
Estimation of Two Component Activities of Binary Liquid Alloys by the Pair Potential Energy Containing a Polynomial of the Partial Radial Distribution Function
Faculty of Metallurgical and Energy Engineering, Kunming University of Science and Technology, Kunming 650093, China
*
Author to whom correspondence should be addressed.
Metals 2023, 13(10), 1773; https://doi.org/10.3390/met13101773
Submission received: 26 August 2023
/
Revised: 9 October 2023
/
Accepted: 17 October 2023
/
Published: 19 October 2023
(This article belongs to the Section Computation and Simulation on Metals)
Abstract
:An investigation of partial radial distribution functions and atomic pair potentials within a system has established that the existing potential functions are rooted in the assumption of a static arrangement of atoms, overlooking their distribution and vibration. In this study, Hill’s proposed radial distribution function polynomials are applied for the pure gaseous state to a binary liquid alloy to derive the pair potential energy. The partial radial distribution functions of 36 binary liquid alloy from literatures were used to obtain the binary model parameters of four thermodynamic models for validation. Results show that the regular solution model (RSM) and molecular interaction volume model (MIVM) outperform other models when the asymmetric method calculates the partial radial distribution function. RSM demonstrates an average SD of 0.078 and an ARD of 32.2%. Similarly, MIVM exhibits an average SD of 0.095 and an average ARD of 32.2%. Wilson model yields an average SD of 0.124 and an average ARD of 226%. Nonrandom two-liquid (NRTL) model exhibits an average SD of 0.225 and an average ARD of 911%. On applying the partial radial distribution function symmetry method, MIVM and RSM outperform the other models, with an average SD of 0.143 and an average ARD of 165.9% for MIVM. RSM yields an average SD of 0.117 and an average ARD of 208.3%. Wilson model exhibits average values of 0.133 and 305.6% for SD and ARD, respectively. NRTL model shows an average SD of 0.200 and an average ARD of 771.8%. Based on this result, the influence of the symmetry degree on the thermodynamic model is explored by examining the symmetry degree as defined by the experimental activity curves of the two components.
1. Introduction
Pair potentials play a fundamental role in comprehending the static and dynamic properties of gases, liquids, and solids [1]. The main objective of studying pair potentials is to develop accurate and reliable models that describe interatomic interactions and enable simulation, prediction, and explanation of the behavior and properties of atomic systems. Various computational simulation studies, such as for protein folding, drug–receptor interactions, and material property prediction, are conducted using atomic pair potentials to ascertain the underlying mechanisms of atomic interactions, explore novel material properties, and optimize drug design. Materials science and surface science researchers have investigated atomic potentials to thoroughly understand phenomena such as material structure, stability, crystal defects, and surface adsorption. Understanding interatomic interactions is essential in material design, catalyst development, and comprehending material interfaces and surface phenomena. Atomic pair potential models encompass widely used semiempirical potentials such as the Lennard–Jones potential [2], Morse potential [3], and Born–Mayer potential [4], as well as atomic force fields, quantum mechanical descriptions, and statistical mechanics methods. Different factors affect the selection and examination of these models, including application needs and accuracy considerations [5,6,7]. Accurate pair potentials provide a comprehensive description of energy, geometric structure, and spectral properties of molecules and serve as the basis for investigating collision and chemical-reaction dynamics, such as those for atomic collisions. Thus, an in-depth study of potential pair functions holds significant physical implications and offers broad applications. This study reveals a direct correlation between the radial distribution function (RDF) and atomic pair potential. This paper proposes a linkage between the radial distribution function and the atomic pair potential energy. This is applied to a binary liquid alloy system using a polynomial expression for the radial distribution function of a pure gas. The group element activity is estimated based on the symmetries outlined in this paper, utilizing the radial distribution functions of 36 binary liquid alloys from the literature into the model.
2. Thermodynamic Model and Symmetry
2.1. Miedema Model
In 1973, Miedema et al. suggested a semi-empirical theoretical model that effectively characterizes the heat of mixing in binary alloys. This model encompasses solid solubility, the mesostable positioning of metal ions following ion injection, surface aggregation phenomena, surface energy, the formation energy of single vacancies in metals and metal compounds, as well as the development of nonphenolic alloys. For any metal binary alloy, the expression is [8]
The activity coefficient is:
In the above equation, and represent the mole fractions of species i and j respectively. and denote the molar volumes of i and j, while and stand for the electron densities, and are electronegative. The variables ,,,, and are empirical constants. For a binary alloy composed of a transition group metal and a multivalent non-transition group metal, the formula is transformed to:
2.2. Regular Solution Model (RSM)
Hildebrand suggested the RSM in 1929 [9]. According to this model, the molar excess volume and mixing entropy are zero, and the molar excess mixing Gibbs free energy is equal to the molar excess mixing enthalpy [10]. The expression for the molar excess Gibbs free energy for a binary system is as follows; the activity coefficient of the component is .
Here, and are the mole fractions of components and , respectively, and is the interaction parameter between components and . is only related to temperature and does not change with the composition of components.
2.3. Wilson Model
In 1964, Wilson [11] suggested a semiempirical and semitheoretical model based on the local concept. This model assumes that “local concentrations” (represented as volume fractions) primarily determine molecular interactions. These concentrations are defined in relation to the Boltzmann distribution probability term for energy. The excess free energy associated with the concentrations can be expressed as follows.
Here, and are the interaction parameters between components and , which are only related to temperature and do not change with the composition of components [12].
2.4. Nonrandom Two-Liquid (NRTL) Model
The NRTL model, suggested by Renon and Prausnitz in 1968 [13], has been extensively used in correlating thermodynamic data, computing thermodynamic properties, and predicting phase equilibrium for diverse fluid systems in chemical processes. This model, based on the concept of local concentration, permits the determination of molar excess Gibbs free energy. The activity coefficient of the component is .
Here, and are energy parameters characterizing the interaction between components i and j; is related to nonrandomness in the mixture, independent of temperature and composition of a solution. Moreover, the characteristic parameter of a solution depends on the solution type. When the mixture is entirely random, many binary system experimental data show that varies from 0.2 to 0.47. In this study, = 0.3.
2.5. Molecular Interaction Volume Model (MIVM)
In 2000, Tao suggested the molecular interaction volume model [14], which applied statistical thermodynamics and fluid phase equilibrium theory to describe the motion of liquid molecules. This model yields the following expression:
The activity coefficients of the components is .
Here, and are the first coordination numbers of and pure substances, respectively, and and are the molar volumes of and , respectively. and are the interaction parameters of and , respectively. is the Boltzmann constant, and T is the temperature.
2.6. Symmetry
Figure 1 shows the molar fraction of the two components as the symmetry axis of their activity curve. The symmetry degree of the activity curve can be defined as the average absolute value of the activity difference between the two components at the same concentration. In this context, represents the measure of symmetry.
The symmetry increases as the S value decreases. At , the binary liquid alloy exhibits complete symmetry, where and denote the experimental activity of components i and j, respectively, and m represents the number of experimental activities. This definition also applies to similar geometric figures. The binary liquid alloy with low symmetry is shown in Figure 2. Table 1 presents the symmetry degrees of the activity curves for the 36 binary liquid alloys based on the definitions mentioned above.
3. Pair Potential Energy Polynomials of the Binary Liquid
In an extremely diluted pure gas, the correlation between the interatomic potential energy and RDF can be expressed as follows [15]:
However, in practical scenarios, the RDF and interatomic potential energy strongly depend on the number density of the system. The relation between the RDF and pair potential energy can be expanded using a polynomial expression in terms of the number density :
It is seen formula Equation (17), that there is connection between the RDF and pair potential energy. In the case of , 3 atoms in the pure gas system, labeled 1, 2 and 3, are shown in Figure 3a. These three atoms have six interactions: 1-1, 1-2, 1-3, 2-2, 2-3, 3-3, which influence each other. The radial distribution function for 1-2 is , i.e., the distribution of atom 1 centred on atom 2, but influenced by atom 3. This form is centred on atom 3 and influences atom 1 and atom 2 in .
Consequently, the expansion based on is . Equation (18) is compared with Equation (17).
The above is the influence of atomic interactions in pure gases; assume let the interaction of atoms is applied to the liquid metal system, and Equations (16) and (17) are applied to the liquid metal. The interactions between atoms in a pure liquid metal are similar to those in the pure gaseous state, both being interactions between the same atoms. However, there are differences in the binary liquid alloy and different interactions between different atoms, as shown in Figure 3b. There are three atomic interactions in binary liquid alloys: , , and . The RDF describes atomic distribution around atom in a binary system. However, the remaining atom influences the atom in the RDF , while the remaining atom influences the atom in the RDF .
Unlike pure gas centered on the remaining atoms 3 affecting the RDF 1–2, as shown in Figure 3a. In binary liquid alloys, the remaining atoms i and j influence the atoms i and j in the radial distribution function . That is, the atom in the RDF influencing is centered on the other atoms , while the atom in the RDF influencing is centered on the other atoms .
