
Genomic Analysis Unveils the Pervasiveness and Diversity of Prophages Infecting Erwinia Species

 , , and
, , and 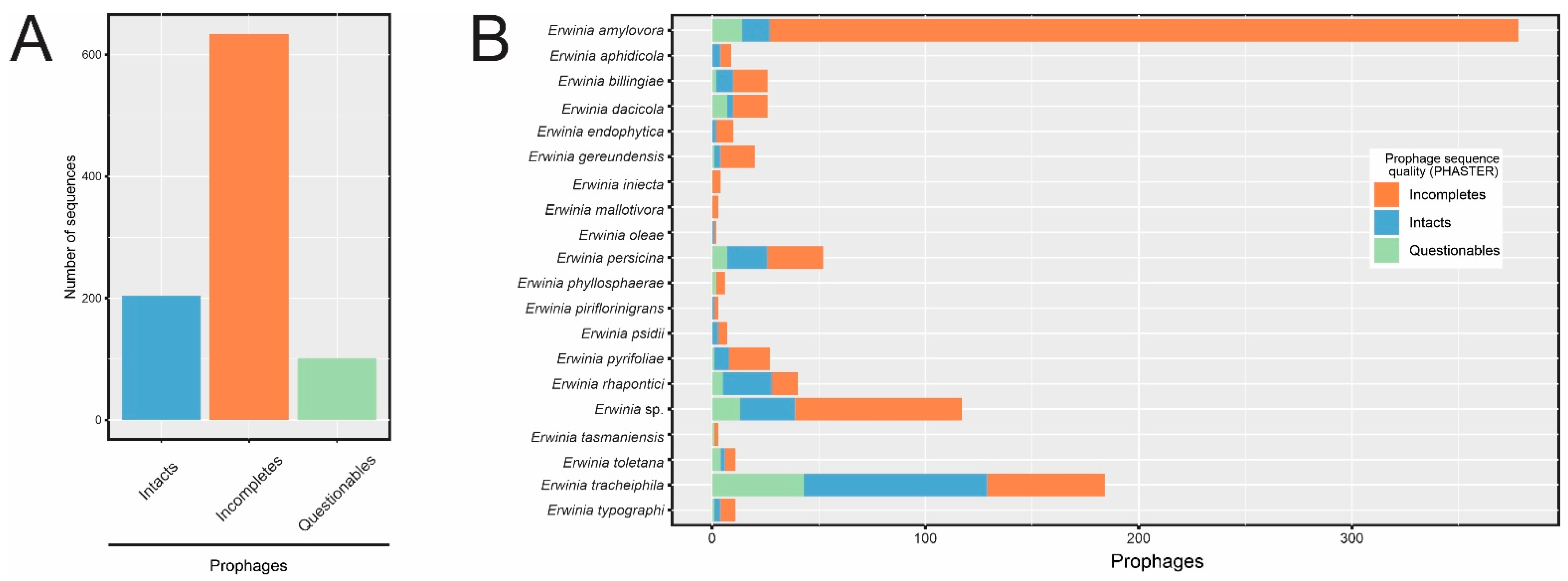{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Genomic Sequences and Quality Assessment
2.2. Identification of Prophage-like Sequences within Erwinia spp. Genomes
2.3. Taxonomic Classification of Putative ERWINIA Prophages
2.4. CRISPR-Cas Systems Identification
2.5. Protospacer Identification
2.6. In Silico Screening for DISARM System in Erwinia spp. Genomes
2.7. In Silico Screening for BREX Types 1–6 in Erwinia spp. Genomes
3. Results
3.1. Acquisition of Erwinia spp. Genome Assemblies and Quality Assessment
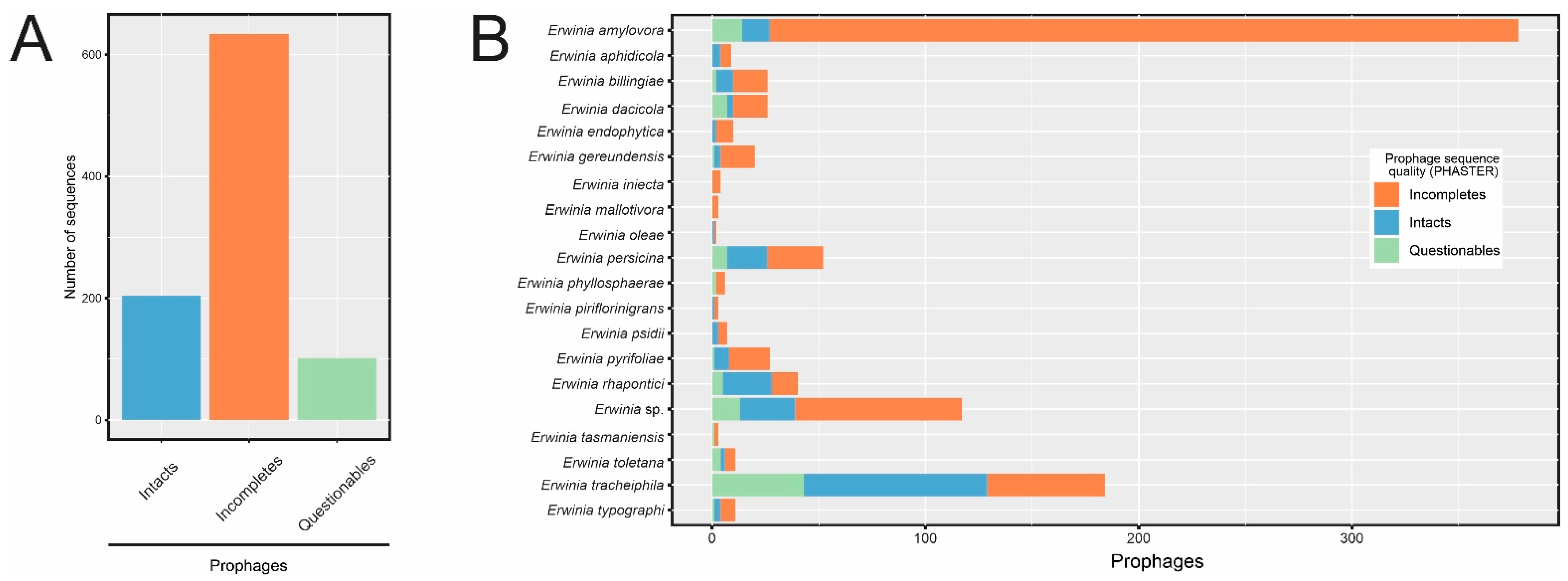
3.2. Prophage-like Sequences Are Pervasive in Erwinia spp. Genomes
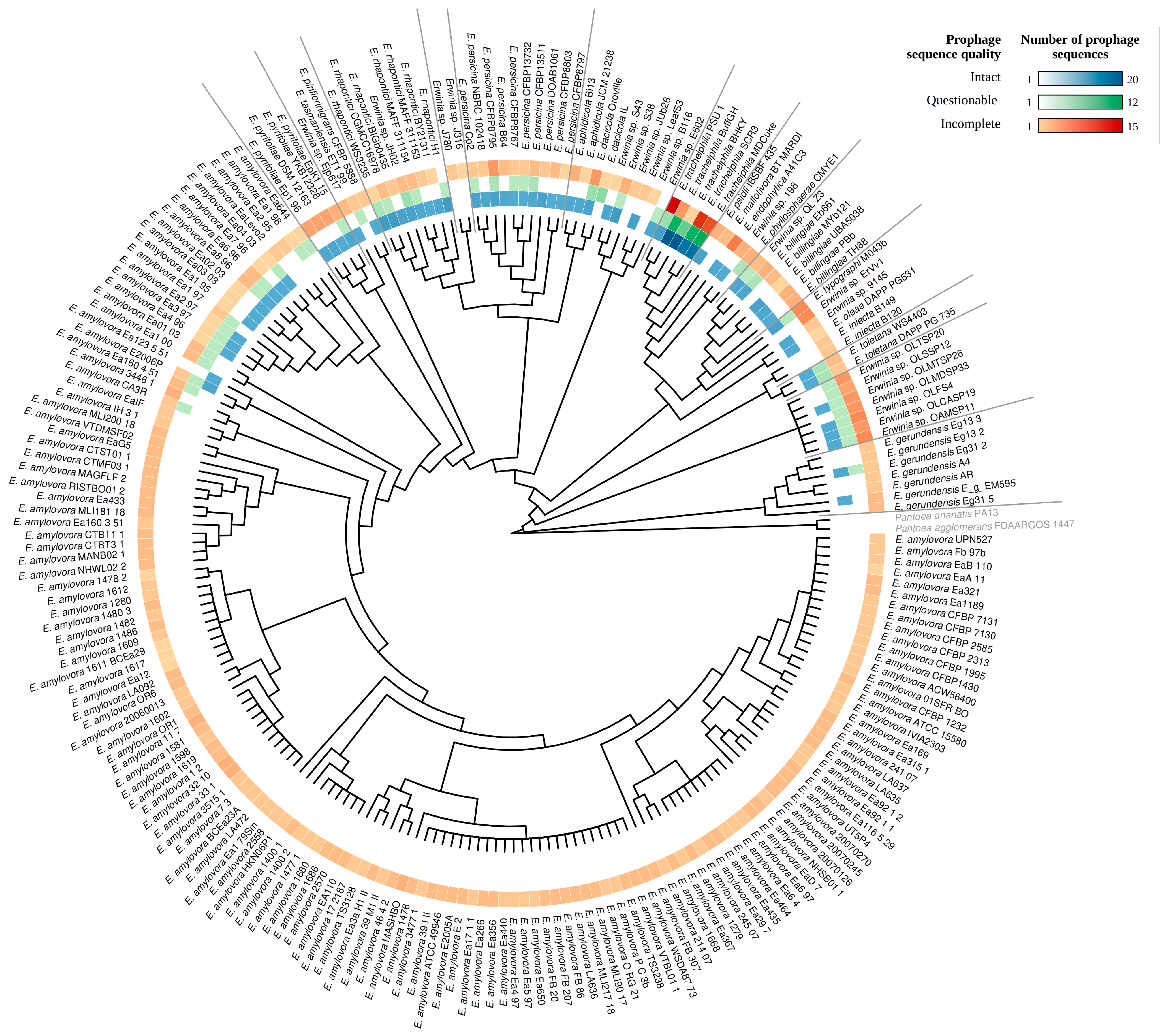
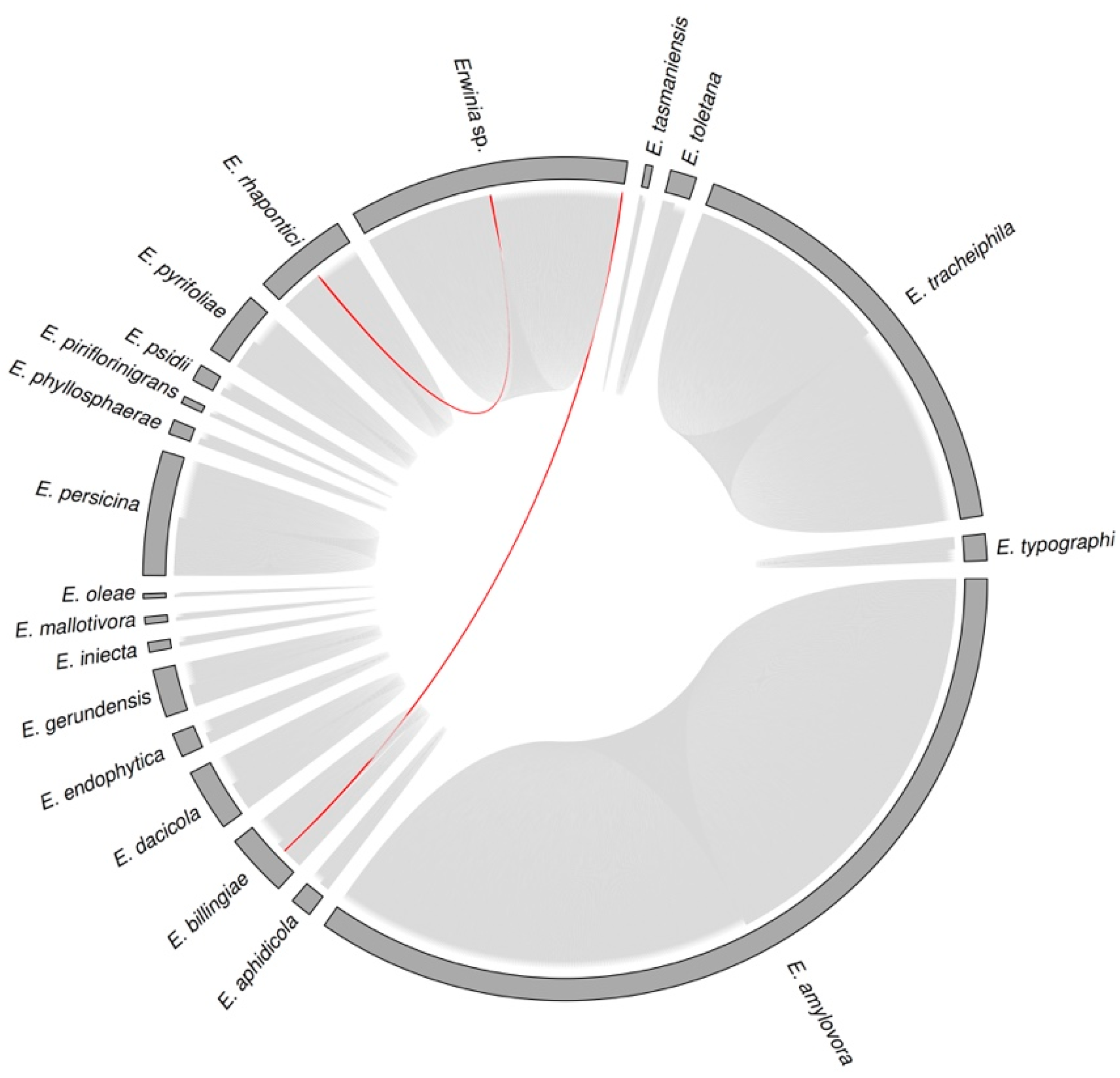
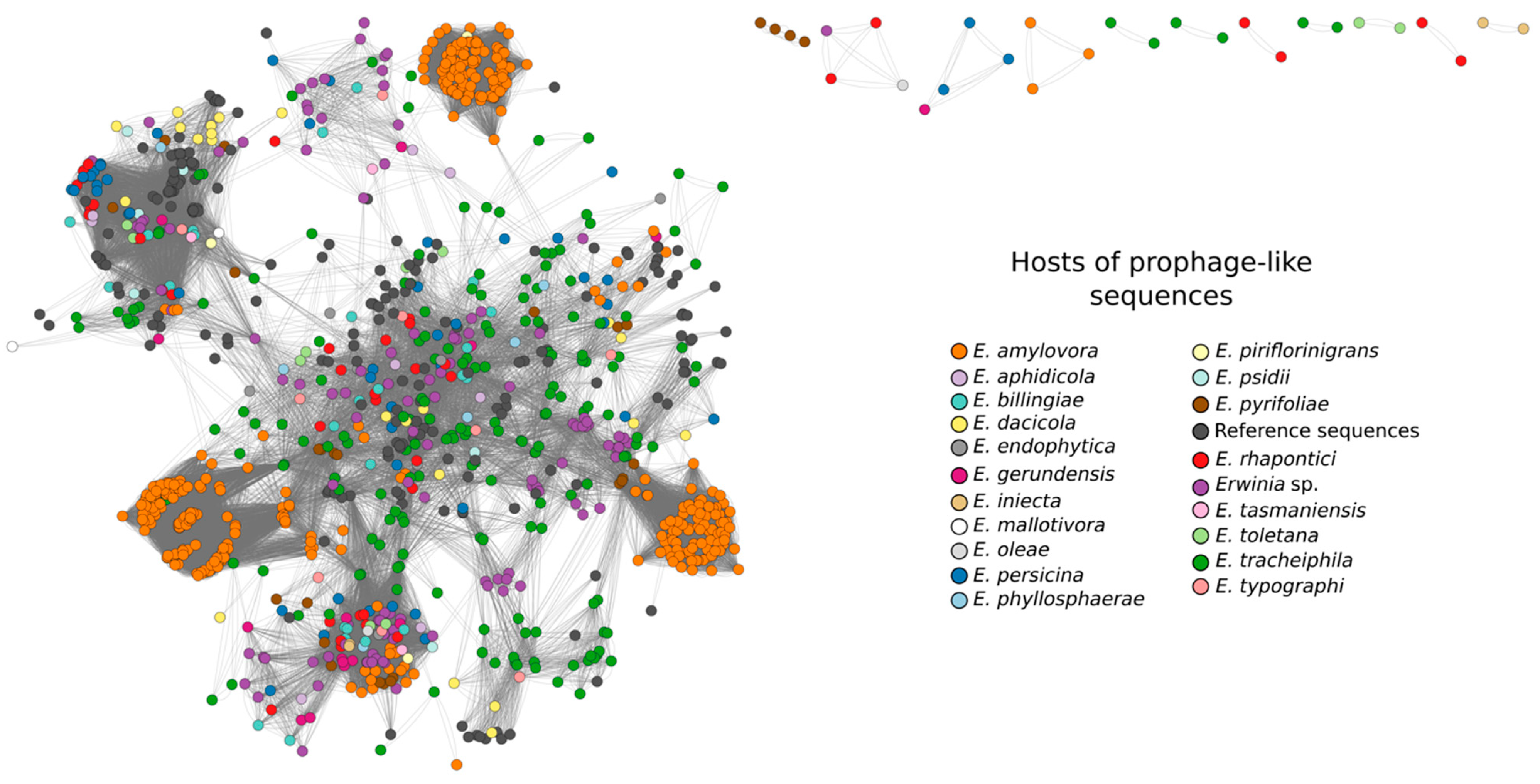
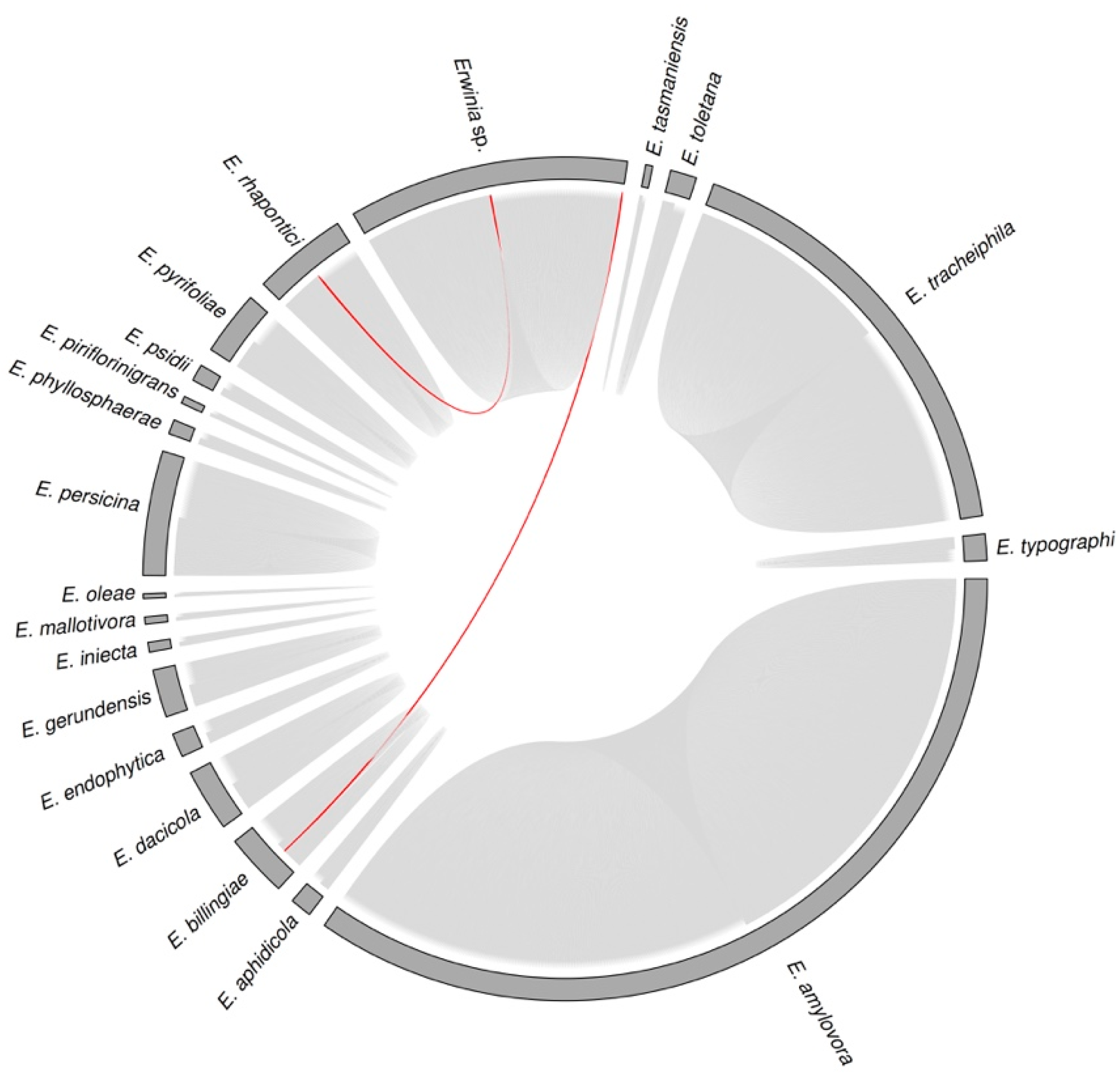
3.3. Sequence Diversity and Taxonomic Classification of the Putative Erwinia Prophages
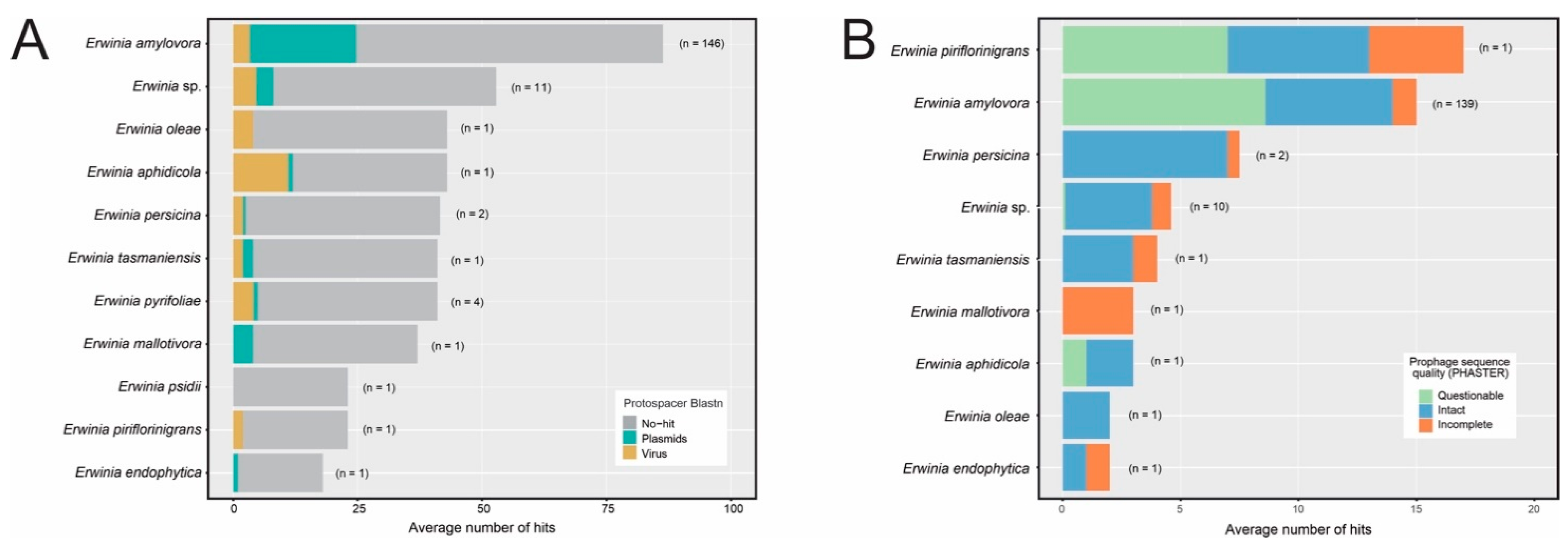
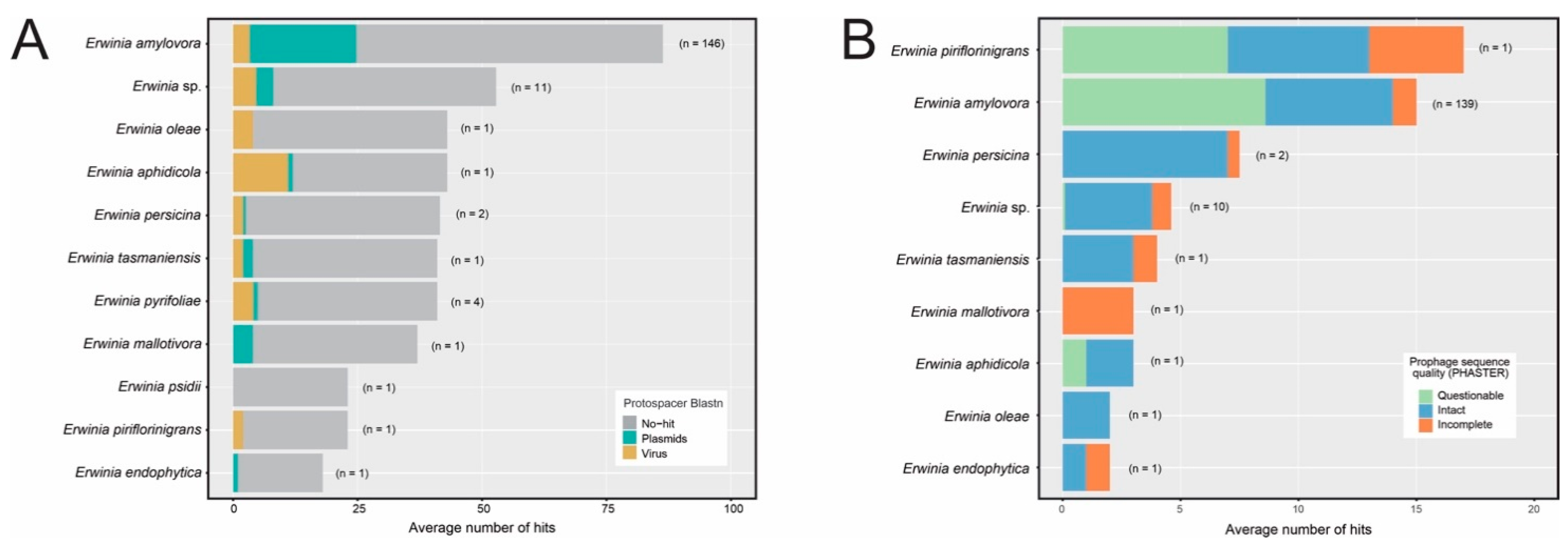
3.4. CRISPR-Cas Systems Are Unevenly Distributed across the Erwinia Genus and Have a High Proportion of Unknown Targets
3.5. BREX and DISARM Anti-Phage Defense Systems Are Rare in Erwinia spp. Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kharadi, R.R.; Schachterle, J.K.; Yuan, X.; Castiblanco, L.F.; Peng, J.; Slack, S.M.; Zeng, Q.; Sundin, G.W. Genetic Dissection of the Erwinia amylovora Disease Cycle. Annu. Rev. Phytopathol. 2021, 59, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Kim, J.-Y.; Park, D.H. Evidence of Greater Competitive Fitness of Erwinia amylovora over E. pyrifoliae in Korean Isolates. Plant Pathol. J. 2022, 38, 355–365. [Google Scholar] [CrossRef] [PubMed]
