The Epigenetic Landscape of Pancreatic Cancer Stem Cells



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
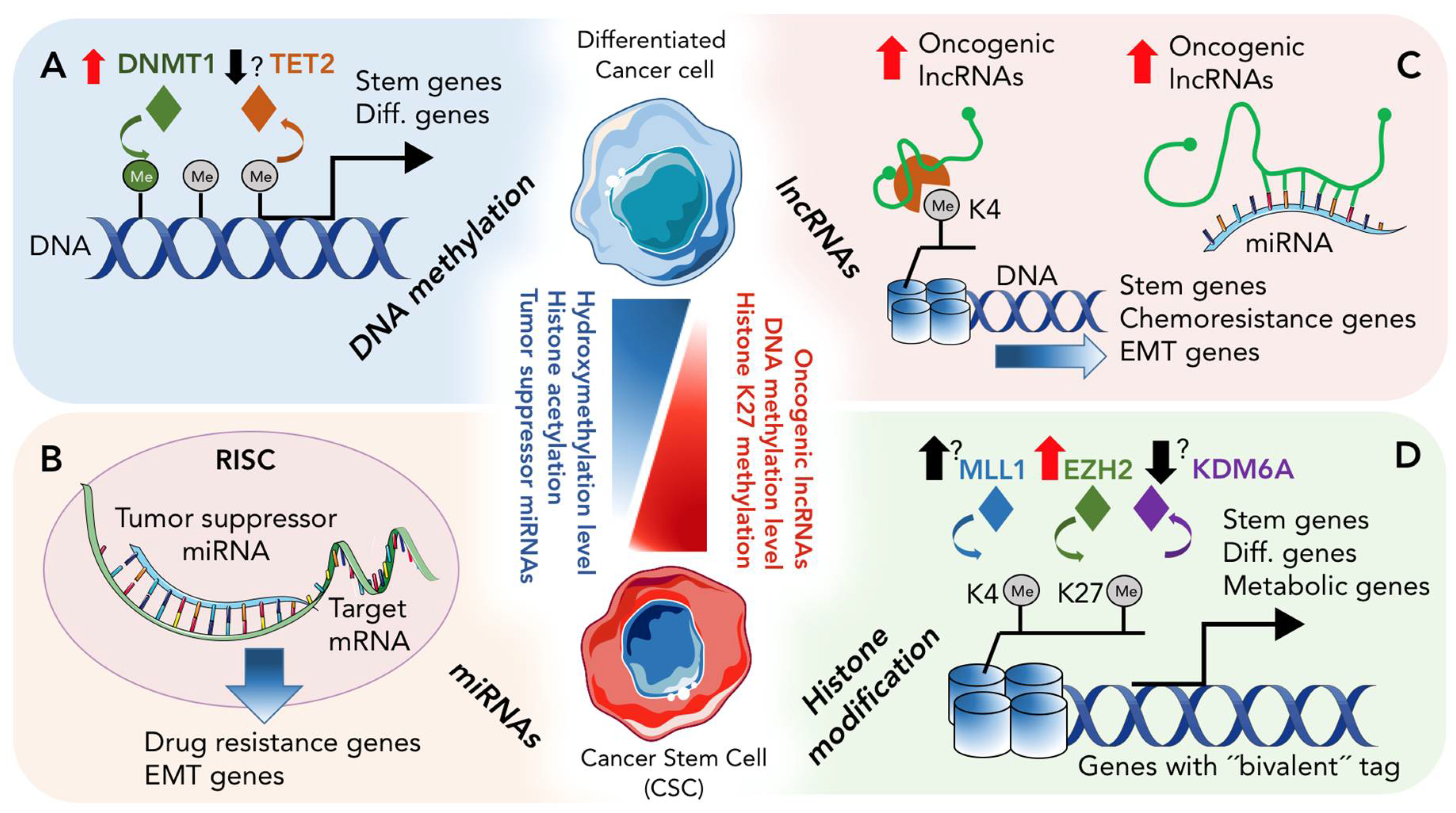
2. Epigenetic Landscape of CSCs
2.1. DNA Methylation and De-Methylation in CSCs
2.2. Histone Modification in PDAC and CSCs
3. Role of Non-Coding RNAs in CSCs
3.1. Role of miRNAs in CSCs
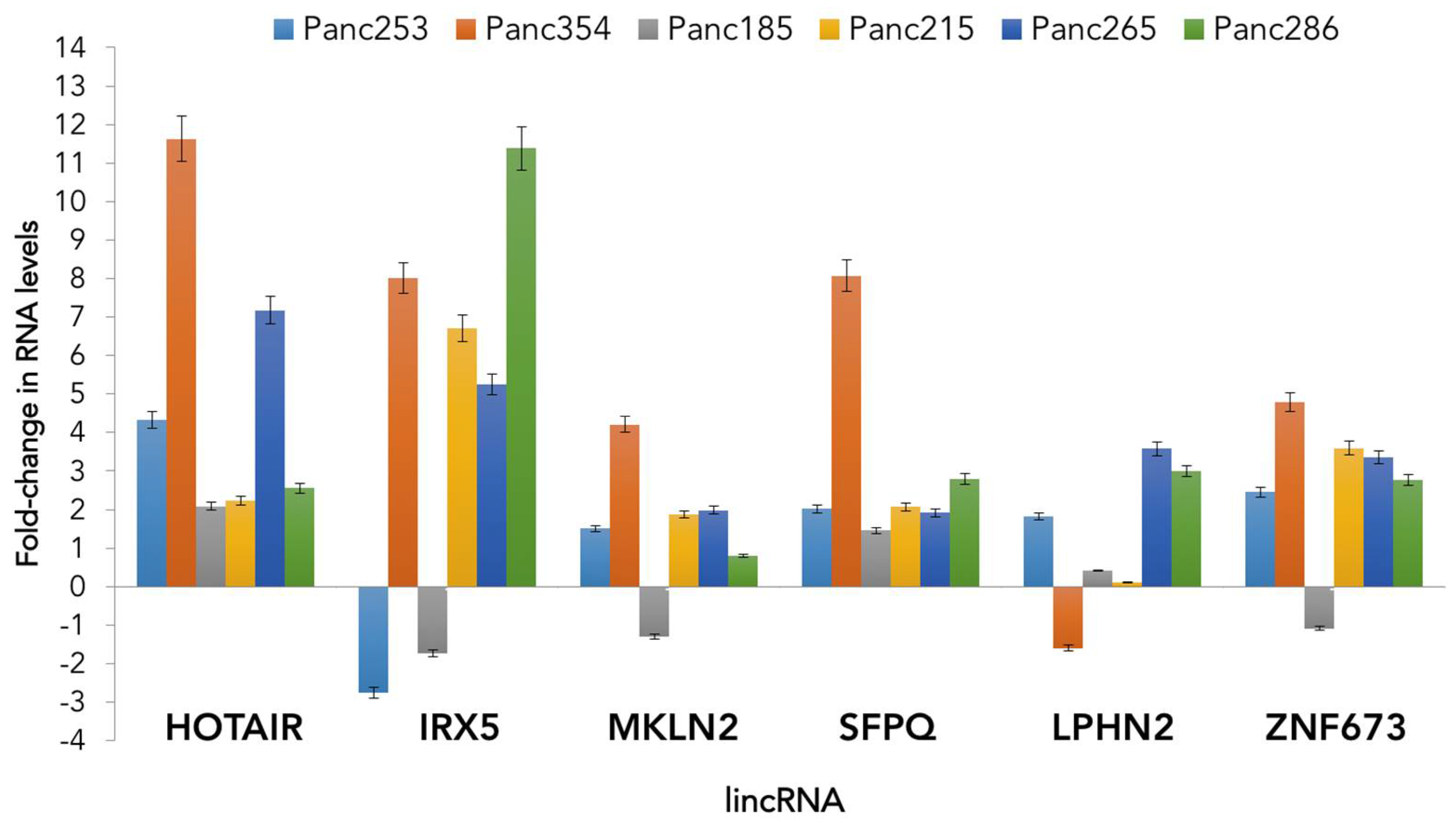
3.2. Role of lncRNA in CSCs
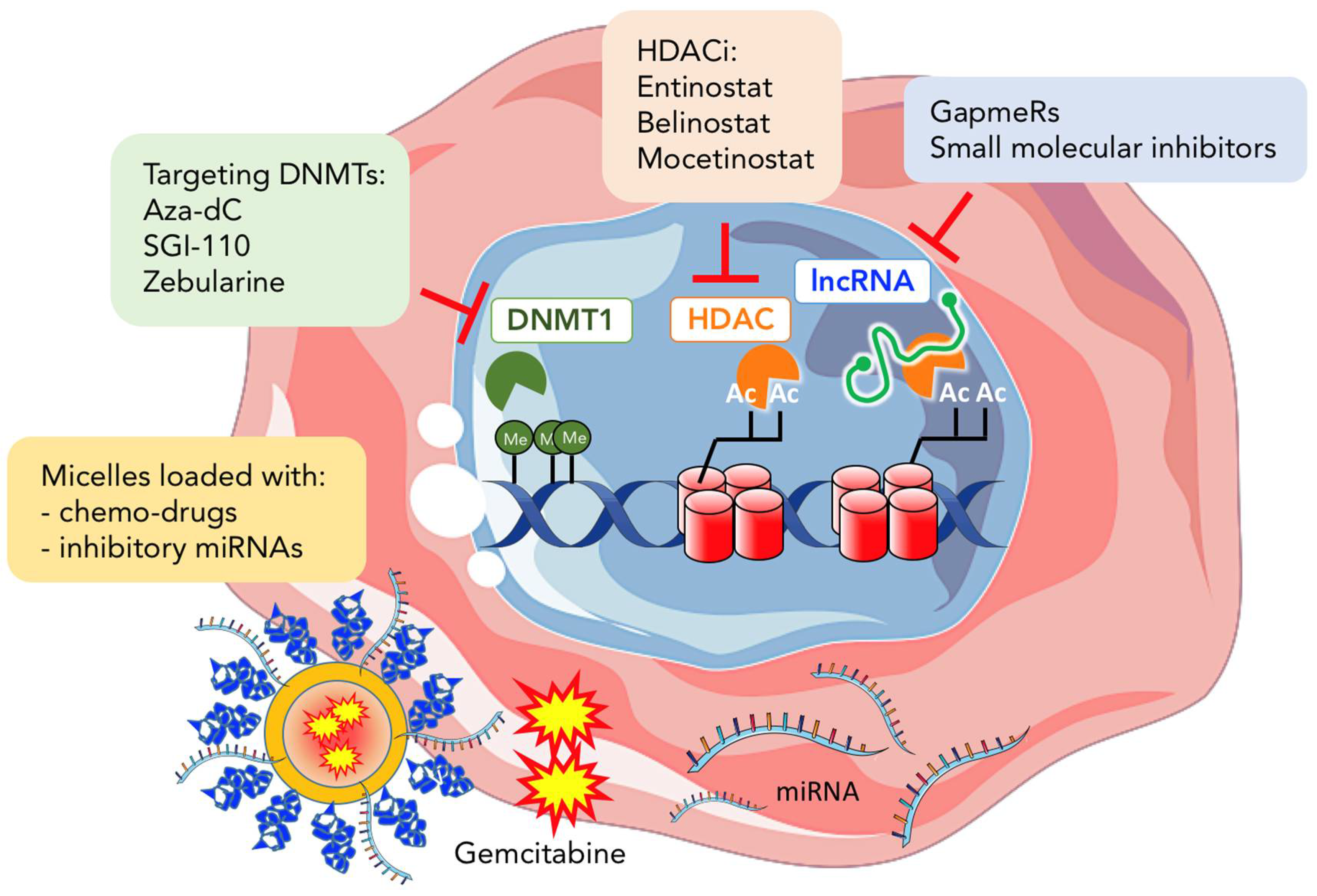
4. Epigenetic Therapy in Cancer
5. Concluding Remarks
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Pogribny, I.P. Epigenetic events in tumorigenesis: Putting the pieces together. Exp. Oncol. 2010, 32, 132–136. [Google Scholar] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E. Looking ahead in cancer stem cell research. Nat. Biotechnol. 2009, 27, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Khiabanian, H.; Rossi, D.; Fabbri, G.; Gattei, V.; Forconi, F.; Laurenti, L.; Marasca, R.; Del Poeta, G.; Foa, R.; et al. Tumor evolutionary directed graphs and the history of chronic lymphocytic leukemia. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michor, F.; Polyak, K. The origins and implications of intratumor heterogeneity. Cancer Prev. Res. 2010, 3, 1361–1364. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.C.; Hollingsworth, R.E.; Hurt, E.M. Cancer stem cell plasticity and tumor hierarchy. World J. Stem. Cells 2015, 7, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furth, J. Transmission of myeloid leukemia of mice: Its relation to myeloma. J. Exp. Med. 1935, 61, 423–446. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into scid mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Hadnagy, A.; Gaboury, L.; Beaulieu, R.; Balicki, D. Sp analysis may be used to identify cancer stem cell populations. Exp. Cell Res. 2006, 312, 3701–3710. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Vargas, H.; Ouzounova, M.; Le Calvez-Kelm, F.; Lambert, M.P.; McKay-Chopin, S.; Tavtigian, S.V.; Puisieux, A.; Matar, C.; Herceg, Z. Methylome analysis reveals jak-stat pathway deregulation in putative breast cancer stem cells. Epigenetics 2011, 6, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Mani, S.; Cros, M.P.; Scoazec, J.Y.; Chemin, I.; Hainaut, P.; Herceg, Z. Epigenetic silencing of SFRP1 activates the canonical WNT pathway and contributes to increased cell growth and proliferation in hepatocellular carcinoma. Tumour Biol. 2012, 33, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Balic, M.; Schwarzenbacher, D.; Stanzer, S.; Heitzer, E.; Auer, M.; Geigl, J.B.; Cote, R.J.; Datar, R.H.; Dandachi, N. Genetic and epigenetic analysis of putative breast cancer stem cell models. BMC Cancer 2013, 13, 358. [Google Scholar] [CrossRef] [PubMed]
- De Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for LGR5(+) stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of cpg and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [PubMed]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNA(AsP) by the DNA methyltransferase homolog DNMT2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Aapola, U.; Kawasaki, K.; Scott, H.S.; Ollila, J.; Vihinen, M.; Heino, M.; Shintani, A.; Kawasaki, K.; Minoshima, S.; Krohn, K.; et al. Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics 2000, 65, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Samaranayake, M.; Pradhan, S. Epigenetic mechanisms in mammals. Cell. Mol. Life Sci. 2009, 66, 596–612. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases DNMT3A and DNMT3B are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Molaro, A.; Dos Santos, C.O.; Thekkat, P.; Song, Q.; Uren, P.J.; Park, J.; Butler, J.; Rafii, S.; McCombie, W.R.; et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol. Cell 2011, 44, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Gopisetty, G.; Xu, J.; Sampath, D.; Colman, H.; Puduvalli, V.K. Epigenetic regulation of CD133/PROM1 expression in glioma stem cells by SP1/MYC and promoter methylation. Oncogene 2013, 32, 3119–3129. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kagara, N.; Huynh, K.T.; Kuo, C.; Okano, H.; Sim, M.S.; Elashoff, D.; Chong, K.; Giuliano, A.E.; Hoon, D.S. Epigenetic regulation of cancer stem cell genes in triple-negative breast cancer. Am. J. Pathol. 2012, 181, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.M.; Tsai, H.C.; Glockner, S.C.; Lin, S.; Ohm, J.E.; Easwaran, H.; James, C.D.; Costello, J.F.; Riggins, G.; Eberhart, C.G.; et al. Abnormal DNA methylation of CD133 in colorectal and glioblastoma tumors. Cancer Res. 2008, 68, 8094–8103. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Mathews, L.A.; Cabarcas, S.M.; Zhang, X.; Yang, A.; Zhang, Y.; Young, M.R.; Klarmann, K.D.; Keller, J.R.; Farrar, W.L. Epigenetic regulation of SOX9 by the nf-kappab signaling pathway in pancreatic cancer stem cells. Stem Cells 2013, 31, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, J.J.; Sinha, A.U.; Zhu, N.; Li, M.; Armstrong, S.A.; Orkin, S.H. Haploinsufficiency of DNMT1 