If the RDF in a binary liquid system can be expanded as a polynomial based on number density, then Equation (18) takes the following form:
Suppose that the subradial distribution function of the principal RDF conforms to , i.e., is the primary RDF, then and conform to and the subradial distribution function is:
Substituting Equation (21) into Equation (20), we obtain:
Then substituting Equation (22) into Equation (18), we obtain:
The relation between the RDF and potential energy can be obtained using Equation (23):
Pair potential energy between molecules can be accurately calculated using the RDF. This function represents the ratio of the probability of finding another atom at a distance to the random distribution [16]. In the double distribution function, for a system with atoms and volume , the probability of a atom appearing in the element is , the probability of an atom appearing at the distance is , and the probability of atomic pairs appearing at a distance is . The double distribution function is given as follows:
In the system, the average potential energy between each atom is:
However, in the RDF of binary systems, the probability of having atom in at and atom in at is . The potential energy is , and the average value of is the sum of all possible times of the probabilities:
Substituting Equation (24) into Equation (27) yields the potential energy between atoms:
The peak value of the RDF signifies the disparity between the local and bulk molar fractions. As the mole fraction increases, the contributions of the second and third RDF peaks diminish while the contribution of the first peak amplifies. The pair potential energy is then calculated by using Equation (28) and the area of the first peak of the skewed RDF. The area under the first peak was computed through graphical integration, as depicted in Figure 4. Notably, this approach differs from Wang’s utilization of the L-PPDF mathematical form with Gaussian fitting, which relies on fitting parameters u and v [17,18]. It is important to emphasize that this study does not incorporate any fitting parameters. The RDF used in this study is defined by three key coordinates: (which represents the starting point of g(r) before reaching zero), (the transverse coordinate of the first peak of g(r)), and (the transverse coordinate of the first valley of g(r)). The asymmetric method of calculating the RDF involves integrating the area between and , while the symmetric method involves integrating twice the area between and (Figure 4). The trapezoidal method [19] is used to compute these areas (Equation (29)). Table 2 lists the references for the partial RDFs of 36 binary liquid alloys.
The RSM has a tunable parameter . The average coordination number is obtained using the pure coordination number of the two components. Additionally, the temperature documented in the literature and the calculated parameter can be referred to calculate the parameter at the desired temperature .
The parameters and of Wilson equation are given. Additionally, the values of parameters and at the desired temperature can be obtained using the temperature from the literature that was employed to calculate the corresponding parameters and .
The NRTL has two parameters and . These parameters obtained using the temperature mentioned in the literature are employed to calculate the parameters and , respectively, at the required temperature .
The MIVM involves parameters and , representing the interaction parameters for i–j and j–i interactions, respectively. Using and values obtained at temperature , the corresponding parameters and can be calculated at the desired temperature .
4. Result Analysis
4.1. Asymmetric Method for Calculating the RDF
The asymmetric method uses the area between and presented in Figure 4 and Equation (28) to determine the parameters for each model, see Table 3. Table 4 demonstrates that among the first 12 systems, the RSM performs better than other models for six binary liquid alloys. The average standard deviation (SD) is 0.027, and the average relative deviation (ARD) is 7.7%. In the case of the 12 binary liquid alloys with moderate symmetry, the RSM outperforms the other three models in six systems, resulting in an average SD of 0.059 and an average ARD of 13.83%. MIVM also performs well, with an average SD of 0.091 and an average ARD of 25.7%. For the 12 binary liquid alloys with low symmetry, the RSM outperforms the other three models in three systems, yielding an average SD of 0.101 and an average ARD of 39.5%. Additionally, the MIVM outperforms the other three models in five binary liquid alloys when the binary liquid alloys have even lower symmetry, with an average SD of 0.131 and an average ARD of 45.3%. Considering the average performance across all 36 binary liquid alloys, the Wilson and NRTL models exhibit the poorest performance, displaying larger SD and ARD values than the other models. As shown in Figure 5, the SD and ARD values generally increase with decreasing symmetry, albeit not significantly. Further analysis of high, medium, and low-symmetry binary liquid alloys reveals a strong correlation between the RSM and symmetry.
4.2. Symmetric Method for Calculating the RDF
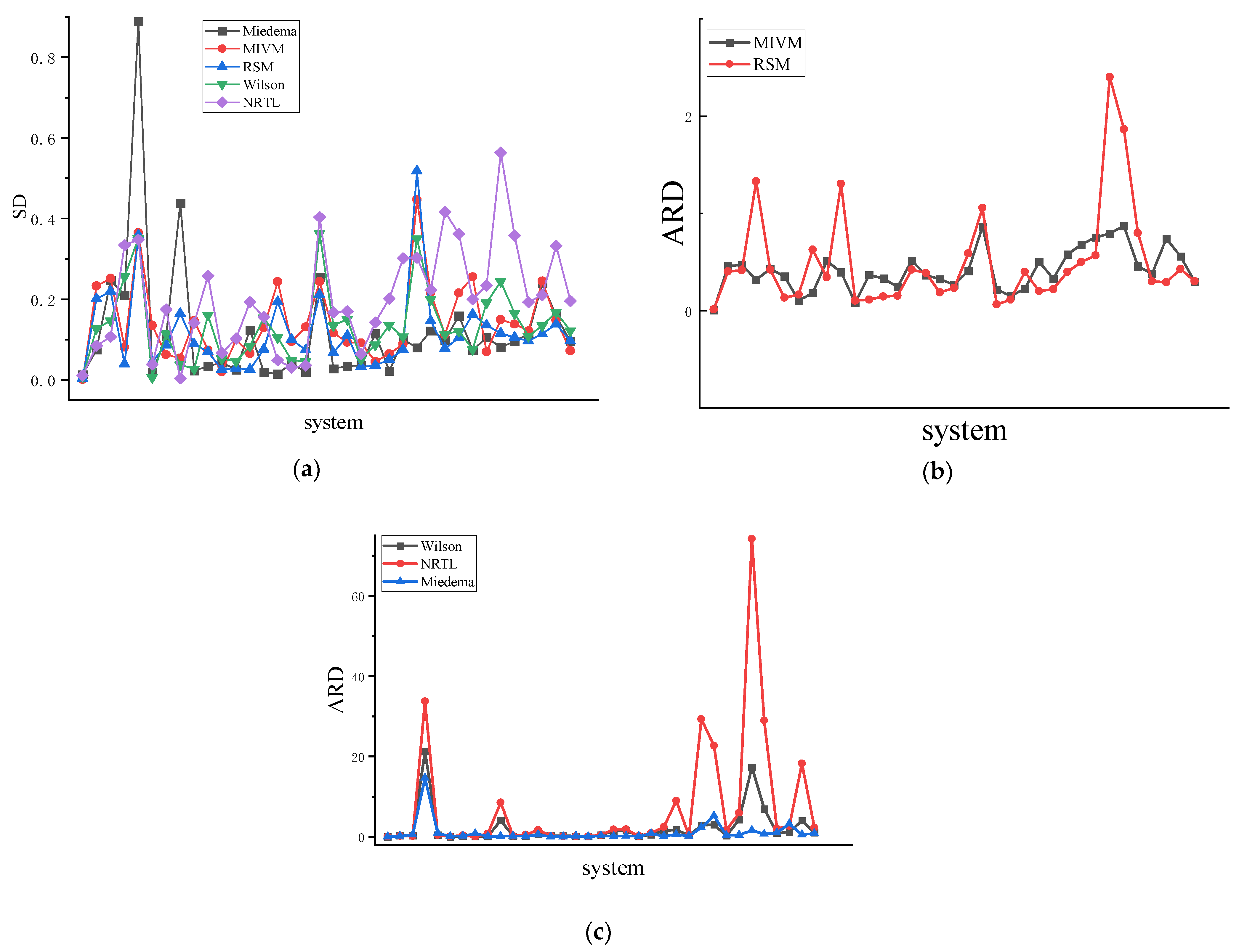
The methodology based on symmetry involves deriving parameters for each model using the region from to (Figure 4) and Equation (28), see Table 5. Analysis of data presented in Table 6 reveals that among the 12 systems characterized by high symmetry, the MIVM outperforms the other three models with an average SD of 0.127 and an average ARD of 30.5%. The RSM exhibits an average SD of 0.111 and an average ARD of 44.6%. In the 12 systems with medium symmetry, the Miedema outperforms the other three models, yielding a average SD of 0.067 and an ARD of 22.8%. By contrast, the MIVM exhibits a average SD of 0.118 and an ARD of 36.7%, and the RSM exhibits a average SD of 0.088 and an ARD of 32.8%. For the 12 systems with low symmetry, the Miedema surpasses the other three models. The RSM exhibits an average SD of 0.116 and an average ARD of 70.2%. Each model exhibits distinct performance characteristics depending on the system representation. As shown in Figure 6, the data comparison indicates an increasing trend in ARD values with decreasing symmetry.
