
Differential Immune Responses and Underlying Mechanisms of Metabolic Reprogramming in Smooth and Rough Variants of Mycobacterium peregrinum Infections


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Conditions and Growth
2.2. Preparation of Single-Cell Bacterial Stocks
2.3. Bone Marrow-Derived Macrophage Culture and Ethics Statement
2.4. Thin-Layer Chromatography
2.5. Antimicrobial Susceptibility Test
2.6. Intracellular Staining (Confocal Microscopy)
2.7. Phagocytosis Assay and Infection Kinetics of M. peregrinum
2.8. Cell Viability Assay and Cytotoxicity Assay
2.9. Measurement of Cytokines via Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. RNA Extraction and Real-Time PCR
2.11. Protein Extraction and Western Blotting Analysis
2.12. Seahorse Extracellular Flux Analysis
2.13. Statistical Analysis
3. Results
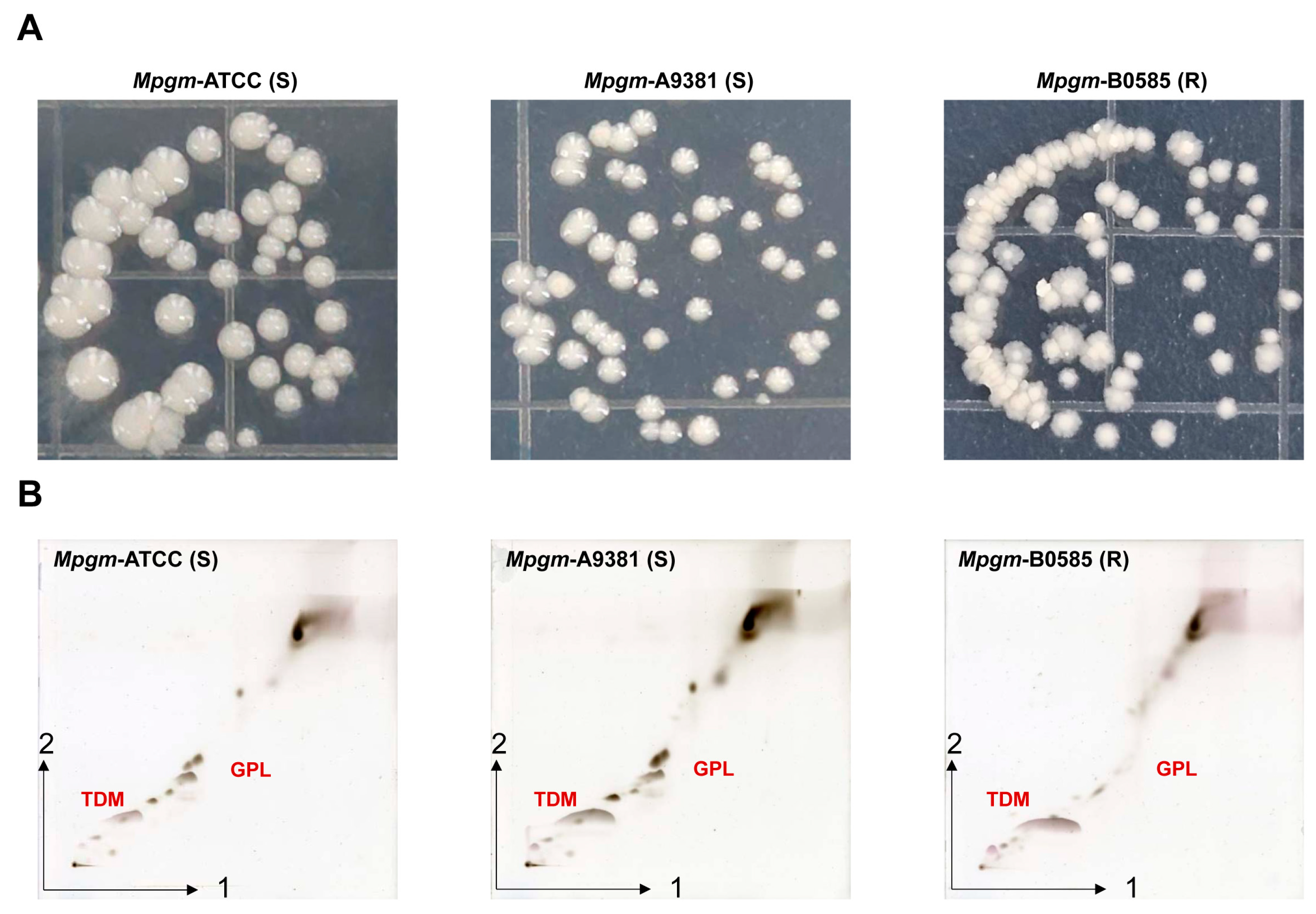
3.1. M. peregrinum Morphology and Lipid Profile Analysis via Thin-Layer Chromatography
3.2. Comparison of the Antimicrobial Susceptibility Results for M. peregrinum
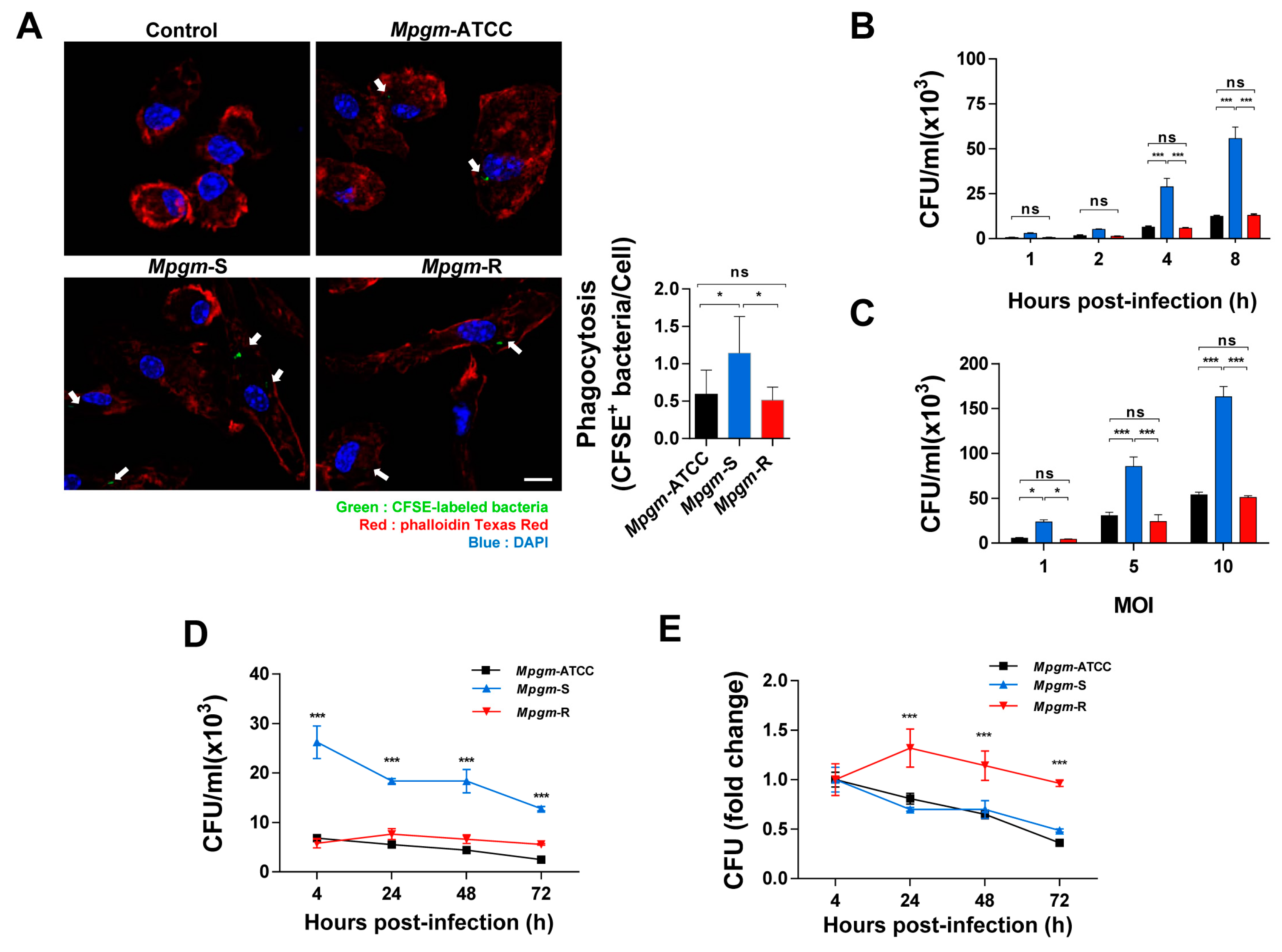
3.3. M. peregrinum Is an Intracellular Bacterium
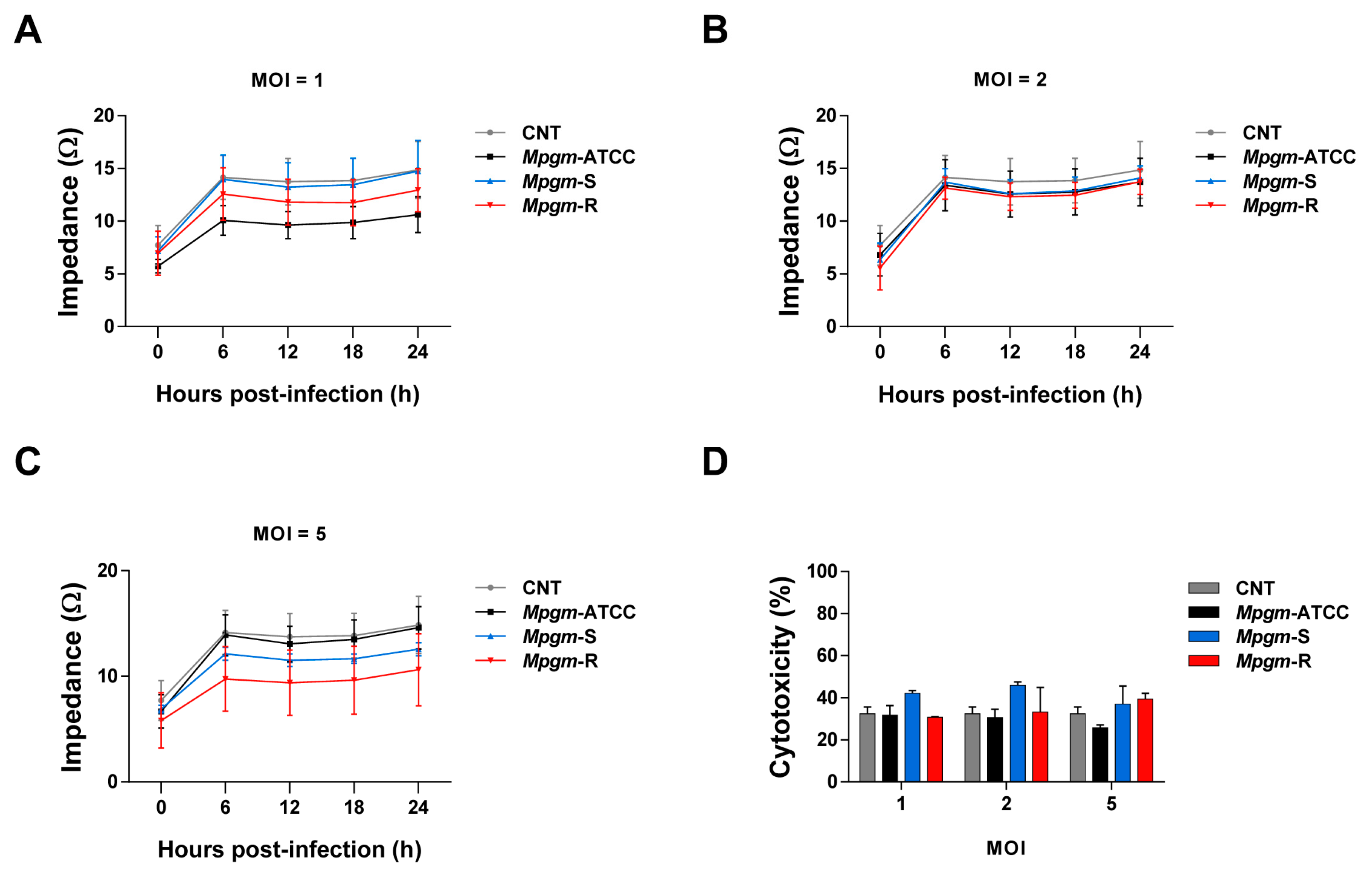
3.4. M. peregrinum Does Not Affect the Viability or Cytotoxicity of BMDMs
3.5. Comparative Analysis of Inflammatory Cytokine Production in BMDMs Infected with M. peregrinum
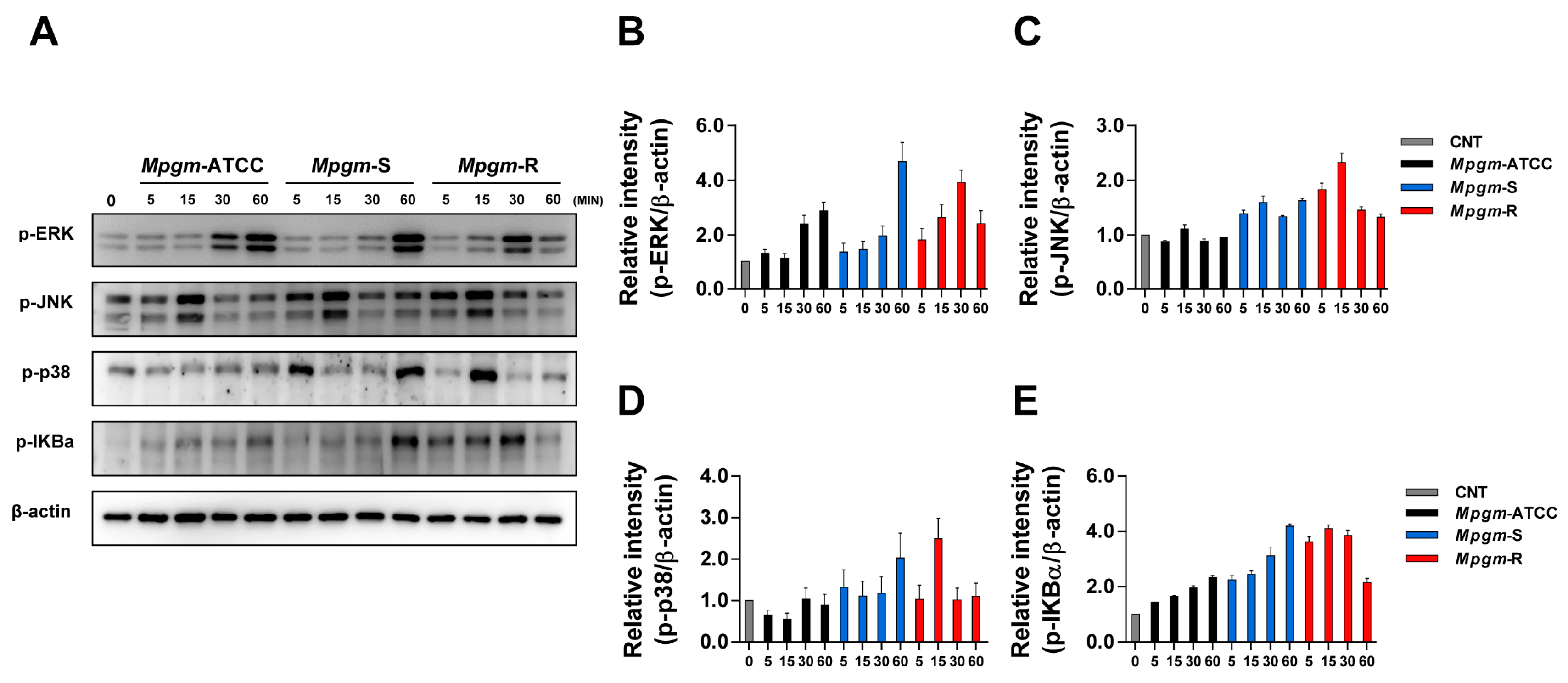
3.6. M. peregrinum Induces Activation of the MAPK and NF-κB Pathways in BMDMs
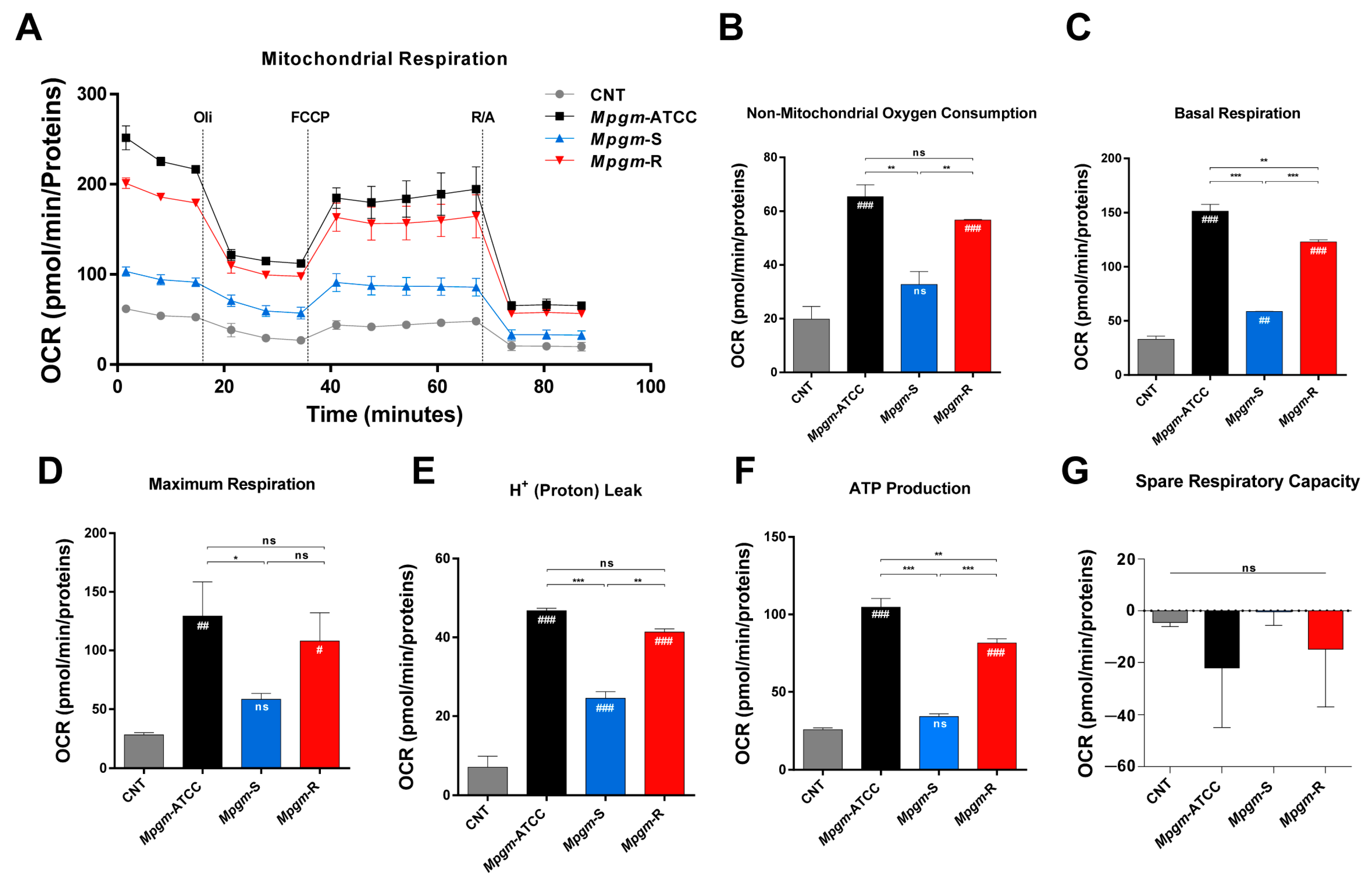
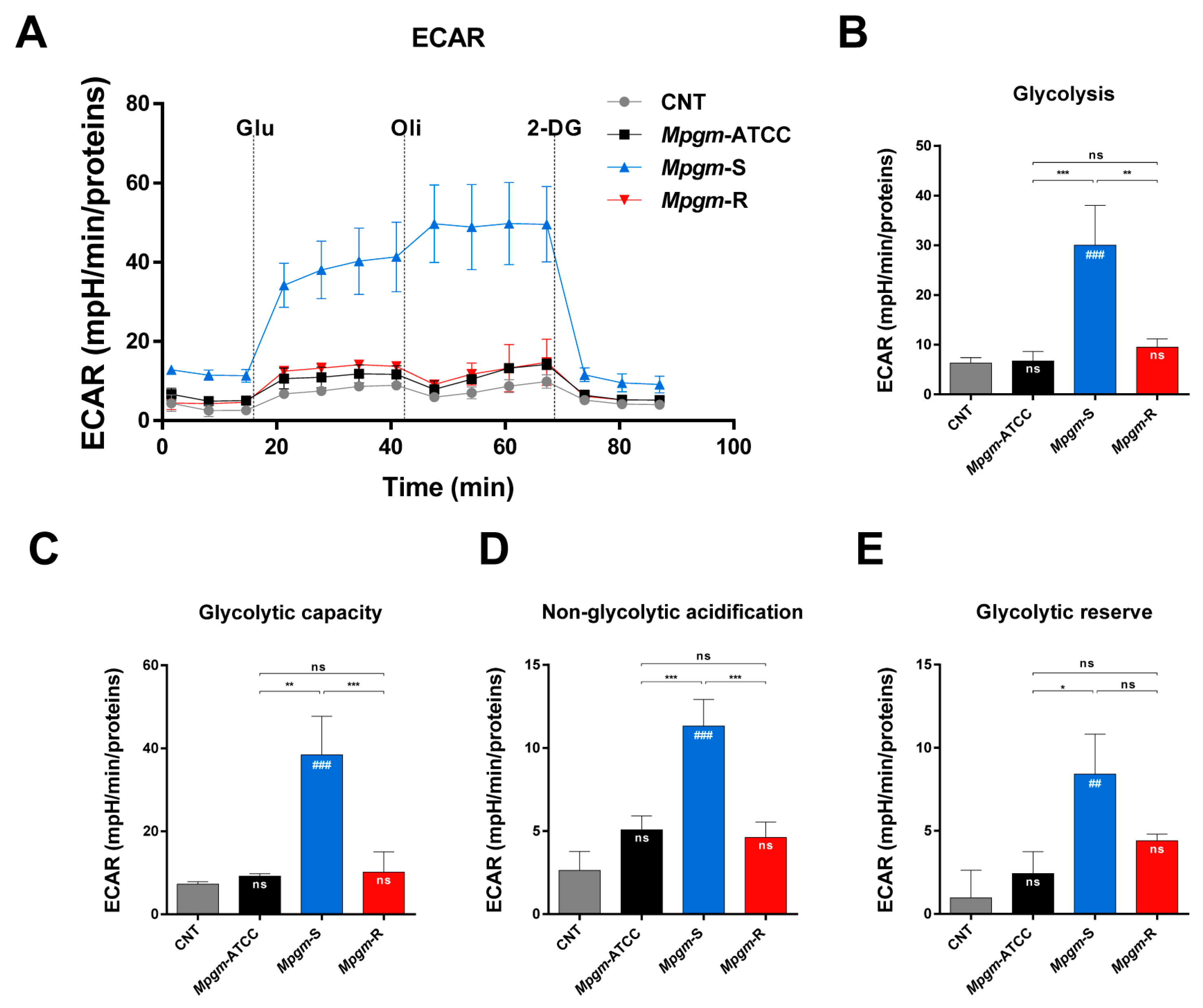
3.7. Metabolic Profile of BMDMs Infected with M. peregrinum