- LaSarre, B.; Olawole, O.I.; Paulsen, A.A.; Halverson, L.J.; Gleason, M.L.; Beattie, G.A. Complete Genome Sequences of Four Strains of Erwinia tracheiphila: A Resource for Studying a Bacterial Plant Pathogen with a Highly Complex Genome. MPMI 2022, 35, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Montoya-Estrada, C.N.; Hermenegildo, P.S.; Alvarez-Romero, P.I.; Badel, J.L.; Guimarães, L.M.S.; Alfenas, A.C. Genetic diversity and aggressiveness of Erwinia psidii on Eucalyptus spp. in Brazil. Plant Pathol. 2019, 68, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Norelli, J.L.; Jones, A.L.; Aldwinckle, S.H. Fire Blight Management in the Twenty-first Century. Plant Dis. 2003, 87, 756–765. [Google Scholar] [CrossRef] [Green Version]
- Dagher, F.; Olishevska, S.; Philion, V.; Zheng, J.; Déziel, E. Development of a novel biological control agent targeting the phytopathogen Erwinia amylovora. Heliyon 2020, 6, e05222. [Google Scholar] [CrossRef]
- Schwarz, C.; Mathieu, J.; Gomez, A.L.G.; Yu, P.; Alvarez, P.J.J. Renaissance for Phage-Based Bacterial Control. Environ. Sci. Technol. 2022, 56, 4691–4701. [Google Scholar] [CrossRef]
- Wendling, C.C.; Refardt, D.; Hall, A.R. Fitness benefits to bacteria of carrying prophages and prophage-encoded antibiotic-resistance genes peak in different environments. Evolution 2021, 75, 515–528. [Google Scholar] [CrossRef]