5. Conclusions
This study uses polynomials to describe the partial RDF in pure gases and extends this approach to binary liquid systems. The primary aim of this study is to characterize the atomic distribution and unravel interatomic interactions, which are essential for accurate thermodynamic calculations. The RDF exhibits irregularities when the symmetric method is used instead of the asymmetric one for RDF calculation. Notably, the estimation of binary liquid alloy activity favors using the asymmetric method, especially when considering the average results obtained from both methods for the 36 binary liquid alloy systems investigated in this study. We selected and compared five thermodynamic models based on their symmetry degree. Data analysis reveals that the RSM exhibits the highest dependency on the symmetry degree. Conversely, the MIVM demonstrates superior adaptability to symmetric and asymmetric systems.
Author Contributions
D.T.: Theoretical guidance, review; J.H.: Conceptualization, Writing—original draft, Writing—review & editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China under Grant No. 51464022.
Data Availability Statement
Data available on request from the authors.
Conflicts of Interest
The authors declare that they have no known competing financial interest or personal relationship that could have appeared to influence the work reported in this paper.
References
- Xie, J.C.; Kar, T.; Xie, R.-H. An Accurate Pair Potential Function for Diatomic Systems. Chem. Phys. Lett. 2014, 591, 69–77. [Google Scholar] [CrossRef]
- Lennard-Jones, J.E. Cohesion. Proc. Phys. Soc. 1931, 43, 461. [Google Scholar] [CrossRef]
- Morse, P.M. Diatomic Molecules According to the Wave Mechanics. II. Vibrational Levels. Phys. Rev. 1929, 34, 57–64. [Google Scholar] [CrossRef]
- Born, M.; Mayer, J.E. Zur Gittertheorie der Ionenkristalle. Z. Phys. 1932, 75, 1–18. [Google Scholar] [CrossRef]
- Ter Horst, M.A.; Schatz, G.C.; Harding, L.B. Potential Energy Surface and Quasiclassical Trajectory Studies of the CN+H2 Reaction. J. Chem. Phys. 1996, 105, 558–571. [Google Scholar] [CrossRef]
- Hirst, D.M. Ab Initio Potential Energy Surfaces for Excited States of the NO2+ Molecular Ion and for the Reaction of N+ with O2. J. Chem. Phys. 2001, 115, 9320–9330. [Google Scholar] [CrossRef]
- Grandinetti, F.; Vinciguerra, V. Adducts of NF2+ with Diatomic and Simple Polyatomic Ligands: A Computational Investigation on the Structure, Stability, and Thermochemistry. Int. J. Mass Spectrom. 2002, 216, 285–299. [Google Scholar] [CrossRef]
- Fan, T.X.; Yang, G.J.; Chen, J.Q.; Zhang, D. Model Prediction of Thermodynamics Activity in Multicomponent Liquid Alloy. Key Eng. Mater. 2006, 313, 19–24. [Google Scholar] [CrossRef]
- Hildebrand, J.H. Solubility. XII. Regular Solutions. J. Am. Chem. Soc. 1929, 51, 66–80. [Google Scholar] [CrossRef]
- Hildebrand, J.H.; Prausnitz, J.M.; Scott, R.L. (Eds.) Regular and Related Solutions: The Solubility of Gases Liquids, and Solids; Van Nostrand Reinhold Company: New York, NY, USA, 1970. [Google Scholar]
- Wilson, G.M. Vapor-Liquid Equilibrium. XI. A New Expression for the Excess Free Energy of Mixing. J. Am. Chem. Soc. 1964, 86, 127–130. [Google Scholar] [CrossRef]
- Nagata, I. Correlation of ternary liquid-liquid equilibria using a modification of the wilson equation. Fluid Phase Equilibria 1992, 72, 1–14. [Google Scholar] [CrossRef]
- Renon, H.; Prausnitz, J.M. Local Compositions in Thermodynamic Excess Functions for Liquid Mixtures. AIChE J. 1968, 14, 135–144. [Google Scholar] [CrossRef]
- Tao, D.P. A New Model of Thermodynamics of Liquid Mixtures and Its Application to Liquid Alloys. Thermochim. Acta 2000, 363, 105–113. [Google Scholar] [CrossRef]
- Hill, T.L. Statistical Mechanics: Principles and Selected Applications; Courier Corporation: Chelmsford, MA, USA, 1957. [Google Scholar]
- Hu, Y. Molecular Thermodynamics of Fluids; Higher Education Press: Beijing, China, 1982. [Google Scholar]
- Wang, C.; Chen, X.; Tao, D. Estimation of Component Activities and Molar Excess Gibbs Energy of 19 Binary Liquid Alloys from Partial Pair Distribution Functions in Literature. Metals 2023, 13, 996. [Google Scholar] [CrossRef]
- Wang, C.; Chen, X.; Tao, D. Development of a Non-Integral Form of Coordination Number Equation Based on Pair Distribution Function and Gaussian Function. Metals 2023, 13, 384. [Google Scholar] [CrossRef]
- Department of Mathematics, Tongji University. Advanced Mathematics, 6th ed.; Higher Education Press: Beijing, China, 2007. [Google Scholar]
- Chanda, S.; Ahmed, A.Z.Z.; Bhuiyan, G.M.; Barman, S.K.; Sarker, S. A Test of Distribution Function Method in the Case of Liquid Transition Metals Alloys. J. Non-Cryst. Solids 2011, 357, 3774–3780. [Google Scholar] [CrossRef]
- Trybula, M.; Jakse, N.; Gasior, W.; Pasturel, A. Structural and Physicochemical Properties of Liquid Al–Zn Alloys: A Combined Study Based on Molecular Dynamics Simulations and the Quasi-Lattice Theory. J. Chem. Phys. 2014, 141, 224504. [Google Scholar] [CrossRef]
- Hongri, C.; Xiufang, B.; Hui, L.; Li, W. Molecular Dynamics Simulation on Structures of Cu–Ni Alloy. China J. Chem. Phys. 2002, 15, 288–294. [Google Scholar]
- Belova, I.V.; Ahmed, T.; Sarder, U.; Yi Wang, W.; Kozubski, R.; Liu, Z.-K.; Holland-Moritz, D.; Meyer, A.; Murch, G.E. Computer Simulation of Thermodynamic Factors in Ni–Al and Cu–Ag Liquid Alloys. Comput. Mater. Sci. 2019, 166, 124–135. [Google Scholar] [CrossRef]
- Wang, H.; Wei, B. Understanding Atomic-Scale Phase Separation of Liquid Fe–Cu Alloy. Chin. Sci. Bull. 2011, 56, 3416–3419. [Google Scholar] [CrossRef]
- Goto, R.; Shimojo, F.; Munejiri, S.; Hoshino, K. Structural and Electronic Properties of Liquid Ge–Sn Alloys: Ab Initio Molecular-Dynamics Simulations. J. Phys. Soc. Jpn. 2004, 73, 2746–2752. [Google Scholar] [CrossRef]
- Song, B.; Xu, N.; Jiang, W.; Yang, B.; Chen, X.; Xu, B.; Kong, L.; Liu, D.; Dai, Y. Study on Azeotropic Point of Pb–Sb Alloys by Ab-Initio Molecular Dynamic Simulation and Vacuum Distillation. Vacuum 2016, 125, 209–214. [Google Scholar] [CrossRef]
- Qin, J.; Pan, S.; Qi, Y.; Gu, T. The Structure and Thermodynamic Properties of Liquid Al–Si Alloys by Ab Initio Molecular Dynamics Simulation. J. Non-Cryst. Solids 2016, 433, 31–37. [Google Scholar] [CrossRef]
- Kbirou, M.; Mazroui, M.; Hasnaoui, A. Atomic Packing and Fractal Behavior of Al-Co Metallic Glasses. J. Alloys Compd. 2018, 735, 464–472. [Google Scholar] [CrossRef]
- Canales, M.; González, D.J.; González, L.E.; Padró, J.A. Static Structure and Dynamics of the Liquid Li–Na and Li–Mg Alloys. Phys. Rev. E 1998, 58, 4747–4757. [Google Scholar] [CrossRef]