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bi, S.; Hu, F.-S.; Yu, H.-Y.; Xu, K.-J.; Zheng, B.-W.; Ji, Z.-K.; Li, J.-J.; Deng, M.; Hu, H.-Y.; Sheng, J.-F. Nontuberculous mycobacterial osteomyelitis. Infect. Dis. 2015, 47, 673–685. [Google Scholar] [CrossRef]
- Lee, W.J.; Kang, S.M.; Sung, H.; Won, C.H.; Chang, S.E.; Lee, M.W.; Kim, M.N.; Choi, J.H.; Moon, K.C. Non-tuberculous mycobacterial infections of the skin: A retrospective study of 29 cases. J. Dermatol. 2010, 37, 965–972. [Google Scholar] [CrossRef]
- Jarzembowski, J.A.; Young, M.B. Nontuberculous Mycobacterial Infections. Arch. Pathol. Lab. Med. 2008, 132, 1333–1341. [Google Scholar] [CrossRef]
- To, K.; Cao, R.; Yegiazaryan, A.; Owens, J.; Venketaraman, V. General Overview of Nontuberculous Mycobacteria Opportunistic Pathogens: Mycobacterium avium and Mycobacterium abscessus. J. Clin. Med. 2020, 9, 2541. [Google Scholar] [CrossRef] [PubMed]
- Brown-Elliott, B.A.; Wallace, R.J. Clinical and Taxonomic Status of Pathogenic Nonpigmented or Late-Pigmenting Rapidly Growing Mycobacteria. Clin. Microbiol. Rev. 2002, 15, 716–746. [Google Scholar] [CrossRef] [PubMed]
- Lazo-Vasquez, A.F.; Gonzales-Zamora, J.A. Mycobacterium peregrinum Pacemaker Pocket Infection: A Case Report and Review of the Literature. Case Rep. Infect. Dis. 2020, 2020, 8831026. [Google Scholar] [CrossRef] [PubMed]
- Lao, C.K.; Wu, T.-S.; Lin, K.-Y.; Lee, M.-H. Disseminated Mycobacterium peregrinum and Mycobacterium avium infection in a patient with AIDS: A case report and review of literature. J. Clin. Tuberc. Other Mycobact. Dis. 2022, 27, 100314. [Google Scholar] [CrossRef]
