
An ARMS-Multiplex PCR Targeting SARS-CoV-2 Omicron Sub-Variants


Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples, RNA Isolation and Reverse Transcription
2.2. Sequence Retrieval and Mutation Targeting
2.3. Design of Primers and PCR
3. Results
3.1. Sequence Retrieval and Verification
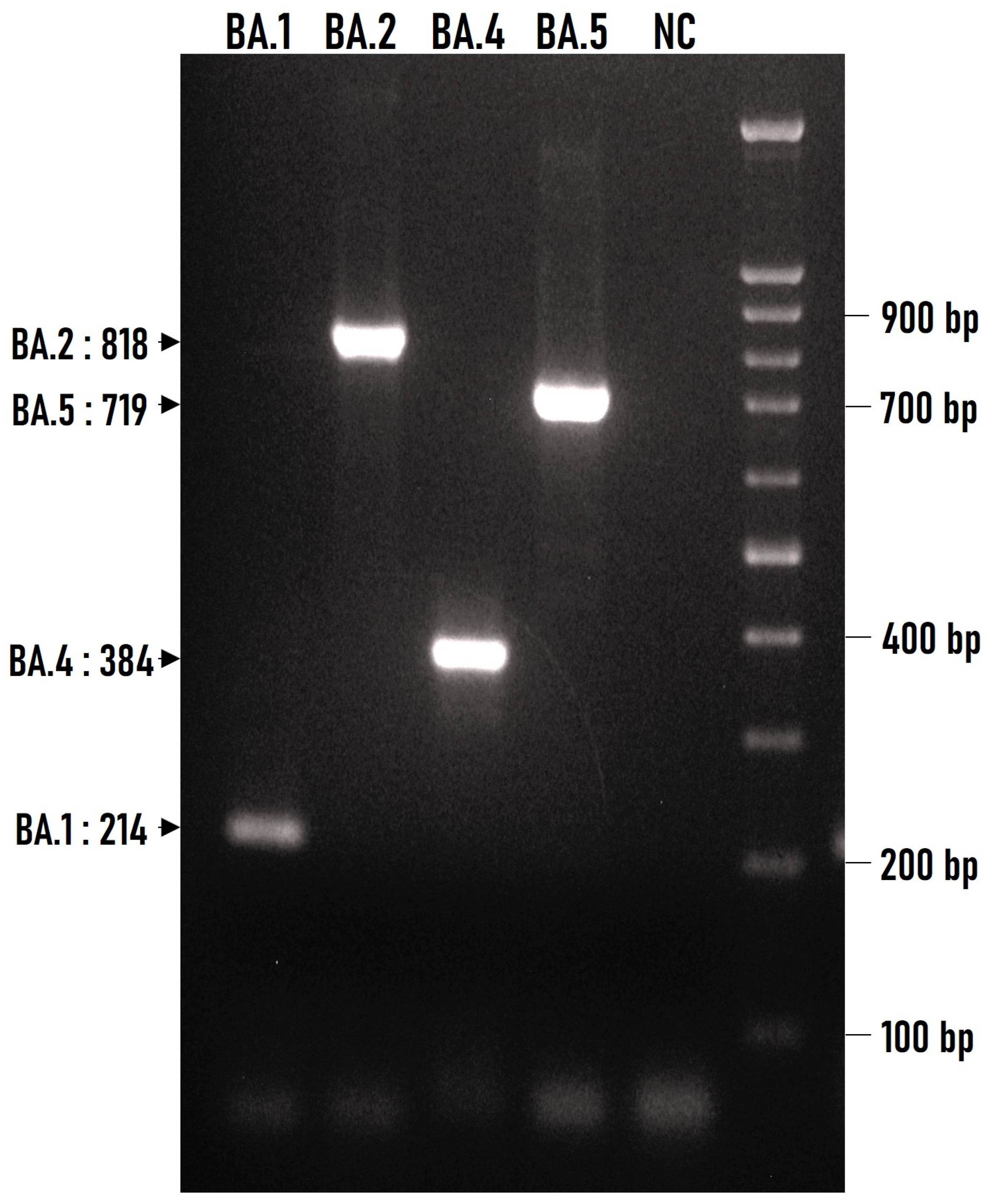
3.2. Discrimination of Basic Omicron Sub-Variants by Multiplex ARMS-PCR
3.3. The Potential of Omicron Specific Multiplex ARMS-PCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weekly Epidemiological Update on COVID-19-13 July 2023. Edition 151. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---13-july-2023 (accessed on 24 July 2023).