- Pu, Z.; Zhang, H.; Li, Y.; Yang, B. Study on Sn-Sb Alloy by Ab-Initio Molecular Dynamic Simulation. IOP Conf. Ser. Mater. Sci. Eng. 2018, 394, 032098. [Google Scholar] [CrossRef]
- Jakse, N.; Pasturel, A. Local Order and Dynamic Properties of Liquid and Undercooled CuxZr1−x Alloys by Ab Initio Molecular Dynamics. Phys. Rev. B 2008, 78, 214204. [Google Scholar] [CrossRef]
- Korkmaz, Ş.; Korkmaz, S.D. Structure and Inter-Diffusion Coefficients of Liquid NaxK1−x Alloys. J. Phase Equilib. Diffus. 2010, 31, 15–21. [Google Scholar] [CrossRef]
- Ishii, Y.; Takanaga, T. Atomic and Electronic Structures and Dynamics in Liquid Alloys near Eutectic Point. J. Phys. Soc. Jpn. 2000, 69, 3334–3341. [Google Scholar] [CrossRef]
- Wang, C.C.; Wong, C.H. Short-to-Medium Range Order of Al–Mg Metallic Glasses Studied by Molecular Dynamics Simulations. J. Alloys Compd. 2011, 509, 10222–10229. [Google Scholar] [CrossRef]
- Korkmaz, S.D.; Korkmaz, Ş. A Study for Structure and Inter-Diffusion Coefficient of Liquid K1−xCsx Metal Alloys. Phys. Chem. Liq. 2011, 49, 801–810. [Google Scholar] [CrossRef]
- Costa Cabral, B. First Principles Molecular Dynamics of a Liquid Li–Na Alloy. J. Mol. Struct. Theochem 1999, 463, 145–149. [Google Scholar] [CrossRef]
- Bai, Y.W.; Zhao, X.L.; Bian, X.F.; Song, K.K.; Zhao, Y. Structure Evolution of Au50Cu50 Alloy from Melt to the Disordered Solid Solution. Mater. Sci. Forum 2020, 993, 273–280. [Google Scholar] [CrossRef]
- Shi, L.; Jia, L.; Ning, P.; Sun, X.; Wang, C.; Ma, Y.; Wang, F.; Qu, T.; Li, K. Vacuum Distillation and Ab Initio Molecular Dynamic Simulation of Al–Li Alloys. Vacuum 2023, 210, 111877. [Google Scholar] [CrossRef]
- Yang, S.J.; Hu, L.; Wang, L.; Wei, B. Molecular Dynamics Simulation of Liquid Structure for Undercooled Zr-Nb Alloys Assisted with Electrostatic Levitation Experiments. Chem. Phys. Lett. 2018, 701, 109–114. [Google Scholar] [CrossRef]
- Özdemir Kart, S.; Tomak, M.; Uludoğan, M.; Çağın, T. Liquid Properties of Pd–Ni Alloys. J. Non-Cryst. Solids 2004, 337, 101–108. [Google Scholar] [CrossRef]
- Liu, D.; Qin, J.Y.; Gu, T.K. The Structure of Liquid Mg–Cu Binary Alloys. J. Non-Cryst. Solids 2010, 356, 1587–1592. [Google Scholar] [CrossRef]
- Rao, R.V.G.; Venkatesh, R. Investigations of the Dynamic Properties of the Liquid Phase of Glassy Ca–Al Alloys. Phys. Stat. Sol. B 1992, 170, 39–46. [Google Scholar] [CrossRef]
- Mendelev, M.I.; Kramer, M.J.; Hao, S.G.; Ho, K.M.; Wang, C.Z. Development of Interatomic Potentials Appropriate for Simulation of Liquid and Glass Properties of NiZr2 Alloy. Philos. Mag. 2012, 92, 4454–4469. [Google Scholar] [CrossRef]
- Roik, O.S.; Yakovenko, O.M.; Kazimirov, V.P.; Sokol’skii, V.E.; Golovataya, N.V.; Kashirina, Y.O. Structure of Liquid Al Sn Alloys. J. Mol. Liq. 2021, 330, 115570. [Google Scholar] [CrossRef]
- Roik, O.S.; Samsonnikov, O.V.; Kazimirov, V.P.; Sokolskii, V.E.; Galushko, S.M. Medium-Range Order in Al-Based Liquid Binary Alloys. J. Mol. Liq. 2010, 151, 42–49. [Google Scholar] [CrossRef]
- Chen, F.; Cao, C.; Zhong, Q.; Liu, J.; Yang, L.; Chen, Z. Ab Initio Molecular Dynamics Study on Local Structure and Dynamic Properties of Liquid Ni62Nb38 Alloy. Mater. Today Commun. 2021, 27, 102207. [Google Scholar] [CrossRef]
- Gruner, S.; Kaban, I.; Kleinhempel, R.; Hoyer, W.; Jóvári, P.; Delaplane, R.G. Short-Range Order and Atomic Clusters in Liquid Cu–Sn Alloys. J. Non-Cryst. Solids 2005, 351, 3490–3496. [Google Scholar] [CrossRef]
- Jakse, N.; Nguyen, T.L.T.; Pasturel, A. Local Order and Dynamic Properties of Liquid Au x Si1−x Alloys by Molecular Dynamics Simulations. J. Chem. Phys. 2012, 137, 204504. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-Y.; Kim, H.; Hwang, G.S. A Comparative First-Principles Study of the Structure, Energetics, and Properties of Li–M (M = Si, Ge, Sn) Alloys. J. Phys. Chem. C 2011, 115, 20018–20026. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Qin, J.; Dong, B.; Pan, S. The Reassessment of the Structural Transition Regions along the Liquidus of Fe–Si Alloys and a Possible Liquid–Liquid Structural Transition in FeSi2 Alloy. Phys. Lett. A 2018, 382, 2655–2661. [Google Scholar] [CrossRef]
- Bhuiyan, G.M.; Ali, I.; Rahman, S.M.M. Atomic Transport Properties of AgIn Liquid Binary Alloys. Phys. B Condens. Matter 2003, 334, 147–159. [Google Scholar] [CrossRef]
- Weber, H.; Schumacher, M.; Jóvári, P.; Tsuchiya, Y.; Skrotzki, W.; Mazzarello, R.; Kaban, I. Experimental and Ab Initio Molecular Dynamics Study of the Structure and Physical Properties of Liquid GeTe. Phys. Rev. B 2017, 96, 054204. [Google Scholar] [CrossRef]
- Peng, H.L.; Voigtmann, T.; Kolland, G.; Kobatake, H.; Brillo, J. Structural and Dynamical Properties of Liquid Al–Au Alloys. Phys. Rev. B 2015, 92, 184201. [Google Scholar] [CrossRef]
- Guo, F.; Tian, Y.; Qin, J.; Xu, R.; Zhang, Y.; Zheng, H.; Lv, T.; Qin, X.; Tian, X.; Sun, Y. Structure of Liquid Cu–Sb Alloys by Ab Initio Molecular Dynamics Simulations, High Temperature X-ray Diffraction, and Resistivity. J Mater Sci 2013, 48, 4438–4445. [Google Scholar] [CrossRef]
- Franke, P.; Neuschütz, D. (Eds.) Binary Systems. Part 1_Elements and Binary Systems from Ag-Al to Au-Tl; Lehrstuhl für Theoretische Hüttenkunde, Rheinisch-Westfälische Technische Hochschule Aachen; Landolt-Börnstein—Group IV Physical Chemistry; Springer: Berlin/Heidelberg, Germany, 2002; Volume 19B1, ISBN 978-3-540-65327-1. [Google Scholar]
- Franke, P.; Neuschütz, D. (Eds.) Binary Systems. Part 2: Elements and Binary Systems from B–C to Cr–Zr; Landolt-Börnstein—Group IV Physical Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; Volume 19B2, ISBN 978-3-540-20205-9. [Google Scholar]
- Franke, P.; Neuschütz, D. (Eds.) Binary Systems. Part 3: Binary Systems from Cs-K to Mg-Zr; Landolt-Börnstein—Group IV Physical Chemistry; Springer: Berlin/Heidelberg, Germany, 2005; Volume 19B3, ISBN 978-3-540-23119-6. [Google Scholar]
- Franke, P.; Neuschütz, D. (Eds.) Binary Systems. Part 4: Binary Systems from Mn-Mo to Y-Zr; Springer: Berlin/Heidelberg, Germany, 2006; Volume 19B4. [Google Scholar]
- Franke, P.; Neuschütz, D. (Eds.) Binary Systems. Part 5: Binary Systems Supplement 1: Phase Diagrams, Phase Transition Data, Integral and Partial Quantities of Alloys; Landolt-Börnstein—Group IV Physical Chemistry; Springer: Berlin/Heidelberg, Germany, 2007; Volume 19B5, ISBN 978-3-540-45279-9. [Google Scholar]
Figure 1.
(a) Schematic diagram of symmetrical activity curves, (b) Schematic diagram of asymmetric activity curves.
Figure 1.
(a) Schematic diagram of symmetrical activity curves, (b) Schematic diagram of asymmetric activity curves.