- Whang, J.; Back, Y.W.; Lee, K.-I.; Fujiwara, N.; Paik, S.; Choi, C.H.; Park, J.-K.; Kim, H.-J. Mycobacterium abscessus glycopeptidolipids inhibit macrophage apoptosis and bacterial spreading by targeting mitochondrial cyclophilin D. Cell Death Dis. 2017, 8, e3012. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.D.; Herrmann, J.-L.; Kremer, L. Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Microbiol. 2020, 18, 392–407. [Google Scholar] [CrossRef] [PubMed]
- Brown-Elliott, B.A.; Woods, G.L. Antimycobacterial Susceptibility Testing of Nontuberculous Mycobacteria. J. Clin. Microbiol. 2019, 57, 10. [Google Scholar] [CrossRef]
- Wilson, J.W.; Schurr, M.J.; Leblanc, C.L.; Ramamurthy, R.; Buchanan, K.L.; Nickerson, C.A. Mechanisms of bacterial pathogenicity. Postgrad. Med. J. 2002, 78, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.A.; Grinstein, S. Phagocytosis: Receptors, signal integration, and the cytoskeleton. Immunol. Rev. 2014, 262, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Cho, S.-N.; Lim, Y.-J.; Choi, J.-A.; Lee, J.; Go, D.; Song, C.-H. Phagocytosis influences the intracellular survival of Mycobacterium smegmatis via the endoplasmic reticulum stress response. Cell Biosci. 2018, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.J.; Peters, K.N.; Behar, S.M. Macrophages clean up: Efferocytosis and microbial control. Curr. Opin. Microbiol. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Podinovskaia, M.; Lee, W.; Caldwell, S.; Russell, D.G. Infection of macrophages with Mycobacterium tuberculosis induces global modifications to phagosomal function. Cell. Microbiol. 2013, 15, 843–859. [Google Scholar] [CrossRef] [PubMed]
- Weinrauch, Y.; Zychlinsky, A. The Induction of Apoptosis by Bacterial Pathogens. Annu. Rev. Microbiol. 1999, 53, 155–187. [Google Scholar] [CrossRef]
- Hay, S.; Kannourakis, G. A time to kill: Viral manipulation of the cell death program. J. Gen. Virol. 2002, 83, 1547–1564. [Google Scholar] [CrossRef] [PubMed]