- Taylor, V.L.; Fitzpatrick, A.D.; Islam, Z.; Maxwell, K.L. The Diverse Impacts of Phage Morons on Bacterial Fitness and Virulence. Adv. Virus Res. 2019, 103, 1–31. [Google Scholar]
- Thompson, D.W.; Casjens, S.R.; Sharma, R.; Grose, J.H. Genomic comparison of 60 completely sequenced bacteriophages that infect Erwinia and/or Pantoea bacteria. Virology 2019, 535, 59–73. [Google Scholar] [CrossRef]
- Zrelovs, N.; Dislers, A.; Kazaks, A. Novel Erwinia persicina Infecting Phage Midgardsormr38 Within the Context of Temperate Erwinia Phages. Front. Microbiol. 2020, 11, 1245. [Google Scholar] [CrossRef] [PubMed]
- Medina-Aparicio, L.; Dávila, S.; Rebollar-Flores, J.E.; Calva, E.; Hernández-Lucas, I. The CRISPR-Cas system in Enterobacteriaceae. Pathog. Dis. 2018, 76, fty002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofir, G.; Melamed, S.; Sberro, H.; Mukamel, Z.; Silverman, S.; Yaakov, G.; Doron, S.; Sorek, R. DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat Microbiol. 2018, 3, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldfarb, T.; Sberro, H.; Weinstock, E.; Cohen, O.; Doron, S.; Charpak-Amikam, Y.; Afik, S.; Ofir, G.; Sorek, R. BREX is a novel phage resistance systemwidespread in microbial genomes. EMBO J. 2015, 34, 169–183. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [Green Version]
