Recent Advances in Chromatin Mechanisms Controlling Pancreatic Carcinogenesis


Abstract
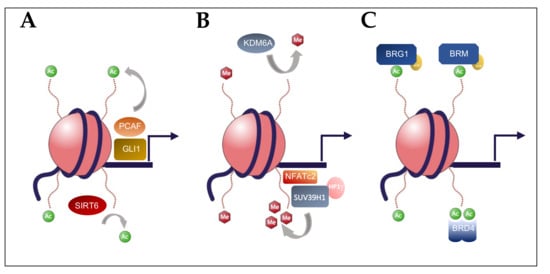
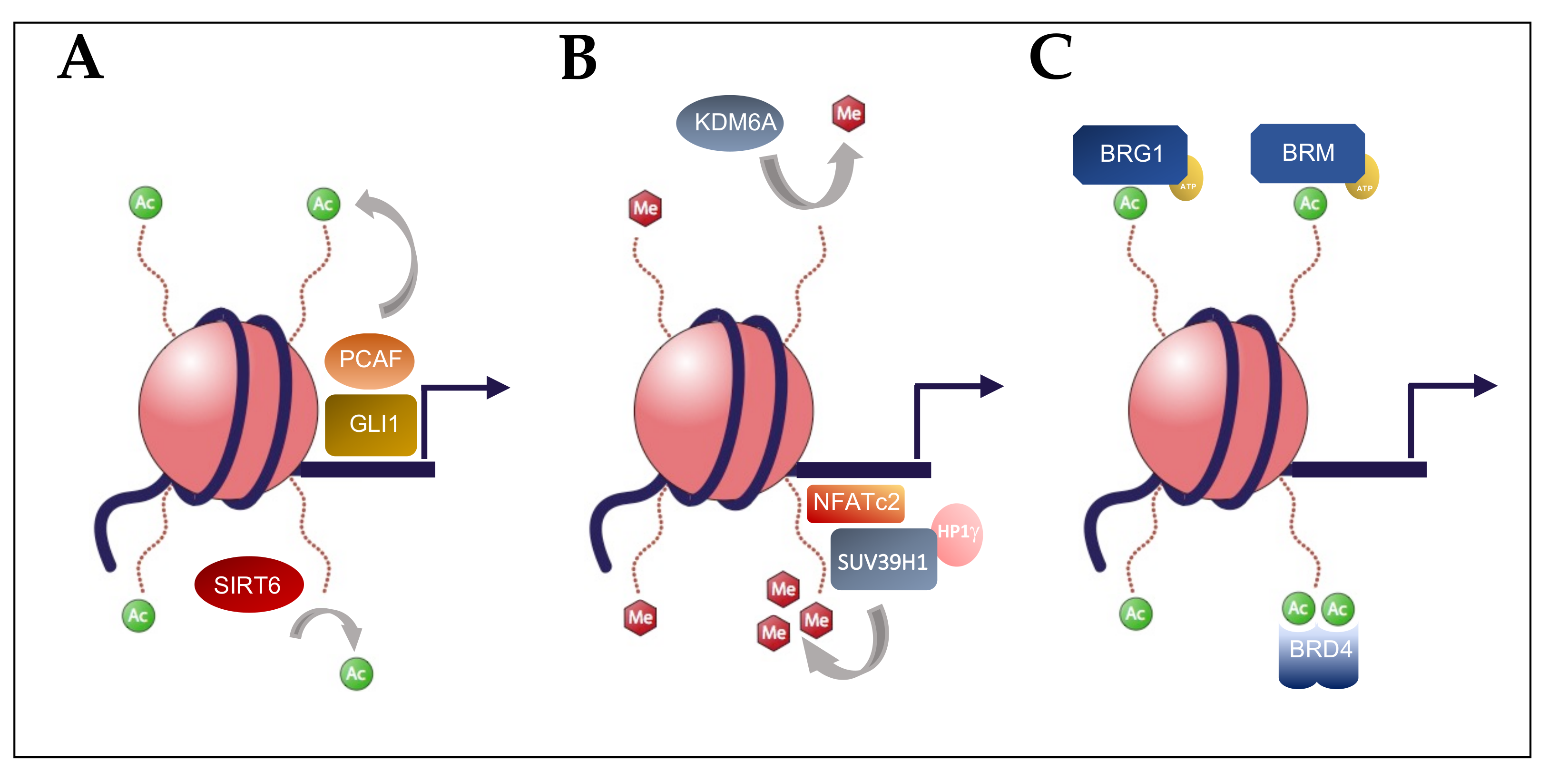
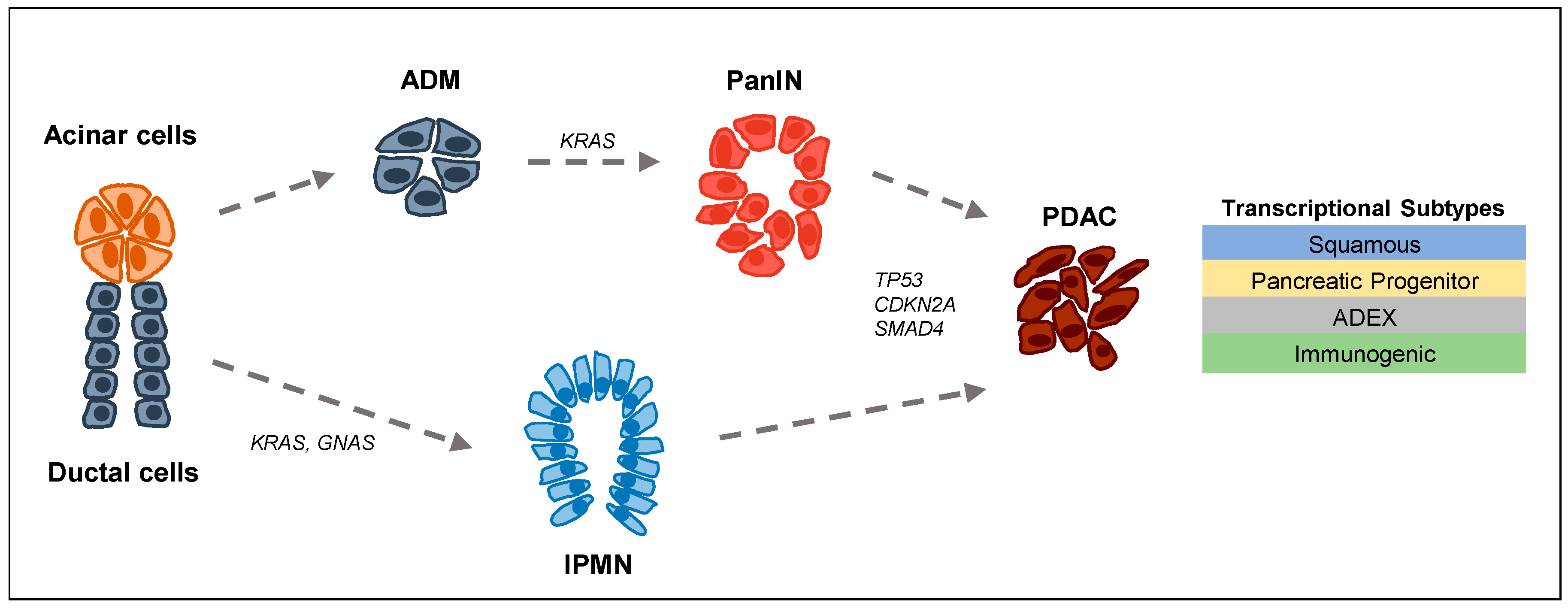
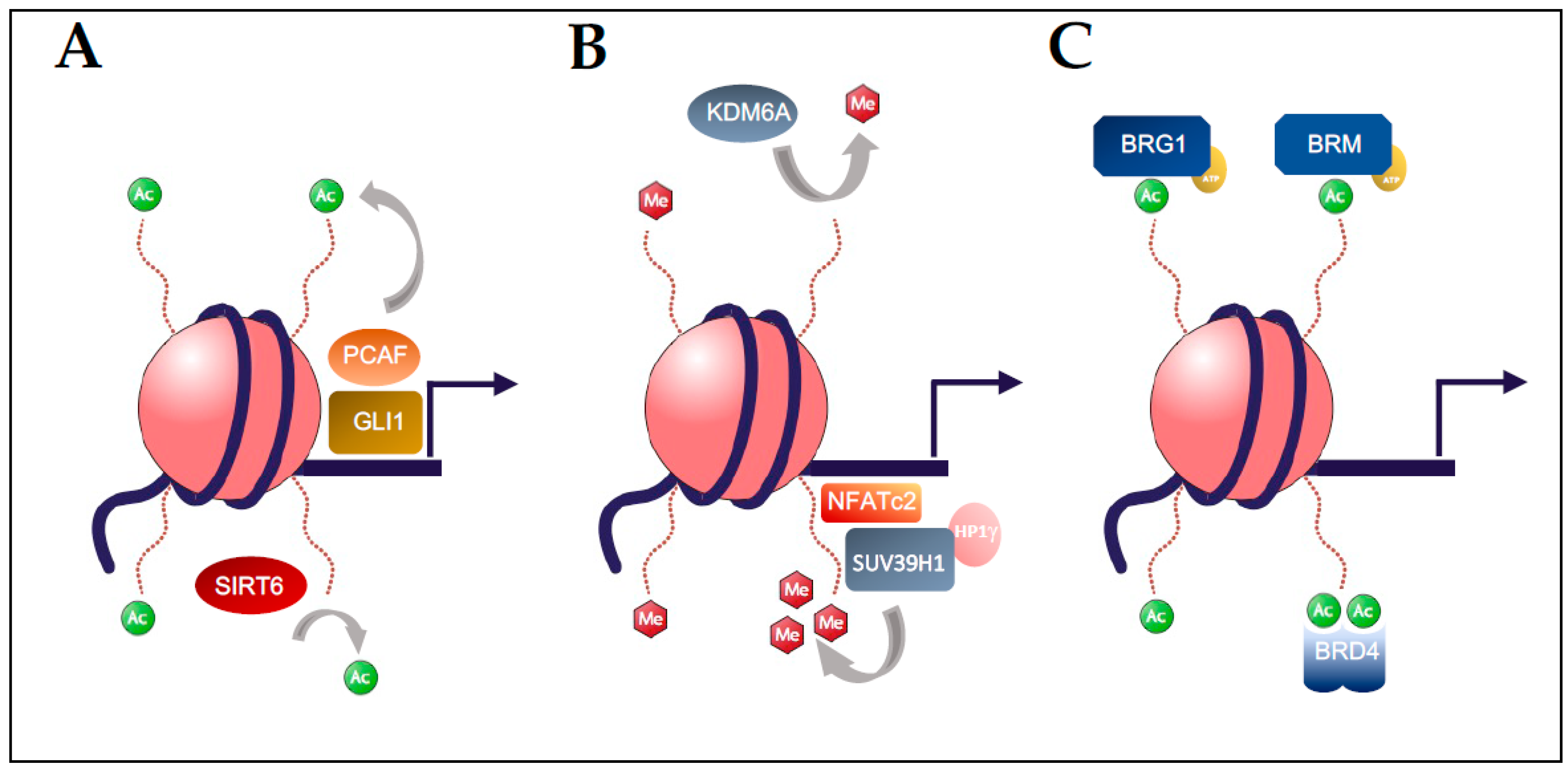
:
{kind=link}
{kind=link}
{kind=link}
1. Tumor Biology and Epigenetics in PDAC
2. Histone Acetylation in Pancreatic Carcinogenesis
3. Histone Methylation in Pancreatic Carcinogenesis
4. BET Bromodomain Regulation in Pancreatic Carcinogenesis
5. Chromatin Remodelers (SWI/SNF) in Pancreatic Carcinogenesis
6. Outlook
Funding
Conflicts of Interest
References
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-del Castillo, C.; Adsay, N.V. Intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology 2010, 139, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.F.; Dubois, C.L.; Morris, J.P.T.; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of SOX9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Bardeesy, N.; Mizukami, Y. Diversity of precursor lesions for pancreatic cancer: The genetics and biology of intraductal papillary mucinous neoplasm. Clin. Transl. Gastroenterol. 2017, 8, e86. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.K.; Grimmond, S.M.; Biankin, A.V. Pancreatic cancer genomics. Curr. Opin. Genet. Dev. 2014, 24, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004, 32, 959–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, N.; Goodman, R.H. Creb-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 2001, 276, 13505–13508. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojanovic, N.; Hassan, Z.; Wirth, M.; Wenzel, P.; Beyer, M.; Schafer, C.; Brand, P.; Kroemer, A.; Stauber, R.H.; Schmid, R.M.; et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 2017, 36, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of bet recruitment to chromatin as an effective treatment for mll-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Imbalzano, A.N.; Khavari, P.A.; Kingston, R.E.; Green, M.R. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 1994, 370, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Linhart, T.; Schlengemann, K.; Reutlinger, K.; Wegele, J.; Adler, G.; Singh, G.; Hofmann, L.; Kunsch, S.; Buch, T.; et al. NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells. Gastroenterology 2010, 138, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Nye, M.D.; Almada, L.L.; Fernandez-Barrena, M.G.; Marks, D.L.; Elsawa, S.F.; Vrabel, A.; Tolosa, E.J.; Ellenrieder, V.; Fernandez-Zapico, M.E. The transcription factor GLI1 interacts with SMAD proteins to modulate transforming growth factor β-induced gene expression in a p300/CREB-binding protein-associated factor (PCAF)-dependent manner. J. Biol. Chem. 2014, 289, 15495–15506. [Google Scholar] [CrossRef] [PubMed]
- Malatesta, M.; Steinhauer, C.; Mohammad, F.; Pandey, D.P.; Squatrito, M.; Helin, K. Histone acetyltransferase PCAF is required for hedgehog-GLI-dependent transcription and cancer cell proliferation. Cancer Res. 2013, 73, 6323–6333. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Kramer, O.H.; Schmid, R.M.; Saur, D. Acetylation as a transcriptional control mechanism-HDACS and HATS in pancreatic ductal adenocarcinoma. J. Gastrointest. Cancer 2011, 42, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed e box binding zinc finger protein SIP1 downregulates e-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef]
- Von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009, 137, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Schafer, C.; Goder, A.; Beyer, M.; Kiweler, N.; Mahendrarajah, N.; Rauch, A.; Nikolova, T.; Stojanovic, N.; Wieczorek, M.; Reich, T.R.; et al. Class I histone deacetylases regulate p53/NF-ĸb crosstalk in cancer cells. Cell. Signal. 2017, 29, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, C.; Zwaans, B.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Kugel, S.; Sebastian, C.; Fitamant, J.; Ross, K.N.; Saha, S.K.; Jain, E.; Gladden, A.; Arora, K.S.; Kato, Y.; Rivera, M.N.; et al. SIRT6 suppresses pancreatic cancer through control of lin28b. Cell 2016, 165, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Moss, E.G.; Tang, L. Conservation of the heterochronic regulator lin-28, its developmental expression and microrna complementary sites. Dev. Biol. 2003, 258, 432–442. [Google Scholar] [CrossRef]