- James, E.R.; Green, D.R. Manipulation of apoptosis in the host–parasite interaction. Trends Parasitol. 2004, 20, 280–287. [Google Scholar] [CrossRef]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Cell death and infection: A double-edged sword for host and pathogen survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef]
- Park, H.-S.; Back, Y.W.; Jang, I.-T.; Lee, K.-I.; Son, Y.-J.; Choi, H.-G.; Dang, T.B.; Kim, H.-J. Mycobacterium tuberculosis Rv2145c Promotes Intracellular Survival by STAT3 and IL-10 Receptor Signaling. Front. Immunol. 2021, 12, 666293. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-M.; Yang, C.-S.; Yuk, J.-M.; Lee, J.-Y.; Kim, K.H.; Shin, S.J.; Takahara, K.; Lee, S.J.; Jo, E.-K. Mycobacterium abscessus activates the macrophage innate immune response via a physical and functional interaction between TLR2 and dectin-1. Cell. Microbiol. 2008, 10, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Escoll, P.; Buchrieser, C. Metabolic reprogramming: An innate cellular defence mechanism against intracellular bacteria? Curr. Opin. Immunol. 2019, 60, 117–123. [Google Scholar] [CrossRef]
- Ramond, E.; Jamet, A.; Coureuil, M.; Charbit, A. Pivotal Role of Mitochondria in Macrophage Response to Bacterial Pathogens. Front. Immunol. 2019, 10, 2461. [Google Scholar] [CrossRef] [PubMed]
- Marie, I.; Heliot, P.; Roussel, F.; Hervé, F.; Muir, J.F.; Levesque, H. Fatal Mycobacterium peregrinum pneumonia in refractory polymyositis treated with infliximab. Rheumatology 2005, 44, 1201–1202. [Google Scholar] [CrossRef] [PubMed]