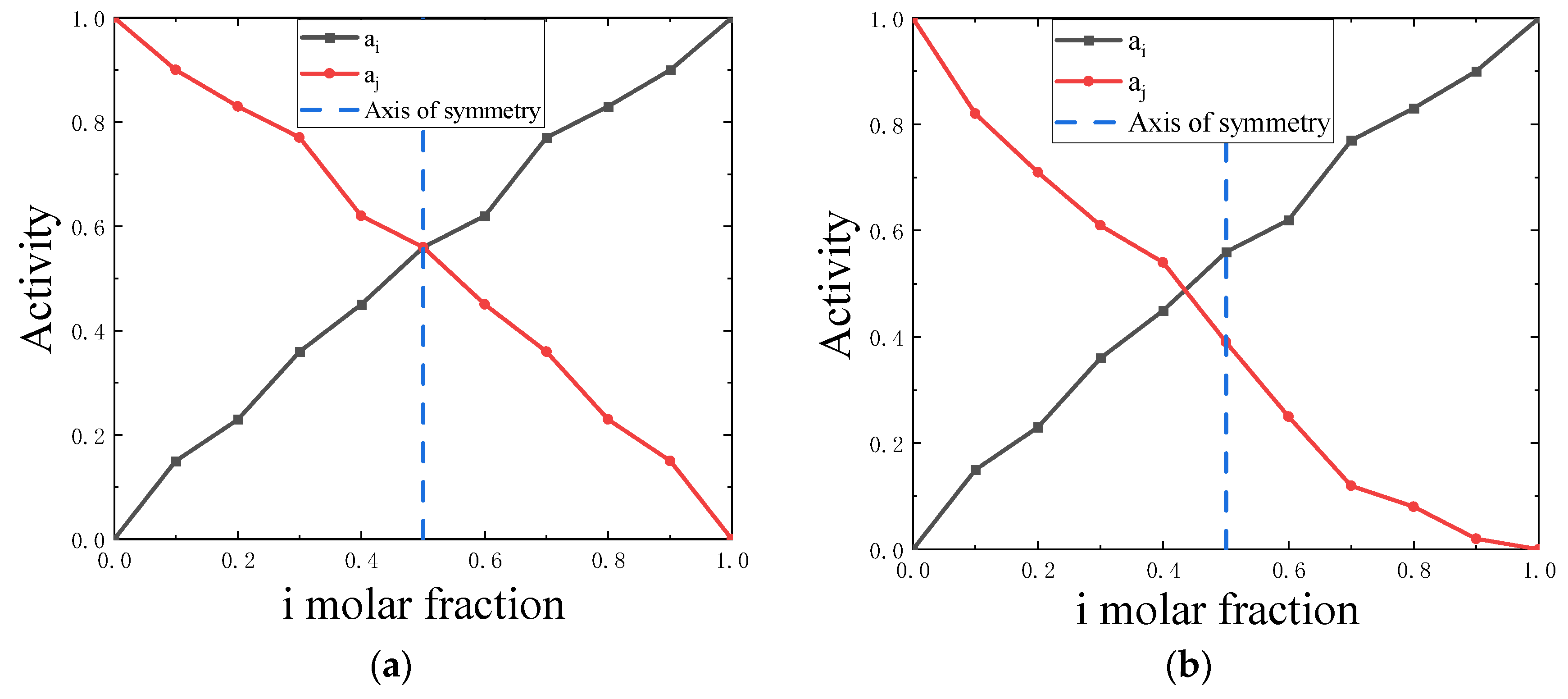
Figure 2.
(a) Schematic diagram of symmetrical Al-Zn activity curves, (b) Schematic diagram of asymmetrical Cu-Sb activity curves.
Figure 2.
(a) Schematic diagram of symmetrical Al-Zn activity curves, (b) Schematic diagram of asymmetrical Cu-Sb activity curves.

Figure 3.
(a) Schematic diagram of pure gases or pure liquid atomics interactions, (b) Schematic diagram of binary liquid alloy atomics interactions.
Figure 3.
(a) Schematic diagram of pure gases or pure liquid atomics interactions, (b) Schematic diagram of binary liquid alloy atomics interactions.
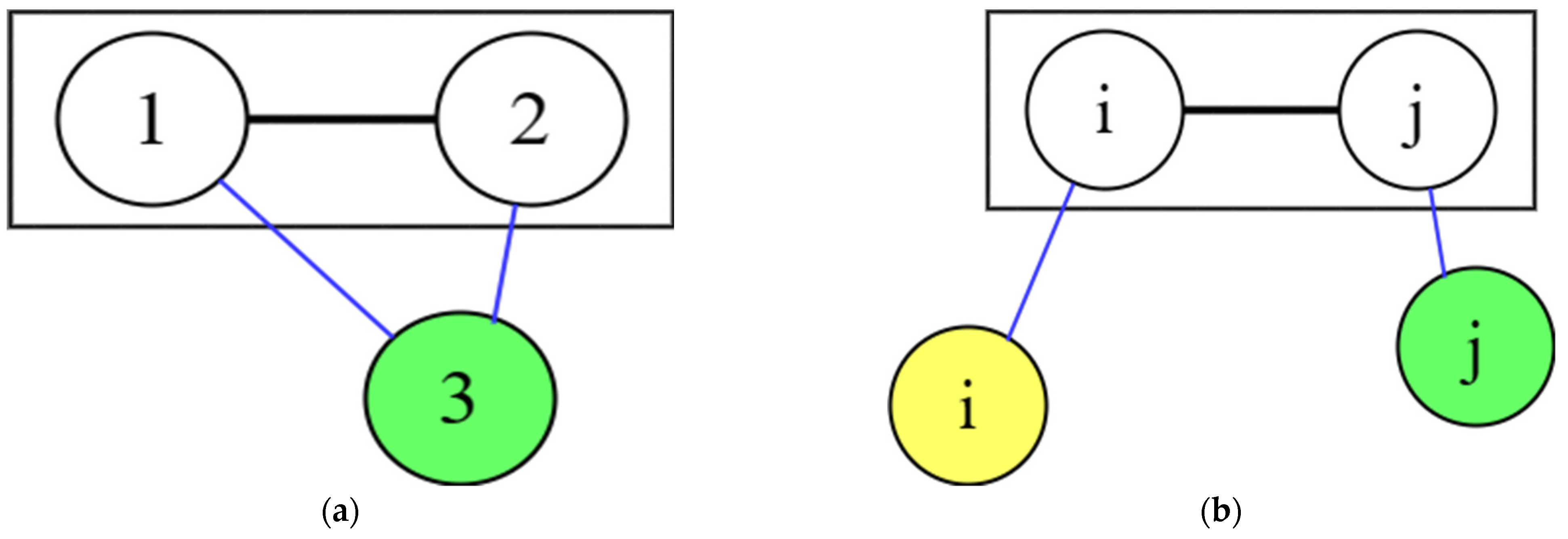
Figure 4.
(a) Schematic diagram of the asymmetric method of graphical integration of a radial distribution function, (b) Schematic diagram of the symmetric method of graphical integration of a radial distribution function.
Figure 4.
(a) Schematic diagram of the asymmetric method of graphical integration of a radial distribution function, (b) Schematic diagram of the symmetric method of graphical integration of a radial distribution function.

Figure 5.
(a) is the SD of 36 binary liquid alloys, (b) is the ARD of MIVM and RSM models of 36 binary liquid alloys, (c) is the ARD of the Wilson equation, NRTL equation, and Miedema of 36 binary liquid alloys.
Figure 5.
(a) is the SD of 36 binary liquid alloys, (b) is the ARD of MIVM and RSM models of 36 binary liquid alloys, (c) is the ARD of the Wilson equation, NRTL equation, and Miedema of 36 binary liquid alloys.
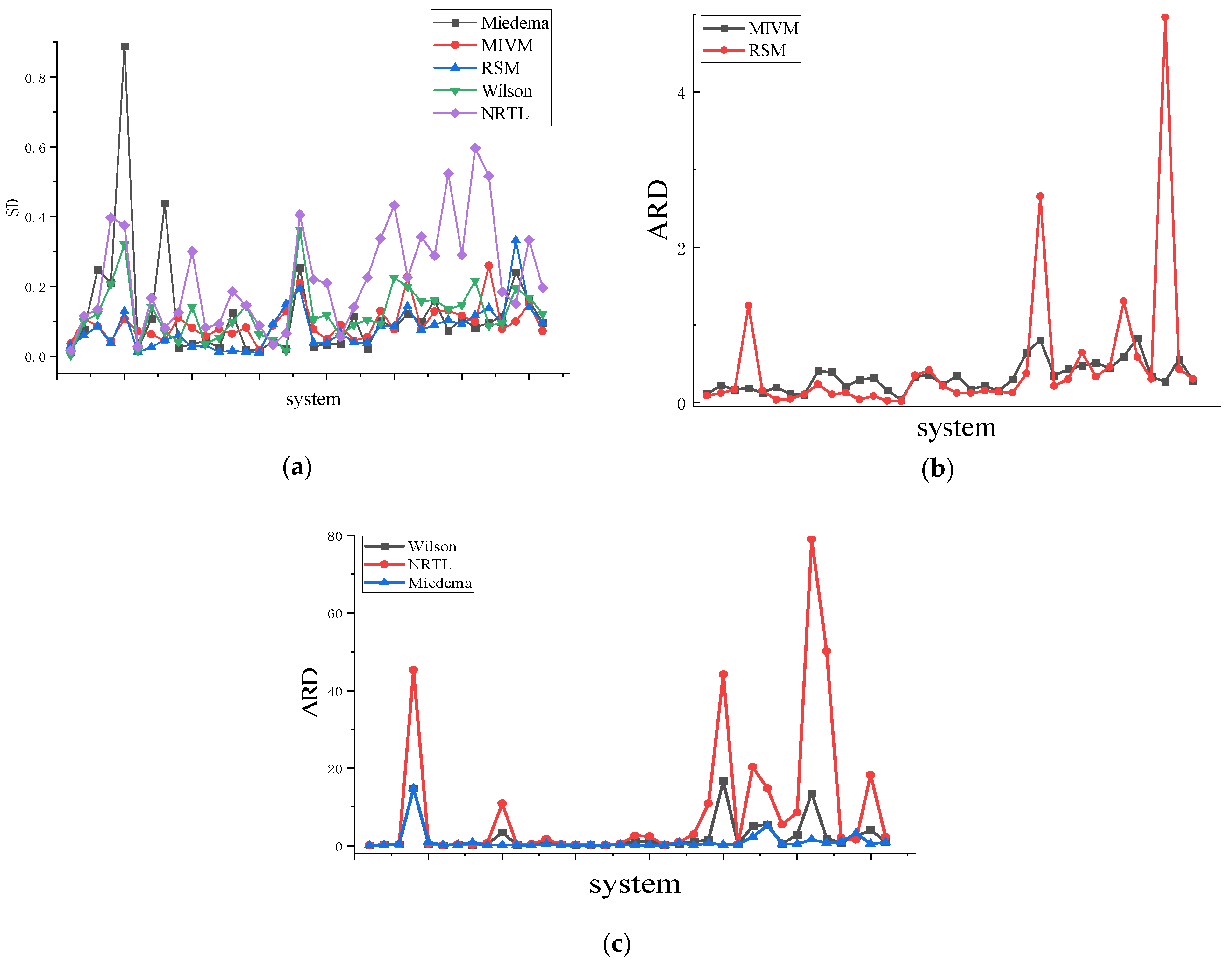
Figure 6.