- King, C.E.; Cuatrecasas, M.; Castells, A.; Sepulveda, A.R.; Lee, J.S.; Rustgi, A.K. Lin28b promotes colon cancer progression and metastasis. Cancer Res. 2011, 71, 4260–4268. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Katsaros, D.; Shaverdashvili, K.; Qian, B.; Wu, Y.; de la Longrais, I.A.; Preti, M.; Menato, G.; Yu, H. Pluripotent factor lin-28 and its homologue lin-28b in epithelial ovarian cancer and their associations with disease outcomes and expression of let-7a and IGF-II. Eur. J. Cancer 2009, 45, 2212–2218. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.R.; Powers, J.T.; Einhorn, W.; Hoshida, Y.; Ng, T.L.; Toffanin, S.; O'Sullivan, M.; Lu, J.; Phillips, L.A.; Lockhart, V.L.; et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat. Genet. 2009, 41, 843–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, I.; Joo, C.; Cho, J.; Ha, M.; Han, J.; Kim, V.N. Lin28 mediates the terminal uridylation of let-7 precursor microrna. Mol. Cell 2008, 32, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Park, S.M.; Shomron, N.; Hedegaard, M.M.; Vinther, J.; Andersen, J.S.; Feig, C.; Xu, J.; Burge, C.B.; Peter, M.E. Identification of let-7-regulated oncofetal genes. Cancer Res. 2008, 68, 2587–2591. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and HMGA2 enhances oncogenic transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Cao, R.; Viatour, P.; Sage, J.; Zhang, Y.; Xiong, Y. PRB family proteins are required for H3K27 trimethylation and polycomb repression complexes binding to and silencing P16INK4α tumor suppressor gene. Genes Dev. 2007, 21, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, S.; Glesel, E.; Singh, G.; Chen, N.M.; Reutlinger, K.; Zhang, J.; Billadeau, D.D.; Fernandez-Zapico, M.E.; Gress, T.M.; Singh, S.K.; et al. Restricted heterochromatin formation links NFATC2 repressor activity with growth promotion in pancreatic cancer. Gastroenterology 2012, 142, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Workman, J.L. The heterochromatin protein 1 (HP1) family: Put away a bias toward HP1. Mol. Cells 2008, 26, 217–227. [Google Scholar] [PubMed]