- Short, W.R.; Emery, C.; Bhandary, M.; O’Donnell, J.A. Misidentification of Mycobacterium peregrinum, the Causal Organism of a Case of Bacteremia and Automatic Implantable Cardioverter Defibrillator-Associated Infection, Due to Its Unusual Acid-Fast Staining Characteristics. J. Clin. Microbiol. 2005, 43, 2015–2017. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Olivero, I.A.; Guevara, A.; Escalona, A.; Oliver, M.; Pérez-Alfonzo, R.; Piquero, J.; Zerpa, O.; de Waard, J.H. Soft-tissue infections due to non-tuberculous mycobacteria following mesotherapy. What is the price of beauty. Enferm. Infecc. Microbiol. Clin. 2006, 24, 302–306. [Google Scholar] [CrossRef]
- Santos, A.; Cremades, R.; Rodriguez, J.C.; Ruiz, M.; Royo, G.; Garcia-Pachon, E. Mycobacterium peregrinum: Bactericidal activity of antibiotics alone and in combination. J. Infect. Chemother. 2008, 14, 262–263. [Google Scholar] [CrossRef]
- Howard, S.T.; Rhoades, E.; Recht, J.; Pang, X.; Alsup, A.; Kolter, R.; Lyons, C.R.; Byrd, T.F. Spontaneous reversion of Mycobacterium abscessus from a smooth to a rough morphotype is associated with reduced expression of glycopeptidolipid and reacquisition of an invasive phenotype. Microbiology 2006, 152, 1581–1590. [Google Scholar] [CrossRef]
- Medjahed, H.; Gaillard, J.-L.; Reyrat, J.-M. Mycobacterium abscessus: A new player in the mycobacterial field. Trends Microbiol. 2010, 18, 117–123. [Google Scholar] [CrossRef]
- Marras, T.K.; Daley, C.L. Epidemiology of human pulmonary infection with mycobacteria nontuberculous. Clin. Chest Med. 2002, 23, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Griffith, D.E.; Aksamit, T.; Brown-Elliott, B.A.; Catanzaro, A.; Daley, C.; Gordin, F.; Holland, S.M.; Horsburgh, R.; Huitt, G.; Iademarco, M.F.; et al. An Official ATS/IDSA Statement: Diagnosis, Treatment, and Prevention of Nontuberculous Mycobacterial Diseases. Am. J. Respir. Crit. Care Med. 2007, 175, 367–416. [Google Scholar] [CrossRef] [PubMed]