- Jang, B.H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Pons, J.C.; Paez-Espino, D.; Riera, G.; Ivanova, N.; Kyrpides, N.C.; Llabrés, M. VPF-Class: Taxonomic assignment and host prediction of uncultivated viruses based on viral protein families. Bioinformatics 2021, 37, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Páez-Espino, D.; Chen, I.M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Reddy, T.; Nayfach, S.; Schulz, F.; Call, L.; et al. IMG/VR v3: An integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 2021, 49, D764–D775. [Google Scholar] [CrossRef]
- Parcey, M.; Gayder, S.; Morley-Senkler, V.; Bakkeren, G.; Úrbez-Torres, J.R.; Ali, S.; Castle, A.J.; Svircev, A.M. Comparative genomic analysis of Erwinia amylovora reveals novel insights in phylogenetic arrangement, plasmid diversity, and streptomycin resistance. Genomics 2020, 112, 3762–3772. [Google Scholar] [CrossRef]
- Koskella, B.; Brockhurst, M.A. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef] [Green Version]
- Almpanis, A.; Swain, M.; Gatherer, D.; McEwan, N. Correlation between bacterial G+C content, genome size and the G+C content of associated plasmids and bacteriophages. Microb. Genom. 2018, 4, e000168. [Google Scholar] [CrossRef]
- Pratama, A.A.; Bolduc, B.; Zayed, A.A.; Zhong, Z.; Guo, J.; Vik, D.R.; Gazitúa, M.C.; Wainaina, J.M.; Roux, S.; Sullivan, M.B. Expanding standards in viromics: In silico evaluation of dsDNA viral genome identification, classification, and auxiliary metabolic gene curation. PeerJ 2021, 9, e11447. [Google Scholar] [CrossRef]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Krupovic, M.; Daly, R.A.; Borges, A.L.; Nayfach, S.; Schulz, F.; Sharrar, A.; Carnevali, P.B.M.; Cheng, J.F.; Ivanova, N.N.; et al. Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nat. Microbiol. 