants and requirement for cowpea chlorotic mottle virus 3a and coat genes for systemic infection. Proc. Natl. Acad. Sci. USA 1990, 87, 1820–1824. [Google Scholar] [CrossRef]
- Ranjith-Kumar, C.T.; Kim, Y.C.; Gutshall, L.; Silverman, C.; Khandekar, S.; Sarisky, R.T.; Kao, C.C. Mechanism of de novo initiation by the hepatitis C virus RNA-dependent RNA polymerase: Role of divalent metals. J. Virol. 2002, 76, 12513–12525. [Google Scholar] [CrossRef] [PubMed]
- Sztuba-Solinska, J.; Fanning, S.W.; Horn, J.R.; Bujarski, J.J. Mutations in the coat protein-binding cis-acting RNA motifs debilitate RNA recombination of Brome mosaic virus. Virus Res. 2012, 170, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Weber, P.H.; Bujarski, J.J. Multiple functions of capsid proteins in (+) stranded RNA viruses during plant-virus interactions. Virus Res. 2015, 196, 140–149. [Google Scholar] [CrossRef]
- Shrestha, N.; Weber, P.H.; Burke, S.V.; Wysocki, W.P.; Duvall, M.R.; Bujarski, J.J. Next generation sequencing reveals packaging of host RNAs by brome mosaic virus. Virus Res. 2018, 252, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; Duvall, M.R.; Bujarski, J.J. Variability among the Isolates of Broad Bean Mottle Virus and Encapsidation of Host RNAs. Pathogens 2022, 11, 817. [Google Scholar] [CrossRef] [PubMed]
- Routh, A.; Johnson, J.E. Discovery of functional genomic motifs in viruses with ViReMa—A Virus Recombination Mapper-for analysis of next-generation sequencing data. Nucleic Acids Res. 2014, 42, e11. [Google Scholar] [CrossRef] [PubMed]
- Ni, P.; Vaughan, R.C.; Tragesser, B.; Hoover, H.; Kao, C.C. The plant host can affect the encapsidation of brome mosaic virus (BMV) RNA: BMV virions are surprisingly heterogeneous. J. Mol. Biol. 2014, 426, 1061–1076. [Google Scholar] [CrossRef]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, 1–10. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Chao, M.; Wang, T.C.; Lin, C.C.; Yung-Liang Wang, R.; Lin, W.B.; Lee, S.E.; Cheng, Y.Y.; Yeh, C.T.; Iang, S.B. Analyses of a whole-genome inter-clade recombination map of hepatitis delta virus suggest a host polymerase-driven and viral RNA structure-promoted template-switching mechanism for viral RNA recombination. Oncotarget 2017, 8, 60841–60859. [Google Scholar] [CrossRef] [PubMed]
- Haasnoot, P.C.; Olsthoorn, R.C.; Bol, J.F. The Brome mosaic virus subgenomic promoter hairpin is structurally similar to the iron-responsive element and functionally equivalent to the minus-strand core promoter stem-loop C. RNA 2002, 8, 110–122. [Google Scholar] [CrossRef]
- Knies, J.L.; Dang, K.K.; Vision, T.J.; Hoffman, N.G.; Swanstrom, R.; Burch, C.L. Compensatory evolution in RNA secondary structures increases substitution rate variation among sites. Mol. Biol. Evol. 2008, 25, 1778–1787. [Google Scholar] [CrossRef] [PubMed]
- Shapka, N.; Nagy, P.D. The AU-rich RNA recombination hot spot sequence of Brome mosaic virus is functional in tombusviruses: Implications for the mechanism of RNA recombination. J. Virol. 2004, 78, 2288–2300. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.D.; Pogany, J. The dependence of viral RNA replication on co-opted host factors. Nat. Rev. Microbiol. 2012, 10, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Martin, D.P.; Weeks, K.M.; Negroni, M. RNA structures facilitate recombination-mediated gene swapping in HIV-1. J. Virol. 2010, 84, 12675–12682. [Google Scholar] [CrossRef]