- Belisle, J.T.; Brennan, P.J. Chemical basis of rough and smooth variation in mycobacteria. J. Bacteriol. 1989, 171, 3465–3470. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, S.; Ren, H.; Chen, J.; Li, J.; Wang, Y.; Hua, Y.; Wang, X.; Huang, N. HMGN2 regulates non-tuberculous mycobacteria survival via modulation of M1 macrophage polarization. J. Cell. Mol. Med. 2019, 23, 7985–7998. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-H.; Park, J.-Y.; Kim, D.-Y.; Lee, T.-S.; Jung, D.-H.; Kim, Y.-J.; Lee, Y.-J.; Lee, Y.-J.; Seo, I.-S.; Song, E.-J.; et al. Type I Interferons Are Involved in the Intracellular Growth Control of Mycobacterium abscessus by Mediating NOD2-Induced Production of Nitric Oxide in Macrophages. Front. Immunol. 2021, 12, 738070. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef]
- Mootoo, A.; Stylianou, E.; Arias, M.; Reljic, R. TNF-α in Tuberculosis: A Cytokine with a Split Personality. Inflamm. Allergy-Drug Targets 2009, 8, 53–62. [Google Scholar] [CrossRef]
- Flesch, I.E.; Kaufmann, S.H. Role of Cytokines in Tuberculosis. Immunobiology 1993, 189, 316–339. [Google Scholar] [CrossRef]
- Shiratsuchi, H.; Johnson, J.L.; Ellner, J.J. Bidirectional effects of cytokines on the growth of Mycobacterium avium within human monocytes. J. Immunol. 1991, 146, 3165–3170. [Google Scholar] [CrossRef]
- Sampaio, E.P.; Elloumi, H.Z.; Zelazny, A.; Ding, L.; Paulson, M.L.; Sher, A.; Bafica, A.L.; Shea, Y.R.; Holland, S.M. Mycobacterium abscessus and M. avium Trigger Toll-Like Receptor 2 and Distinct Cytokine Response in Human Cells. Am. J. Respir. Cell Mol. Biol. 2008, 39, 431–439. [Google Scholar] [CrossRef]