(a) is the SD of 36 binary liquid alloys, (b) is the ARD of MIVM and RSM models of 36 binary liquid alloys, (c) is the ARD of the Wilson equation, NRTL equation, and Miedema of 36 binary liquid alloys.
Figure 6.
(a) is the SD of 36 binary liquid alloys, (b) is the ARD of MIVM and RSM models of 36 binary liquid alloys, (c) is the ARD of the Wilson equation, NRTL equation, and Miedema of 36 binary liquid alloys.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Symmetry of the 36 binary liquid alloys from highest to lowest.
| System | Co-Ni | Al-Zn | Cu-Ni | Al-Ni | Cu-Fe | Ge-Sn | Ag-Cu | Pb-Sb | Al-Si | Al-Co |
| 0 | 0 | 0.0028 | 0.0034 | 0.0048 | 0.007 | 0.0088 | 0.0096 | 0.0102 | 0.0114 | |
| System | Li-Mg | Sb-Sn | Cu-Zr | K-Na | Pb-Sn | Al-Mg | Cs-K | Li-Na | Au-Cu | Al-Li |
| 0.0114 | 0.0115 | 0.0119 | 0.0136 | 0.0158 | 0.0177 | 0.0228 | 0.0336 | 0.0364 | 0.0452 | |
| System | Nb-Zr | Ni-Pd | Cu-Mg | Al-Ca | Ni-Zr | Al-Sn | Al-Cu | Nb-Ni | Cu-Sn | Au-Si |
| 0.0473 | 0.0494 | 0.0597 | 0.0724 | 0.0807 | 0.0823 | 0.1116 | 0.1334 | 0.1342 | 0.139 | |
| System | Li-Sn | Fe-Si | Ag-In | Ge-Te | Al-Au | Cu-Sb | ||||
| 0.14 | 0.1454 | 0.1483 | 0.1548 | 0.160 | 0.208 |
Table 2.
Partial radial distribution functions for 36 binary liquid alloys in literatures.
| System | Co-Ni [20] | Al-Zn [21] | Cu-Ni [22] | Al-Ni [23] | Cu-Fe [24] | Ge-Sn [25] | Ag-Cu [23] | Pb-Sb [26] | Al-Si [27] | Al-Co [28] |
| System | Li-Mg [29] | Sb-Sn [30] | Cu-Zr [31] | K-Na [32] | Pb-Sn [33] | Al-Mg [34] | Cs-K [35] | Li-Na [36] | Au-Cu [37] | Al-Li [38] |
| System | Nb-Zr [39] | Ni-Pd [40] | Cu-Mg [41] | Al-Ca [42] | Ni-Zr [43] | Al-Sn [44] | Al-Cu [45] | Nb-Ni [46] | Cu-Sn [47] | Au-Si [48] |
| System | Li-Sn [49] | Fe-Si [50] | Ag-In [51] | Ge-Te [52] | Al-Au [53] | Cu-Sb [54] |
Table 3.
Parameters of four models of the asymmetric method.
| System | MIVM | RSM | Wilson | NRTL | |||
|---|---|---|---|---|---|---|---|
| Co-Ni | 0.964 | 0.977 | 0.343 | 0.964 | 0.977 | −0.036 | −0.023 |
| Al-Zn | 1.246 | 0.743 | 0.423 | 1.166 | 0.795 | 0.220 | −0.297 |
| Cu-Ni | 1.086 | 0.863 | 0.374 | 0.999 | 0.938 | 0.082 | −0.147 |
| Al-Ni | 1.493 | 1.649 | −5.203 | 0.991 | 2.485 | 0.401 | 0.500 |
| Cu-Fe | 0.943 | 0.805 | 1.512 | 0.943 | 0.805 | −0.059 | −0.217 |
| Ge-Sn | 0.740 | 1.273 | 0.266 | 1.214 | 0.776 | −0.302 | 0.241 |
| Ag-Cu | 0.675 | 1.194 | 1.225 | 0.471 | 1.709 | −0.394 | 0.177 |
| Pb-Sb | 1.558 | 0.517 | 1.046 | 1.457 | 0.553 | 0.443 | −0.660 |
| Al-Si | 1.405 | 1.077 | −1.856 | 1.226 | 1.235 | 0.340 | 0.074 |
| Al-Co | 1.112 | 1.884 | −4.213 | 0.802 | 2.612 | 0.106 | 0.633 |
| Li-Mg | 1.357 | 0.914 | −1.095 | 1.469 | 0.844 | 0.305 | −0.090 |
| Sb-Sn | 0.769 | 1.545 | −0.847 | 0.735 | 1.618 | −0.262 | 0.435 |
| Cu-Zr | 0.908 | 1.669 | −2.277 | 1.782 | 0.851 | −0.096 | 0.512 |
| K-Na | 0.700 | 1.176 | 1.018 | 0.366 | 2.252 | −0.357 | 0.162 |
| Pb-Sn | 1.044 | 0.912 | 0.267 | 0.932 | 1.021 | 0.043 | −0.092 |
| Al-Mg | 0.820 | 1.123 | 0.459 | 1.162 | 0.793 | −0.198 | 0.116 |
| Cs-K | 1.204 | 0.635 | 1.310 | 0.801 | 0.955 | 0.185 | −0.454 |
| Li-Na | 0.764 | 0.999 | 1.347 | 1.416 | 0.539 | −0.270 | −0.001 |
| Au-Cu | 1.163 | 1.444 | −2.877 | 0.812 | 2.068 | 0.151 | 0.368 |
| Al-Li | 1.136 | 1.379 | −2.356 | 1.485 | 1.055 | 0.128 | 0.321 |
| Nb-Zr | 0.814 | 1.255 | −0.110 | 1.048 | 0.975 | −0.206 | 0.227 |
| Ni-Pd | 1.168 | 1.087 | −1.307 | 1.590 | 0.798 | 0.153 | 0.080 |
| Cu-Mg | 1.312 | 1.258 | −2.781 | 2.575 | 0.641 | 0.272 | 0.230 |
| Al-Ca | 2.202 | 1.259 | −5.761 | 5.826 | 0.476 | 0.789 | 0.230 |
| Ni-Zr | 1.522 | 1.730 | −5.373 | 3.247 | 0.811 | 0.420 | 0.548 |
| Al-Sn | 0.624 | 1.440 | 0.598 | 1.024 | 0.877 | −0.471 | 0.365 |
| Al-Cu | 1.568 | 1.530 | −5.209 | 2.192 | 1.152 | 0.476 | 0.450 |
| Nb-Ni | 1.772 | 1.071 | −3.558 | 1.070 | 1.774 | 0.572 | 0.069 |
| Cu-Sn | 1.987 | 0.849 | −2.904 | 0.874 | 1.932 | 0.687 | −0.163 |
| Au-Si | 1.224 | 1.684 | −3.129 | 1.034 | 1.993 | 0.202 | 0.521 |
| Li-Sn | 3.396 | 2.186 | −10.225 | 4.263 | 1.742 | 1.223 | 0.782 |
| Fe-Si | 5.678 | 1.776 | −9.822 | 4.695 | 2.148 | 1.737 | 0.574 |
| Ag-In | 1.595 | 0.925 | −2.227 | 2.476 | 0.596 | 0.467 | −0.078 |
| Ge-Te | 0.409 | 1.891 | 1.285 | 0.912 | 0.848 | −0.894 | 0.637 |
| Al-Au | 1.680 | 1.660 | −5.740 | 2.287 | 1.219 | 0.519 | 0.507 |
| Cu-Sb | 1.804 | 0.881 | −2.315 | 0.757 | 2.098 | 0.590 | −0.127 |
Table 4.