- Quarleri, J.; Galvan, V.; Delpino, M.V. Omicron variant of the SARS-CoV-2: A quest to define the consequences of its high mutational load. GeroScience 2022, 44, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev. Med. Virol. 2022, 32, 2381. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.; Rais, M.A.; Mohanty, A.; Chopra, H.; Chandran, D.; Bin Emran, T.; Dhama, K. Omicron (B.1.1.529) variant and its subvariants and lineages may lead to another COVID-19 wave in the world? -An overview of current evidence and counteracting strategies. Int. J. Surg. Open 2023, 55, 100625. [Google Scholar] [CrossRef] [PubMed]
- One Year since the Emergence of COVID-19 Virus Variant Omicron. Available online: https://www.who.int/news-room/feature-stories/detail/one-year-since-the-emergence-of-omicron (accessed on 3 June 2023).
- Parums, D.V. Editorial: Revised World Health Organization (WHO) Terminology for Variants of Concern and Variants of Interest of SARS-CoV-2. Med. Sci. Monit. 2021, 27, e933622-1–e933622-2. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: World Health Organization (WHO) Variants of Concern Lineages Under Monitoring (VOC-LUM) in Response to the Global Spread of Lineages and Sublineages of Omicron, or B.1.1.529, SARS-CoV-2. Med. Sci. Monit. 2022, 28, e937676-1–e937676-3. [Google Scholar] [CrossRef]
- Statement on the Update of WHO ‘s Working Definitions and Tracking System for SARS-CoV-2 Variants of Concern and Variants of Interest. Available online: https://www.who.int/news/item/16-03-2023-statement-on-the-update-of-who-s-working-definitions-and-tracking-system-for-sars-cov-2-variants-of-concern-and-variants-of-interest (accessed on 9 June 2023).
- Newton, C.R.; Graham, A.; Heptinstall, L.E.; Powell, S.J.; Summers, C.; Kalsheker, N.; Smith, J.C.; Markham, A.F. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989, 11, 2503–2516. [Google Scholar] [CrossRef]
- Cheng, Y.; Ji, C.; Zhou, H.Y.; Zheng, H.; Wu, A. Web Resources for SARS-CoV-2 Genomic Database, Annotation, Analysis and Variant Tracking. Viruses 2023, 15, 1158. [Google Scholar] [CrossRef]
- Paton, R.S.; Overton, C.E.; Ward, T. The rapid replacement of the SARSCoV-2 Delta variant by Omicron (B.1.1.529) in England. Sci. Transl. Med. 2022, 14, 395. [Google Scholar] [CrossRef]
- Wang, S.; Xu, X.; Wei, C.; Li, S.; Zhao, J.; Zheng, Y.; Liu, X.; Zeng, X.; Yuan, W.; Peng, S. Molecular Evolutionary Characteristics of SARS-CoV-2 Emerging in the United States. J. Med. Virol. 2022, 94, 310–317. [Google Scholar] [CrossRef]
- Chaguza, C.; Coppi, A.; Earnest, R.; Ferguson, D.; Kerantzas, N.; Warner, F.; Young, P.H.; Breban, M.I.; Billig, K.; Koch, R.T.; et al. Rapid emergence of SARS-CoV-2 Omicron variant is associated with an infection advantage over Delta in vaccinated persons. Med 2022, 3, 325–334e4. [Google Scholar] [CrossRef]
- Callaway, E. COVID ‘variant soup’ is making winter surges hard to predict. Nature 2022, 611, 213–214. [Google Scholar] [CrossRef]
- Chakraborty, C.; Bhattacharya, M.; Sharma, A.R.; Dhama, K. Recombinant SARS-CoV-2 Variants XD, XE, and XF: The Emergence of Recombinant Variants Requires an Urgent Call for Research—Correspondence. Int. J. Surg. 2022, 102, 106670. [Google Scholar] [CrossRef]
- Akash, K.; Sharma, A.; Kumar, D.; Singh, S.K.; Gupta, G.; Chellappan, D.K.; Dua, K.; Nagraik, R. Molecular Aspects of Omicron, Vaccine Development, and Recombinant Strain XE: A Review. J. Med. Virol. 2022, 94, 4628–4643. [Google Scholar] [CrossRef]
- ECDC De-Escalates BA.2, BA.4 and BA.5 from Its List of Variants of Concern. Available online: https://www.ecdc.europa.eu/en/news-events/ecdc-de-escalates-ba2-ba4-and-ba5-its-list-variants-concern (accessed on 4 June 2023).