- Nagy, P.D.; Bujarski, J.J. Efficient system of homologous RNA recombination in brome mosaic virus: Sequence and structure requirements and accuracy of crossovers. J. Virol. 1995, 69, 131–140. [Google Scholar] [CrossRef]
- Annamalai, P.; Rao, A.L. In vivo packaging of brome mosaic virus RNA3, but not RNAs 1 and 2, is dependent on a cis-acting 3’ tRNA-like structure. J. Virol. 2007, 81, 173–181. [Google Scholar] [CrossRef]
- Baumstark, T.; Ahlquist, P. The brome mosaic virus RNA3 intergenic replication enhancer folds to mimic a tRNA TpsiC-stem loop and is modified in vivo. RNA 2001, 7, 1652–1670. [Google Scholar]
- Sibert, B.S.; Navine, A.K.; Pennington, J.; Wang, X.; Ahlquist, P. Cowpea chlorotic mottle bromovirus replication proteins support template-selective RNA replication in Saccharomyces cerevisiae. PLoS ONE 2018, 13, e0208743. [Google Scholar] [CrossRef] [PubMed]
- Becher, P.; Tautz, N. RNA recombination in pestiviruses: Cellular RNA sequences in viral genomes highlight the role of host factors for viral persistence and lethal disease. RNA Biol. 2011, 8, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.A. Exchange of Genetic Sequences Between Viruses and Hosts. Curr. Top. Microbiol. Immunol. 2017, 407, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Gorzer, I.; Guelly, C.; Trajanoski, S.; Puchhammer-Stockl, E. The impact of PCR-generated recombination on diversity estimation of mixed viral populations by deep sequencing. J. Virol. Methods 2010, 169, 248–252. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GenBank ID | RNA Segment | Length (nts) | Encoded Proteins and nts Positions |
|---|---|---|---|
| BMV | |||
| NC_002026.1 | RNA1 | 3234 | 1a (transferase-helicase): 75–960 |
| NC_002027.1 | RNA2 | 2865 | 2a (RdRp): 104–2572 |
| NC_002028.2 | RNA3 | 2111 | 3a (movement): 92-1003; coat: 1245–1814 |
| Host: H. vulgare 1 | 248,180 mRNAs | Total 478,475,198 | |
| 1347 rRNAs | Total 1,491,619 | ||
| CCMV | |||
| AF325739.1 | RNA1 | 3174 | 1a: 71–2950 |
| AF325740.1 | RNA2 | 2773 | 2a: 109–2535 |
| AF325741.1 | RNA3 | 2175 | 3a: 239–1147, coat: 1362–1934 |
| Host: V. unguiculata 2 | 42,287 mRNAs | Total 83,574,877 | |
| 22 rRNAs | Total 12,051 | ||
| Read Mapping a | BMV | CCMV |
|---|---|---|
| Total no. reads | 108,930,036 | 108,937,068 |
| Mapped to RNA1 (%) | 24,647,631 (22.63%) | 37,785,834 (34.69%) |
| Mapped to RNA2 (%) | 43,755,190 (40.17%) | 17,519,764 (16.08%) |
| Mapped to RNA3 (%) | 9,022,371 (8.28%) | 18,284,856 (16.78%) |
| Mapped to host mRNAs (%) | 75,289 (0.07%) | 43,928 (0.04%) |
| Mapped to host rRNAs (%) | 348 (0%) | 110,387 (0.1%) |
| Unmapped reads (%) | 31,429,207 (28.85%) | 35,192,299 (32.31%) |
| ViReMa Analyzed Reads a | BMV + (Host mRNAs and rRNAs) | CCMV + (Host mRNAs and rRNAs) |
|---|---|---|
| Total reads analyzed | 31,429,207 | 35,192,299 |
| Recombinations | 440,975 | 644,596 |
| 1.40% | 1.83% | |
| Nucleotide substitutions (≥2 nt) | 391,820 | 426,030 |
| 1.25% | 1.21% | |
| Micro-insertions (≤4 nt) | 1616 | 81,264 |
| 0.01% | 0.23% | |
| Single mapping reads with pads | 25,268,730 | 28,884,471 |
| 80.40% | 82.08% | |
| Ambiguous recombinations | 4,000,122 | 3,326,635 |
| 12.73% | 9.45% | |
| Completely unmapped | 1,322,860 | 1,828,745 |
| 4.21% | 5.20% |
| BMV | ||||
|---|---|---|---|---|
| Column is 5′ & Row is 3′ | RNA1 | RNA2 | RNA3 | Total |
| RNA1 | 1183 | 78 | 22 | 1283 |
| RNA2 | 176 a | 2046 | 28 | 2250 |
| RNA3 | 37 | 24 | 415 | 476 |
| Total | 1396 | 2148 | 465 | 4009 |