- Quesniaux, V.J.; Nicolle, D.M.; Torres, D.; Kremer, L.; Guérardel, Y.; Nigou, J.; Puzo, G.; Erard, F.; Ryffel, B. Toll-Like Receptor 2 (TLR2)-Dependent-Positive and TLR2-Independent-Negative Regulation of Proinflammatory Cytokines by Mycobacterial lipomannans. J. Immunol. 2004, 172, 4425–4434. [Google Scholar] [CrossRef]
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. To Eat and to Be Eaten: Mutual Metabolic Adaptations of Immune Cells and Intracellular Bacterial Pathogens upon Infection. Front. Cell. Infect. Microbiol. 2017, 7, 316. [Google Scholar] [CrossRef]
- Olive, A.J.; Sassetti, C.M. Metabolic crosstalk between host and pathogen: Sensing, adapting and competing. Nat. Rev. Microbiol. 2016, 14, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, T.; Chen, W. Modulation of Macrophage Immunometabolism: A New Approach to Fight Infections. Front. Immunol. 2022, 13, 780839. [Google Scholar] [CrossRef] [PubMed]
- Cumming, B.M.; Addicott, K.W.; Adamson, J.H.; Steyn, A.J. Mycobacterium tuberculosis induces decelerated bioenergetic metabolism in human macrophages. eLife 2018, 7, e39169. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIC (μg/mL) | ||||||
|---|---|---|---|---|---|---|
| Antibiotic | Mpgm-ATCC | Interpretation | Mpgm-S | Interpretation | Mpgm-R | Interpretation |
| Clarithromycin | ≤0.5 | S | ≤0.5 | S | ≤0.5 | S |
| Amikacin | ≤2 | S | ≤2 | S | ≤2 | S |
| Moxifloxacin | ≤0.125 | S | ≤0.125 | S | ≤0.125 | S |
| Linezolid | 2 | S | 2 | S | 4 | S |
| Streptomycin | 32 | – | 8 | – | 4 | – |
| Ciprofloxacin | ≤0.25 | S | ≤0.25 | S | ≤0.25 | S |
| Doxycycline | ≥16 | R | ≥16 | R | 16 | R |