2019, 4, 1895–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade-Domínguez, A.; Kolter, R.; Shapiro, L.R. Complete Genome Sequence of EtG, the First Phage Sequenced from Erwinia tracheiphila. Genome Announc. 2018, 6, e00127-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, R.; Macchi, G.; Caproni, A.; Sicurella, M.; Buratto, M.; Salvatori, F.; Pappadà, M.; Manfredini, S.; Baldisserotto, A.; Marconi, P. Control of Erwinia amylovora Growth by Moringa oleifera Leaf Extracts: In Vitro and in Planta Effects. Plants 2022, 11, 957. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.R.; Paulson, J.N.; Arnold, B.J.; Scully, E.D.; Zhaxybayeva, O.; Pierce, N.E.; Rocha, J.; Klepac-Ceraj, V.; Holton, K.; Kolter, R. An introduced crop plant is driving diversification of the virulent bacterial pathogen Erwinia tracheiphila. MBio Ecol. Evol. Sci. 2018, 9, e01307-18. [Google Scholar] [CrossRef] [Green Version]
- Borruso, L.; Salomone-Stagni, M.; Polsinelli, I.; Schmitt, A.O.; Benini, S. Conservation of Erwinia amylovora pathogenicity-relevant genes among Erwinia genomes. Arch. Microbiol. 2017, 199, 1335–1344. [Google Scholar] [CrossRef] [Green Version]
- Bondy-Denomy, J.; Davidson, A.R. When a virus is not a parasite: The beneficial effects of prophages on bacterial fitness. J. Microbiol. 2014, 52, 235–242. [Google Scholar] [CrossRef]
- López-Leal, G.; Camelo-Valera, L.C.; Hurtado-Ramírez, J.M.; Verleyen, J.; Castillo-Ramírez, S.; Reyes-Muñoz, A. Mining of Thousands of Prokaryotic Genomes Reveals High Abundance of Prophages with a Strictly Narrow Host Range. mSystems 2022, 7, 0032622. [Google Scholar] [CrossRef]