- Gilbert, N.; Boyle, S.; Sutherland, H.; de Las Heras, J.; Allan, J.; Jenuwein, T.; Bickmore, W.A. Formation of facultative heterochromatin in the absence of HP1. EMBO J. 2003, 22, 5540–5550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, A.L.; Oulad-Abdelghani, M.; Ortiz, J.A.; Remboutsika, E.; Chambon, P.; Losson, R. Heterochromatin formation in mammalian cells: Interaction between histones and HP1 proteins. Mol. Cell 2001, 7, 729–739. [Google Scholar] [CrossRef]
- Abbosh, P.H.; Montgomery, J.S.; Starkey, J.A.; Novotny, M.; Zuhowski, E.G.; Egorin, M.J.; Moseman, A.P.; Golas, A.; Brannon, K.M.; Balch, C.; et al. Dominant-negative histone H3 lysine 27 mutant derepresses silenced tumor suppressor genes and reverses the drug-resistant phenotype in cancer cells. Cancer Res. 2006, 66, 5582–5591. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Ochiai, A. Enhancer of zeste homolog 2 downregulates E-cadherin by mediating histone H3 methylation in gastric cancer cells. Cancer Sci. 2008, 99, 738–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.M.; Neesse, A.; Dyck, M.L.; Steuber, B.; Koenig, A.O.; Lubeseder-Martellato, C.; Winter, T.; Forster, T.; Bohnenberger, H.; Kitz, J.; et al. Context-dependent epigenetic regulation of nuclear factor of activated T cells 1 in pancreatic plasticity. Gastroenterology 2017, 152, 1507–1520. [Google Scholar] [CrossRef] [PubMed]
- Van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, L.; Emre, N.C.; Kruhlak, M.J.; Chung, H.J.; Steidl, C.; Slack, G.; Wright, G.W.; Lenz, G.; Ngo, V.N.; Shaffer, A.L.; et al. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell 2010, 18, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhu, W.; Xu, W.; Zhang, B.; Shi, S.; Ji, S.; Liu, J.; Long, J.; Liu, C.; Liu, L.; et al. LSD1 sustains pancreatic cancer growth via maintaining HIF1α-dependent glycolytic process. Cancer Lett. 2014, 347, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Tzatsos, A.; Paskaleva, P.; Ferrari, F.; Deshpande, V.; Stoykova, S.; Contino, G.; Wong, K.K.; Lan, F.; Trojer, P.; Park, P.J.; et al. KDM2B promotes pancreatic cancer via polycomb-dependent and -independent transcriptional programs. J. Clin. Investig. 