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat. Commun. 2023, 14, 2800. [Google Scholar] [CrossRef]
- Scarpa, F.; Azzena, I.; Locci, C.; Casu, M.; Fiori, P.L.; Ciccozzi, A.; Angeletti, S.; Imperia, E.; Giovanetti, M.; Maruotti, A.; et al. Molecular In-Depth on the Epidemiological Expansion of SARS-CoV-2 XBB.1.5. Microorganisms 2023, 11, 912. [Google Scholar] [CrossRef]
- Yamasoba, D.; Uriu, K.; Plianchaisuk, A.; Kosugi, Y.; Pan, L.; Zahradnik, J.; Genotype to Phenotype Japan (G2P-Japan) Consortium; Ito, J.; Sato, K. Virological characteristics of the SARS-CoV-2 omicron XBB.1.16 variant. Lancet Infect. Dis. 2023, 23, 655–656. [Google Scholar] [CrossRef]
- Hansen, C.H.; Friis, N.U.; Bager, P.; Stegger, M.; Fonager, J.; Fomsgaard, A.; Gram, M.A.; Christiansen, L.E.; Ethelberg, P.S.; Legarth, R.; et al. Risk of reinfection, vaccine protection, and severity of infection with the BA.5 omicron subvariant: A nation-wide population-based study in Denmark. Lancet Infect. Dis. 2023, 23, 167–176. [Google Scholar] [CrossRef]
- Kang, S.W.; Park, H.; Kim, J.Y.; Lim, S.Y.; Lee, S.; Bae, J.Y.; Kim, J.; Chang, E.; Bae, S.; Jung, J.; et al. Comparison of the clinical and virological characteristics of SARS-CoV-2 Omicron BA.1/BA.2 and omicron BA.5 variants: A prospective cohort study. J. Infect. 2023, 86, 148–151. [Google Scholar] [CrossRef]
- Abdul Aziz, N.; Nash, S.G.; Zaidi, A.; Nyberg, T.; Groves, N.; Hope, R.; Bernal, J.L.; Dabrera, G.; Thelwall, S. Risk of severe outcomes among SARS-CoV-2 Omicron BA.4 and BA.5 cases compared to BA.2 cases in England. J. Infect. 2023, 87, 8–11. [Google Scholar] [CrossRef]
- Ciuffreda, L.; Lorenzo-Salazar, J.M.; García-Martínez de Artola, D.; Gill-Campesino, H.; Alcoba-Florez, J.; Rodriguez-Perez, H.; Inigo-Campos, A.; Salas-Hernadez, J.; Rodriguez-Nunez, J.; Munoz-Barrera, A.; et al. Reinfection rate and disease severity of the BA.5 Omicron SARS-CoV-2 lineage compared to previously circulating variants of concern in the Canary Islands (Spain). Emerg. Microbes Infect. 2023, 12, 2202281. [Google Scholar] [CrossRef]
- Wolter, N.; Jassat, W.; Walaza, S.; Welch, R.; Moultrie, H.; Groome, M.J.; Gyamfi Amoako, D.; Everatt, J.; Bhiman, J.N.; Scheepers, C.; et al. Clinical severity of SARS-CoV-2 Omicron BA.4 and BA.5 lineages compared to BA.1 and Delta in South Africa. Nat. Commun. 2022, 13, 5860. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.Y.; Fall, A.; Norton, J.M.; Eldesouki, R.E.; Abdullah, O.; Han, L.; Yunker, M.; Mostafa, H.H. Severity outcomes associated with SARS-CoV-2 XBB variants, an observational analysis. J. Clin. Virol. 2023, 165, 105500. [Google Scholar] [CrossRef] [PubMed]
- Corbisier, P.; Petrillo, M.; Marchini, A.; Querci, M.; Buttinger, G.; Bekliz, M.; Spiess, K.; Polacek, C.; Fomsgaard, A.; Van den Eede, G. A Qualitative RT-PCR Assay for the Specific Identification of the SARS-CoV-2 B.1.1.529 (Omicron) Variant of Concern. J. Clin. Virol. 2022, 152, 105191. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.; Boes, S.; McCullough, M.; Gribschaw, J.; Marsh, J.W.; Harrison, L.H.; Wells, A. First detection of SARS-CoV-2 Omicron BA.4 variant in Western Pennsylvania, United States. J. Med. Virol. 2022, 94, 4053–4055. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef]
- Spiess, K.; Gunalan, V.; Marving, E.; Nielsen, S.H.; Jørgensen, M.G.P.; Fomsgaard, A.S.; Nielsen, L.; Alfano-Nunez, A.; Karst, S.M.; Mortensen, S.; et al. Rapid and Flexible RT-qPCR Surveillance Platforms To Detect SARS-CoV-2 Mutations. Microbiol. Spectr. 2023, 11, e0359122. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. Methods for the Detection and Characterisation of SARS-CoV-2 Variants–Second Update as of 2 August 2022. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/Methods-for-the-detection-char-SARS-CoV-2-variants_2nd%20update_final.pdf (accessed on 6 June 2023).