- Roach, D.R.; Sjaarda, D.R.; Sjaarda, C.P.; Ayala, C.J.; Howcroft, B.; Castle, A.J.; Svircev, A.M. Absence of lysogeny in wild populations of Erwinia amylovora and Pantoea agglomerans. Microb. Biotechnol. 2015, 8, 510–518. [Google Scholar] [CrossRef]
- Bobay, L.-M.; Touchon, M.; Rocha, E.P.C. Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. USA 2014, 111, 12127–12132. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Yu, M.; Yang, C.; Deng, C.; Ma, L.; Liu, Y. Effects of abiotic factors on the stability and infectivity of polyvalent coliphage. Water Sci. Technol. 2022, 85, 141–151. [Google Scholar] [CrossRef]
- Shahin, K.; Bouzari, M. Bacteriophage application for biocontrolling Shigella flexneri in contaminated foods. J. Food Sci. Technol. 2018, 55, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Tom, E.F.; Molineux, I.J.; Paff, M.L.; Bull, J.J. Experimental evolution of UV resistance in a phage. PeerJ 2018, 9, e5190. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Ni, P.; Deng, B.; Wang, S.; Xu, W.; Wang, D. Isolation and characterisation of phages against Pseudomonas syringae pv. actinidiae. Acta Agric. Scand. Sect. B Soil Plant Sci. 2019, 69, 199–208. [Google Scholar]
- Klassen, J.L.; Currie, C.R. Gene fragmentation in bacterial draft genomes: Extent, consequences and mitigation. BMC Genom. 2012, 13, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohlin, J.; Snipen, L.; Hardy, S.P.; Kristoffersen, A.B.; Lagesen, K.; Dønsvik, T.; Skjerve, E.; Ussery, D.W. Analysis of intra-genomic GC content homogeneity within prokaryotes. BMC Genom. 