2013, 123, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6α activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to bet inhibitors. Cancer Cell 2018, 33, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Tang, Z.; Chen, C.W.; Shimada, M.; Koche, R.P.; Wang, L.H.; Nakadai, T.; Chramiec, A.; Krivtsov, A.V.; Armstrong, S.A.; et al. A UTX-MLL4-p300 transcriptional regulatory network coordinately shapes active enhancer landscapes for eliciting transcription. Mol. Cell 2017, 67, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Gozdecka, M.; Meduri, E.; Mazan, M.; Tzelepis, K.; Dudek, M.; Knights, A.J.; Pardo, M.; Yu, L.; Choudhary, J.S.; Metzakopian, E.; et al. UTX-mediated enhancer and chromatin remodeling suppresses myeloid leukemogenesis through noncatalytic inverse regulation of ETS and GATA programs. Nat. Genet. 2018, 50, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nahar, S.; Nakagawa, A.; Fernandez-Barrena, M.G.; Mertz, J.A.; Bryant, B.M.; Adams, C.E.; Mino-Kenudson, M.; Von Alt, K.N.; Chang, K.; et al. Regulation of GLI underlies a role for bet bromodomains in pancreatic cancer growth and the tumor microenvironment. Clin. Cancer Res. 2016, 22, 4259–4270. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of bet family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Kumar, K.; Knab, L.M.; Chow, C.R.; Raza, S.S.; Bentrem, D.J.; Ebine, K.; Munshi, H.G. Bet bromodomain inhibitors block growth of pancreatic cancer cells in three-dimensional collagen. Mol. Cancer Ther. 2014, 13, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.L.; Miller, A.L.; Kreitzburg, K.M.; Council, L.N.; Gamblin, T.L.; Christein, J.D.; Heslin, M.J.; Arnoletti, J.P.; Richardson, J.H.; Chen, D.; et al. The bet bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene 2016, 35, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tateishi, K.; Kudo, Y.; Hoshikawa, M.; Tanaka, M.; Nakatsuka, T.; Fujiwara, H.; Miyabayashi, K.; Takahashi, R.; Tanaka, Y.; et al. Stromal remodeling by the bet bromodomain inhibitor jq1 suppresses the progression of human pancreatic cancer. Oncotarget 2016, 7, 61469–61484. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Bian, B.; Bigonnet, M.; Gayet, O.; Loncle, C.; Maignan, A.; Gilabert, M.; Moutardier, V.; Garcia, S.; Turrini, O.; Delpero, J.R.; et al. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the bet bromodomain inhibitor JQ1: Implications for individualized medicine efforts. EMBO Mol. Med. 2017, 9, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Raza, S.S.; Knab, L.M.; Chow, C.R.; Kwok, B.; Bentrem, D.J.; Popovic, R.; Ebine, K.; Licht, J.D.; Munshi, H.G. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the bet bromodomain inhibitor JQ1. Sci. Rep. 2015, 5, 9489. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Cote, J.; Xue, Y.; Zhou, S.; Khavari, P.A.; Biggar, S.R.; Muchardt, C.; Kalpana, G.V.; Goff, S.P.; Yaniv, M.; et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996, 15, 5370–5382. [Google Scholar] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numata, M.; Morinaga, S.; Watanabe, T.; Tamagawa, H.; Yamamoto, N.; Shiozawa, M.; Nakamura, Y.; Kameda, Y.; Okawa, S.; Rino, Y.; et al. The clinical significance of SWI/SNF complex in pancreatic cancer. Int. J. Oncol. 2013, 42, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Segedi, M.; Anderson, L.N.; Espin-Garcia, O.; Borgida, A.; Bianco, T.; Cheng, D.; Chen, Z.; Patel, D.; Brown, M.C.; Xu, W.; et al. BRM polymorphisms, pancreatic cancer risk and survival. Int. J. Cancer 2016, 139, 2474–2481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, F.; Du, C.; Guo, H.; Ma, L.; Liu, X.; Kornmann, M.; Tian, X.; Yang, Y. BRM/SMARCA2 promotes the proliferation and chemoresistance of pancreatic cancer cells by targeting JAK2/STAT3 signaling. Cancer Lett. 2017, 402, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Molin, M.D.; Matthaei, H.; Wu, J.; Blackford, A.; Debeljak, M.; Rezaee, N.; Wolfgang, C.L.; Butturini, G.; Salvia, R.; Bassi, C.; et al. Clinicopathological correlates of activating gnas mutations in intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann. Surg. Oncol. 2013, 20, 3802–3808. [Google Scholar] [CrossRef] [PubMed]