- Chrysostomou, A.C.; Aristokleous, A.; Rodosthenous, J.H.; Christodoulou, C.; Stathi, G.; Kostrikis, L.G. Detection of Circulating SARS-CoV-2 Variants of Concern (VOCs) Using a Multiallelic Spectral Genotyping Assay. Life 2023, 13, 304. [Google Scholar] [CrossRef]
- Specchiarello, E.; Matusali, G.; Carletti, F.; Gruber, C.E.M.; Fabeni, L.; Minosse, C.; Giombini, E.; Rueca, M.; Maggi, F.; Amendola, A.; et al. Detection of SARS-CoV-2 Variants via Different Diagnostics Assays Based on Single-Nucleotide Polymorphism Analysis. Diagnostics 2023, 13, 1573. [Google Scholar] [CrossRef]
- Fan, Y.; Li, X.; Zhang, L.; Wan, S.; Zhang, L.; Zhou, F. SARS-CoV-2 Omicron variant: Recent progress and future perspectives. Signal Transduct. Target Ther. 2022, 7, 141. [Google Scholar] [CrossRef]
- Gartzonika, K.; Bozidis, P.; Priavali, E.; Sakkas, H. Rapid Detection of blaKPC-9 Allele from Clinical Isolates. Pathogens 2021, 10, 487. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Sub-Variant | Mutation and Genetic Locus | Direction | Primer | Position on Wuhan (NC_045512.2) | Sequence | PCR Product (bp) |
|---|---|---|---|---|---|---|
| BA.1 |
A to G (2832) Orf1ab | Forward | 2832 ARMS | 2809–2832 | GAT TGA TAA AGT ACT TCA TGC GAG | 214 |
| Reverse | 3022–2999 | ATG TGA AGC CAA TTT AAA CTC ACC | ||||
| BA.2 |
C to T (9866) Orf1ab | Forward | 9866 ARMS | 9842–9866 | TTG CGT AGT GAT GTG CTC TTA CAT T | 818 |
| Reverse | 10,659–10,637 | ACT GTA ATA GTT GTG TCC GTA CC | ||||
| BA.4 |
G to T (27,788) Orf7a-Orf8 interspace | Forward | 27,788 ARMS | 27,762–27,788 | GAA CTT TCA TTA ATT GAC TAC TAT CTT | 384 |
| Reverse | 28,145–28,119 | TAA ACA GGA AAC TGT ATA ATT ACC GAT | ||||
| BA.5 |
C to T (26,529) M gene | Forward | 25,835–25,859 | GCT GGC ATA CTA ATT GTT ACG ACT A | 719 | |
| Reverse | 26,529 ARMS | 26,553–26,529 | CAA CGG TAA TAG TAC CGT GGG ATT T |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozidis, P.; Petridi, E.; Gartzonika, K. An ARMS-Multiplex PCR Targeting SARS-CoV-2 Omicron Sub-Variants. Pathogens 2023, 12, 1017. https://doi.org/10.3390/pathogens12081017
Bozidis P, Petridi E, Gartzonika K. An ARMS-Multiplex PCR Targeting SARS-CoV-2 Omicron Sub-Variants. Pathogens. 2023; 12(8):1017. https://doi.org/10.3390/pathogens12081017
Chicago/Turabian StyleBozidis, Petros, Eleni Petridi, and Konstantina Gartzonika. 2023. "An ARMS-Multiplex PCR Targeting SARS-CoV-2 Omicron Sub-Variants" Pathogens 12, no. 8: 1017. https://doi.org/10.3390/pathogens12081017
APA StyleBozidis, P., Petridi, E., & Gartzonika, K. (2023). An ARMS-Multiplex PCR Targeting SARS-CoV-2 Omicron Sub-Variants. Pathogens, 12(8), 1017. https://doi.org/10.3390/pathogens12081017