2010, 11, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Salzberg, S.L. SkewIT: The Skew Index Test for large-scale GC Skew analysis of bacterial genomes. PLoS Comput. Biol. 2020, 16, e1008439. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.J.; Svircev, A.M.; Smith, R.; Castle, A.J. Bacteriophages of Erwinia amylovora. Appl. Environ. Microbiol. 2003, 69, 2133–2138. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.P.C.; Bikard, D. Microbial defenses against mobile genetic elements and viruses: Who defends whom from what? PLoS Biol. 2022, 20, e3001514. [Google Scholar] [CrossRef]
- Watson, B.N.J.; Steens, J.A.; Staals, R.H.J.; Westra, E.R.; van Houte, S. Coevolution between bacterial CRISPR-Cas systems and their bacteriophages. Cell Host Microbe 2021, 29, 715–725. [Google Scholar] [CrossRef]
- Cui, Z.; Huntley, R.B.; Zeng, Q.; Steven, B. Temporal and spatial dynamics in the apple flower microbiome in the presence of the phytopathogen Erwinia amylovora. ISME J. 2021, 15, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Geider, K.; Auling, G.; Du, Z.; Jakovljevic, V.; Jock, S.; Völksch, B. Erwinia tasmaniensis sp. nov., a non-phytopathogenic bacterium from apple and pear trees. Int. J. Syst. Evol. 2006, 56, 2937–2943. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Lurz, R.; Kube, M.; Quedenau, C.; Jelkmann, W.; Geider, K. Molecular and physiological properties of bacteriophages from North America and Germany affecting the fire blight pathogen Erwinia amylovora. Microb. Biotechnol. 