Deviations and relative errors of five models in the asymmetric method.
| System | Miedema | MIVM | RSM | Wilson | NRTL | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| SD | ARD/% | SD | ARD/% | SD | ARD/% | SD | ARD/% | SD | ARD/% | |
| Co-Ni | 0.013 | 3.852 | 0.036 | 10.8 | 0.029 | 8.8 | 0.003 | 0.8 | 0.015 | 4.4 |
| Al-Zn | 0.075 | 15.536 | 0.108 | 22 | 0.058 | 12.1 | 0.100 | 20.5 | 0.116 | 23.7 |
| Cu-Ni | 0.247 | 49.697 | 0.085 | 16.5 | 0.085 | 16.5 | 0.120 | 23.1 | 0.134 | 25.7 |
| Al-Ni | 0.210 | 1467.533 | 0.044 | 18.3 | 0.037 | 125 | 0.208 | 1472 | 0.398 | 4520 |
| Cu-Fe | 0.889 | 93.800 | 0.106 | 12.5 | 0.128 | 15.1 | 0.321 | 37.5 | 0.375 | 44 |
| Ge-Sn | 0.019 | 5.384 | 0.071 | 19.4 | 0.011 | 3.1 | 0.016 | 4.1 | 0.027 | 7.5 |
| Ag-Cu | 0.108 | 19.924 | 0.062 | 10.8 | 0.025 | 4.6 | 0.143 | 26.1 | 0.167 | 30.5 |
| Pb-Sb | 0.438 | 79.951 | 0.044 | 9.9 | 0.047 | 10.5 | 0.070 | 15.9 | 0.080 | 18.1 |
| Al-Si | 0.023 | 10.017 | 0.110 | 40.5 | 0.058 | 23.2 | 0.041 | 18.5 | 0.125 | 63.3 |
| Al-Co | 0.034 | 10.163 | 0.080 | 39.2 | 0.027 | 10.5 | 0.142 | 341 | 0.300 | 1085 |
| Li-Mg | 0.043 | 17.882 | 0.056 | 20.7 | 0.032 | 12.3 | 0.035 | 14.2 | 0.082 | 34.9 |
| Sb-Sn | 0.025 | 6.367 | 0.078 | 29.3 | 0.012 | 3.6 | 0.054 | 24.5 | 0.093 | 44.2 |
| Cu-Zr | 0.124 | 53.790 | 0.064 | 31.6 | 0.015 | 8.4 | 0.098 | 74.4 | 0.185 | 162 |
| K-Na | 0.019 | 3.619 | 0.081 | 15.3 | 0.012 | 2 | 0.144 | 27.8 | 0.145 | 28 |
| Pb-Sn | 0.015 | 1.847 | 0.016 | 3.2 | 0.009 | 1.7 | 0.063 | 14.3 | 0.087 | 19.6 |
| Al-Mg | 0.042 | 14.076 | 0.087 | 32.9 | 0.092 | 34.9 | 0.048 | 17 | 0.033 | 11.2 |
| Cs-K | 0.020 | 4.475 | 0.129 | 36 | 0.148 | 41.7 | 0.016 | 3.3 | 0.066 | 16.9 |
| Li-Na | 0.255 | 24.451 | 0.210 | 22.9 | 0.192 | 21.3 | 0.364 | 40.5 | 0.405 | 45.2 |
| Au-Cu | 0.028 | 7.697 | 0.076 | 34.5 | 0.038 | 12.1 | 0.105 | 101 | 0.219 | 255 |
| Al-Li | 0.034 | 17.993 | 0.048 | 17 | 0.037 | 12.2 | 0.118 | 115 | 0.209 | 236 |
| Nb-Zr | 0.036 | 8.347 | 0.089 | 21.1 | 0.066 | 15.1 | 0.058 | 13 | 0.056 | 12.4 |
| Ni-Pd | 0.115 | 72.225 | 0.044 | 15 | 0.039 | 14.1 | 0.090 | 53.7 | 0.140 | 91 |
| Cu-Mg | 0.022 | 8.778 | 0.054 | 30.2 | 0.038 | 12.4 | 0.104 | 103 | 0.226 | 286 |
| Al-Ca | 0.100 | 56.098 | 0.129 | 64.3 | 0.087 | 37.4 | 0.094 | 144 | 0.338 | 1081 |
| Ni-Zr | 0.080 | 20.755 | 0.076 | 80.2 | 0.085 | 266 | 0.225 | 1661 | 0.433 | 4415 |
| Al-Sn | 0.122 | 18.737 | 0.226 | 34.4 | 0.142 | 21.3 | 0.199 | 30.4 | 0.226 | 34.5 |
| Al-Cu | 0.099 | 228.621 | 0.076 | 43 | 0.075 | 30.1 | 0.158 | 516 | 0.343 | 2031 |
| Nb-Ni | 0.159 | 517.211 | 0.128 | 47.2 | 0.089 | 64 | 0.161 | 540 | 0.288 | 1478 |
| Cu-Sn | 0.073 | 38.233 | 0.131 | 51.2 | 0.103 | 33.3 | 0.135 | 48 | 0.524 | 544 |
| Au-Si | 0.106 | 43.309 | 0.116 | 44 | 0.090 | 45.6 | 0.147 | 279 | 0.290 | 847 |
| Li-Sn | 0.081 | 159.055 | 0.098 | 59.1 | 0.117 | 130 | 0.216 | 1347 | 0.596 | 7906 |
| Fe-Si | 0.095 | 67.563 | 0.259 | 82.9 | 0.137 | 58.3 | 0.088 | 186 | 0.516 | 5003 |
| Ag-In | 0.114 | 97.817 | 0.077 | 32.8 | 0.095 | 30.3 | 0.095 | 76 | 0.185 | 191 |
| Ge-Te | 0.240 | 319.161 | 0.099 | 27.3 | 0.333 | 496 | 0.195 | 239 | 0.150 | 148 |
| Al-Au | 0.159 | 53.722 | 0.149 | 55.9 | 0.139 | 42.7 | 0.168 | 405 | 0.333 | 1822 |
| Cu-Sb | 0.095 | 80.468 | 0.072 | 28 | 0.096 | 30.4 | 0.122 | 104 | 0.196 | 229 |
| Ave | 0.121 | 102.7 | 0.095 | 32.2 | 0.078 | 47.4 | 0.124 | 226 | 0.225 | 911 |
Table 5.
Parameters of four models of the symmetric method.
| System | MIVM | RSM | Wilson | NRTL | |||
|---|---|---|---|---|---|---|---|
| Co-Ni | 1.089 | 0.900 | 0.118 | 1.089 | 0.900 | 0.085 | −0.106 |
| Al-Zn | 1.317 | 0.938 | −1.162 | 1.232 | 1.003 | 0.275 | −0.064 |
| Cu-Ni | 1.391 | 0.877 | −1.138 | 1.280 | 0.953 | 0.330 | −0.132 |
| Al-Ni | 1.155 | 2.099 | −5.114 | 0.766 | 3.163 | 0.144 | 0.742 |
| Cu-Fe | 0.890 | 1.145 | −0.103 | 0.890 | 1.145 | −0.117 | 0.135 |
| Ge-Sn | 0.562 | 1.573 | 0.545 | 0.923 | 0.958 | −0.576 | 0.453 |
| Ag-Cu | 1.309 | 0.582 | 1.538 | 0.914 | 0.833 | 0.269 | −0.542 |
| Pb-Sb | 1.231 | 0.810 | 0.017 | 1.151 | 0.866 | 0.207 | −0.211 |
| Al-Si | 1.534 | 1.140 | −2.499 | 1.338 | 1.307 | 0.428 | 0.131 |
| Al-Co | 0.830 | 1.774 | −2.218 | 0.551 | 2.673 | −0.186 | 0.573 |
| Li-Mg | 1.087 | 0.988 | −0.363 | 1.177 | 0.912 | 0.083 | −0.012 |
| Sb-Sn | 0.825 | 1.576 | −1.288 | 0.788 | 1.650 | −0.192 | 0.455 |
| Cu-Zr | 1.206 | 1.206 | 1.206 | 1.206 | 1.206 | 1.206 | 1.206 |
| K-Na | 0.562 | 1.364 | 1.390 | 0.293 | 2.613 | −0.577 | 0.311 |
| Pb-Sn | 1.601 | 0.830 | −1.549 | 1.430 | 0.929 | 0.471 | −0.186 |
| Al-Mg | 0.808 | 1.126 | 0.528 | 1.144 | 0.796 | −0.213 | 0.119 |
| Cs-K | 1.642 | 0.660 | −0.391 | 1.092 | 0.992 | 0.496 | −0.416 |
| Li-Na | 0.728 | 1.069 | 1.245 | 1.350 | 0.577 | −0.317 | 0.067 |
| Au-Cu | 2.423 | 0.515 | −1.229 | 1.692 | 0.737 | 0.885 | −0.664 |
| Al-Li | 0.813 | 1.374 | −0.580 | 1.062 | 1.051 | −0.208 | 0.318 |
| Nb-Zr | 0.671 | 1.414 | 0.279 | 0.864 | 1.098 | −0.399 | 0.346 |
| Ni-Pd | 1.174 | 1.105 | 11.250 | −1.445 | 1.599 | 0.811 | 0.159 |
| Cu-Mg | 0.882 | 1.538 | −1.689 | 1.731 | 0.783 | −0.126 | 0.430 |
| Al-Ca | 2.423 | 0.515 | −1.229 | 1.692 | 0.737 | 0.885 | −0.664 |
| Ni-Zr | 2.150 | 4.152 | −12.151 | 4.587 | 1.947 | 0.766 | 1.424 |
| Al-Sn | 0.649 | 1.393 | 0.566 | 1.065 | 0.849 | −0.433 | 0.332 |
| Al-Cu | 1.963 | 1.534 | −6.647 | 1.472 | 2.180 | 0.714 | 0.453 |
| Nb-Ni | 3.602 | 1.064 | −7.456 | 2.174 | 1.763 | 1.281 | 0.062 |
| Cu-Sn | 4.758 | 0.617 | −5.972 | 2.091 | 1.403 | 1.560 | −0.484 |
| Au-Si | 1.438 | 0.875 | −0.992 | 1.214 | 1.036 | 0.363 | −0.134 |
| Li-Sn | 2.077 | 2.672 | −8.741 | 2.608 | 2.129 | 0.731 | 0.983 |
| Fe-Si | 1.534 | 1.843 | −4.414 | 1.268 | 2.228 | 0.428 | 0.611 |
| Ag-In | 1.066 | 1.467 | −2.560 | 1.654 | 0.946 | 0.064 | 0.384 |
| Ge-Te | 0.636 | 3.025 | −3.272 | 1.419 | 1.356 | −0.452 | 1.107 |
| Al-Au | 1.903 | 0.844 | −2.370 | 0.799 | 2.011 | 0.643 | −0.169 |
| Cu-Sb | 1.743 | 1.017 | −3.207 | 1.743 | 1.017 | 0.556 | 0.017 |
Table 6.