| CCMV | ||||
| Column is 5′ & row is 3′ | RNA1 | RNA2 | RNA3 | Total |
| RNA1 | 1726 | 175 | 234 | 2135 |
| RNA2 | 165 | 949 | 141 | 1255 |
| RNA3 | 247 | 133 | 1213 | 1593 |
| Total | 2138 | 1257 | 1588 | 4983 |
| Indel Types a | Frame Shifts b | BMV | CCMV | ||
|---|---|---|---|---|---|
| Counts | % | Counts | % | ||
| del3 | 1->1 | 178 | 4.89 | 207 | 5.32 |
| ins3 | 1->1 | 506 | 13.89 | 379 | 9.75 |
| del1 | 1->2 | 1560 | 42.83 | 1775 | 45.65 |
| ins2 | 1->2 | 433 | 11.89 | 405 | 10.42 |
| del2 | 1->3 | 411 | 11.29 | 541 | 13.91 |
| ins1 | 1->3 | 554 | 15.21 | 581 | 14.94 |
| Total events | 3642 | 3888 | |||
| BMV | Intra-Recombination Hotspot Position (Number of Events a) | Inter-Recombination Hotspot Position (Number of Events) |
|---|---|---|
| RNA1 | 1054 (13), 1058 (13), 1097 (14), 1360 (14) | 1053 (11), 1054 (22),1058 (15), 1097 (15) |
| RNA2 | 1212 (17), 1274 (11), 1314 (15), 1488 (20), 1559 (24), 1773 (36), 1776 (21), 1792 (15), 1795 (16) | na |
| RNA3 | na | 1200 (23) |
| CCMV | Intra Recombination Hotspot Position (Number of Events) | Inter Recombination Hotspot Position (Number of Events) |
| RNA1 | 815 (21), 877 (13), 888 (12), 1104 (15), 1231 (35), 1103 (12), 60 (22), 1261 (12) | 734 (11), 1104 (14), 1229 (18), 1230 (13), 1231 (82), 1260 (38), 1261 (30) |
| RNA2 | 653 (16), 2016 (40), 2017 (20), 2018 (12) | 852 (11), 2016 (43), 2017 (15) |
| RNA | 359 (15), 906 (11), 950 (26), 1585 (27), 1586 (11), 1731 (15), 1801 (14) | 359 (15), 1584 (25), 1585 (29), 1586 (16), 1801 (18) |
| Recombinations | BMV | BMV (%) | CCMV | CCMV (%) |
|---|---|---|---|---|
| Virus–Virus | 4009 | 95.93 | 4983 | 97.31 |
| mRNA–mRNA | 91 | 2.18 | 113 | 2.21 |
| rRNA–rRNA | 0 | 0.00 | 12 | 0.23 |
| Virus–mRNA | 76 | 1.82 | 8 | 0.16 |
| Virus–rRNA | 3 | 0.07 | 4 | 0.08 |
| mRNA–rRNA | 0 | 0.00 | 1 | 0.02 |
| Substitutions (≥2 nt) | BMV | BMV (%) | CCMV | CCMV (%) |
| Virus–Virus | 4679 | 99.98 | 4114 | 97.77 |
| mRNA–mRNA | 0 | 0 | 0 | 0 |
| rRNA–rRNA | 1 | 0.02 | 94 | 2.23 |
| Micro–insertions (≤4 nt) | BMV | BMV (%) | CCMV | CCMV (%) |
| Virus–Virus | 28 | 100 | 33 | 100% |
| mRNA–mRNA | 0 | 0 | 0 | 0% |
| rRNA–rRNA | 0 | 0 | 0 | 0% |
| Ambiguous recombinations | BMV | BMV (%) | CCMV | CCMV (%) |
| Virus–Virus | 33,924 | 86.33 | 33,960 | 94.27 |
| mRNA–mRNA | 4829 | 12.29 | 1342 | 3.73 |
| rRNA–rRNA | 104 | 0.26 | 679 | 1.88 |
| Virus–mRNA | 243 | 0.62 | 44 | 0.12 |
| Virus–rRNA | 192 | 0.49 | 0 | 0 |
| mRNA–rRNA | 4 | 0.01 | 0 | 0 |
| Single mapping | BMV | BMV (%) | CCMV | CCMV (%) |
| Virus–Virus | 194,281 | 99.62 | 229,656 | 99.43 |
| mRNA–mRNA | 732 | 0.38 | 250 | 0.11 |
| rRNA–rRNA | 0 | 0 | 1056 | 0.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dexheimer, S.; Shrestha, N.; Chapagain, B.S.; Bujarski, J.J.; Yin, Y. Characterization of Variant RNAs Encapsidated during Bromovirus Infection by High-Throughput Sequencing. Pathogens 2024, 13, 96. https://doi.org/10.3390/pathogens13010096
Dexheimer S, Shrestha N, Chapagain BS, Bujarski JJ, Yin Y. Characterization of Variant RNAs Encapsidated during Bromovirus Infection by High-Throughput Sequencing. Pathogens. 2024; 13(1):96. https://doi.org/10.3390/pathogens13010096
Chicago/Turabian StyleDexheimer, Sarah, Nipin Shrestha, Bandana Sharma Chapagain, Jozef J. Bujarski, and Yanbin Yin. 2024. "Characterization of Variant RNAs Encapsidated during Bromovirus Infection by High-Throughput Sequencing" Pathogens 13, no. 1: 96. https://doi.org/10.3390/pathogens13010096