2011, 4, 735–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gayder, S.; Parcey, M.; Nesbitt, D.; Castle, A.J.; Svircev, A.M. Population Dynamics between Erwinia amylovora, Pantoea agglomerans and Bacteriophages: Exploiting Synergy and Competition to Improve Phage Cocktail Efficacy. Microorganisms 2020, 8, 1449. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Malone, L.M.; Birkholz, N.; Fineran, P.C. Conquering CRISPR: How phages overcome bacterial adaptive immunity. Curr. Opin. Biotechnol. 2021, 68, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.A.M.; Pfeifer, E.; Touchon, M.; Rocha, E.P.C. Causes and Consequences of Bacteriophage Diversification via Genetic Exchanges across Lifestyles and Bacterial Taxa. Mol. Biol. Evol. 2021, 38, 2497–2512. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, T.; Rezende, R.R.d.; Lima, T.T.M.; Souza, F.d.O.; Alfenas-Zerbini, P. Genomic Analysis Unveils the Pervasiveness and Diversity of Prophages Infecting Erwinia Species. Pathogens 2023, 12, 44. https://doi.org/10.3390/pathogens12010044
Morgan T, Rezende RRd, Lima TTM, Souza FdO, Alfenas-Zerbini P. Genomic Analysis Unveils the Pervasiveness and Diversity of Prophages Infecting Erwinia Species. Pathogens. 2023; 12(1):44. https://doi.org/10.3390/pathogens12010044
Chicago/Turabian StyleMorgan, Tulio, Rafael Reis de Rezende, Thamylles Thuany Mayrink Lima, Flávia de Oliveira Souza, and Poliane Alfenas-Zerbini. 2023. "Genomic Analysis Unveils the Pervasiveness and Diversity of Prophages Infecting Erwinia Species" Pathogens 12, no. 1: 44. https://doi.org/10.3390/pathogens12010044