- Von Figura, G.; Fukuda, A.; Roy, N.; Liku, M.E.; Morris Iv, J.P.; Kim, G.E.; Russ, H.A.; Firpo, M.A.; Mulvihill, S.J.; Dawson, D.W.; et al. The chromatin regulator Brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2014, 16, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, N.; Malik, S.; Villanueva, K.E.; Urano, A.; Lu, X.; Von Figura, G.; Seeley, E.S.; Dawson, D.W.; Collisson, E.A.; Hebrok, M. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015, 29, 658–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, R.L.; Brennan, J.; Schisler, J.C.; Serber, D.; Patterson, C.; Magnuson, T. Arid1a-DNA interactions are required for promoter occupancy by SWI/SNF. Mol. Cell Biol. 2013, 33, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Alver, B.H.; San Roman, A.K.; Wilson, B.G.; Wang, X.; Agoston, A.T.; Park, P.J.; Shivdasani, R.A.; Roberts, C.W. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 2017, 49, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Rahmanto, Y.S.; Wu, R.C.; Wang, Y.; Wang, Z.; Wang, T.L.; Shih, I.M. Roles of deletion of arid1a, a tumor suppressor, in mouse ovarian tumorigenesis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.L.; Damrauer, J.S.; Raab, J.R.; Schisler, J.C.; Wilkerson, M.D.; Didion, J.P.; Starmer, J.; Serber, D.; Yee, D.; Xiong, J.; et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat. Commun. 2015, 6, 6118. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Fukuda, A.; Ogawa, S.; Maruno, T.; Takada, Y.; Tsuda, M.; Hiramatsu, Y.; Araki, O.; Nagao, M.; Yoshikawa, T.; et al. ARID1A maintains differentiation of pancreatic ductal cells and inhibits development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 2018, in press. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hank, T.; Liss, A.S. Recent Advances in Chromatin Mechanisms Controlling Pancreatic Carcinogenesis. Epigenomes 2018, 2, 11. https://doi.org/10.3390/epigenomes2020011
Hank T, Liss AS. Recent Advances in Chromatin Mechanisms Controlling Pancreatic Carcinogenesis. Epigenomes. 2018; 2(2):11. https://doi.org/10.3390/epigenomes2020011
Chicago/Turabian StyleHank, Thomas, and Andrew S. Liss. 2018. "Recent Advances in Chromatin Mechanisms Controlling Pancreatic Carcinogenesis" Epigenomes 2, no. 2: 11. https://doi.org/10.3390/epigenomes2020011