Deviations and relative errors of five models in the symmetric method.
| System | Miedema | MIVM | RSM | Wilson | NRTL | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| SD | ARD/% | SD | ARD/% | SD | ARD/% | SD | ARD/% | SD | ARD/% | |
| Co-Ni | 0.013 | 3.852 | 0.002 | 0.4 | 0.004 | 1.1 | 0.007 | 2.1 | 0.011 | 3.3 |
| Al-Zn | 0.075 | 15.536 | 0.233 | 45.8 | 0.201 | 40 | 0.128 | 26 | 0.085 | 17.5 |
| Cu-Ni | 0.247 | 49.697 | 0.252 | 46.9 | 0.220 | 41.3 | 0.147 | 28.3 | 0.107 | 20.8 |
| Al-Ni | 0.210 | 1467.533 | 0.081 | 31.7 | 0.039 | 133 | 0.257 | 2130 | 0.335 | 3370 |
| Cu-Fe | 0.889 | 93.800 | 0.365 | 42.8 | 0.359 | 42 | 0.351 | 41.2 | 0.347 | 40.7 |
| Ge-Sn | 0.019 | 5.384 | 0.135 | 35.4 | 0.045 | 13.2 | 0.007 | 1.6 | 0.039 | 10.8 |
| Ag-Cu | 0.108 | 19.924 | 0.063 | 10.2 | 0.087 | 16.3 | 0.115 | 21.2 | 0.175 | 31.8 |
| Pb-Sb | 0.438 | 79.951 | 0.054 | 17.9 | 0.164 | 62.6 | 0.038 | 13.3 | 0.004 | 0.9 |
| Al-Si | 0.023 | 10.017 | 0.147 | 50.8 | 0.089 | 34.2 | 0.027 | 12.1 | 0.142 | 72.6 |
| Al-Co | 0.034 | 10.163 | 0.074 | 39.6 | 0.070 | 130 | 0.161 | 411 | 0.258 | 852 |
| Li-Mg | 0.043 | 17.882 | 0.020 | 7.9 | 0.026 | 10.2 | 0.052 | 21.7 | 0.067 | 28.5 |
| Sb-Sn | 0.025 | 6.367 | 0.100 | 36.5 | 0.029 | 11.3 | 0.046 | 20.7 | 0.103 | 49.6 |
| Cu-Zr | 0.124 | 53.790 | 0.065 | 33 | 0.026 | 14.4 | 0.084 | 62.1 | 0.193 | 171 |
| K-Na | 0.019 | 3.619 | 0.130 | 24.3 | 0.076 | 15.2 | 0.154 | 29.6 | 0.156 | 29.9 |
| Pb-Sn | 0.015 | 1.847 | 0.244 | 51.4 | 0.194 | 42 | 0.105 | 23.7 | 0.049 | 11 |
| Al-Mg | 0.042 | 14.076 | 0.095 | 36.2 | 0.101 | 38.4 | 0.049 | 17.5 | 0.031 | 10.7 |
| Cs-K | 0.020 | 4.475 | 0.132 | 32.3 | 0.074 | 18.8 | 0.046 | 11.5 | 0.036 | 8.9 |
| Li-Na | 0.255 | 24.451 | 0.244 | 26.7 | 0.211 | 23.4 | 0.363 | 40.4 | 0.404 | 45.1 |
| Au-Cu | 0.028 | 7.697 | 0.117 | 40.7 | 0.067 | 58.8 | 0.135 | 137 | 0.168 | 181 |
| Al-Li | 0.034 | 17.993 | 0.093 | 86.2 | 0.110 | 106 | 0.151 | 156 | 0.171 | 182 |
| Nb-Zr | 0.036 | 8.347 | 0.092 | 21.6 | 0.032 | 6.3 | 0.052 | 11.5 | 0.065 | 14.7 |
| Ni-Pd | 0.115 | 72.225 | 0.046 | 15.2 | 0.036 | 11.3 | 0.088 | 52.4 | 0.143 | 93.2 |
| Cu-Mg | 0.022 | 8.778 | 0.065 | 22.3 | 0.051 | 40 | 0.136 | 144 | 0.202 | 245 |
| Al-Ca | 0.100 | 56.098 | 0.089 | 50.3 | 0.075 | 20 | 0.107 | 175 | 0.301 | 901 |
| Ni-Zr | 0.080 | 20.755 | 0.248 | 75.6 | 0.098 | 35.8 | 0.140 | 86.2 | 0.573 | 6633.0 |
| Al-Sn | 0.122 | 18.737 | 0.216 | 32.8 | 0.147 | 21.9 | 0.200 | 30.6 | 0.224 | 34.3 |
| Al-Cu | 0.099 | 228.621 | 0.113 | 57.6 | 0.078 | 39.9 | 0.114 | 289 | 0.417 | 2930 |
| Nb-Ni | 0.159 | 517.211 | 0.216 | 67.7 | 0.104 | 49.9 | 0.121 | 302 | 0.363 | 2270 |
| Cu-Sn | 0.073 | 38.233 | 0.255 | 75.4 | 0.163 | 56.7 | 0.076 | 28 | 0.200 | 167 |
| Au-Si | 0.106 | 43.309 | 0.070 | 79 | 0.136 | 240 | 0.192 | 425 | 0.234 | 591 |
| Li-Sn | 0.081 | 159.055 | 0.150 | 86.9 | 0.116 | 187 | 0.244 | 1730 | 0.564 | 7420 |
| Fe-Si | 0.095 | 67.563 | 0.138 | 45.8 | 0.105 | 79.9 | 0.165 | 693 | 0.358 | 2900 |
| Ag-In | 0.114 | 97.817 | 0.122 | 37.9 | 0.097 | 30.1 | 0.109 | 90.8 | 0.193 | 204 |
| Ge-Te | 0.240 | 319.161 | 0.245 | 73.9 | 0.114 | 29.1 | 0.135 | 118 | 0.211 | 268 |
| Al-Au | 0.159 | 53.722 | 0.149 | 55.9 | 0.139 | 42.7 | 0.168 | 405 | 0.333 | 1820 |
| Cu-Sb | 0.095 | 80.468 | 0.072 | 29.9 | 0.097 | 30 | 0.122 | 105 | 0.196 | 228 |
| Ave | 0.121 | 102.7 | 0.137 | 42.35 | 0.105 | 49.24 | 0.128 | 219.2 | 0.207 | 884.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hang, J.; Tao, D. Estimation of Two Component Activities of Binary Liquid Alloys by the Pair Potential Energy Containing a Polynomial of the Partial Radial Distribution Function. Metals 2023, 13, 1773. https://doi.org/10.3390/met13101773
AMA Style
Hang J, Tao D. Estimation of Two Component Activities of Binary Liquid Alloys by the Pair Potential Energy Containing a Polynomial of the Partial Radial Distribution Function. Metals. 2023; 13(10):1773. https://doi.org/10.3390/met13101773
Chicago/Turabian StyleHang, Jiulong, and Dongping Tao. 2023. "Estimation of Two Component Activities of Binary Liquid Alloys by the Pair Potential Energy Containing a Polynomial of the Partial Radial Distribution Function" Metals 13, no. 10: 1773. https://doi.org/10.3390/met13101773
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.