| Clofazimine | 1 | – | 0.5 | – | ≤0.25 | – |
| Trimethoprim | 32 | R | 8 | R | 4 | R |
| Sulfamethoxazole | 608 | R | 152 | R | 76 | R |
| Cefoxitin | 8 | S | 16 | S | 8 | S |
| Imipenem | ≤1 | S | 2 | S | ≤1 | S |
| Meropenem | ≤1 | S | 4 | S | 2 | S |
| Tobramycin | 4 | I | 4 | I | 4 | I |
| Tigecycline | ≤0.125 | – | ≤0.125 | – | ≤0.125 | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.W.; Kim, H.W.; Yu, A.-R.; Yoon, H.S.; Kang, M.; Park, H.-W.; Lee, S.K.; Whang, J.; Kim, J.-S. Differential Immune Responses and Underlying Mechanisms of Metabolic Reprogramming in Smooth and Rough Variants of Mycobacterium peregrinum Infections. Pathogens 2023, 12, 1446. https://doi.org/10.3390/pathogens12121446
Lee JW, Kim HW, Yu A-R, Yoon HS, Kang M, Park H-W, Lee SK, Whang J, Kim J-S. Differential Immune Responses and Underlying Mechanisms of Metabolic Reprogramming in Smooth and Rough Variants of Mycobacterium peregrinum Infections. Pathogens. 2023; 12(12):1446. https://doi.org/10.3390/pathogens12121446
Chicago/Turabian StyleLee, Ji Won, Ho Won Kim, A-Reum Yu, Hoe Sun Yoon, Minji Kang, Hwan-Woo Park, Sung Ki Lee, Jake Whang, and Jong-Seok Kim. 2023. "Differential Immune Responses and Underlying Mechanisms of Metabolic Reprogramming in Smooth and Rough Variants of Mycobacterium peregrinum Infections" Pathogens 12, no. 12: 1446. https://doi.org/10.3390/pathogens12121446
APA StyleLee, J. W., Kim, H. W., Yu, A.-R., Yoon, H. S., Kang, M., Park, H.-W., Lee, S. K., Whang, J., & Kim, J.-S. (2023). Differential Immune Responses and Underlying Mechanisms of Metabolic Reprogramming in Smooth and Rough Variants of Mycobacterium peregrinum Infections. Pathogens, 12(12), 1446. https://doi.org/10.3390/pathogens12121446