impairs leukemia stem cell function through derepression of bivalent chromatin domains. Genes Dev. 2012, 26, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Pathania, R.; Ramachandran, S.; Elangovan, S.; Padia, R.; Yang, P.; Cinghu, S.; Veeranan-Karmegam, R.; Arjunan, P.; Gnana-Prakasam, J.P.; Sadanand, F.; et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat. Commun. 2015, 6, 6910. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Lin, J.H.; Hsu, T.W.; Su, K.; Li, A.F.; Hsu, H.S.; Hung, S.C. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int. J. Cancer 2015, 136, 547–559. [Google Scholar] [PubMed]
- Zagorac, S.; Alcala, S.; Fernandez Bayon, G.; Bou Kheir, T.; Schoenhals, M.; Gonzalez-Neira, A.; Fernandez Fraga, M.; Aicher, A.; Heeschen, C.; Sainz, B., Jr. Dnmt1 inhibition reprograms pancreatic cancer stem cells via upregulation of the miR-17-92 cluster. Cancer Res. 2016, 76, 4546–4558. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of tet proteins in 5MC to 5HMC conversion, es-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by mll partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.P.; Guo, F.; Yang, H.; Wu, H.P.; Xu, G.F.; Liu, W.; Xie, Z.G.; Shi, L.; He, X.; Jin, S.G.; et al. The role of TET3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011, 477, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, M.L.; Im, K.M.; Misner, K.J.; Tan, W.; Lou, H.; Gold, B.; Wells, D.W.; Bravo, H.C.; Fredrikson, K.M.; Harkins, T.T.; et al. Somatic alterations contributing to metastasis of a castration-resistant prostate cancer. Hum. Mutat. 2013, 34, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, L.; Mao, S.Q.; Li, Z.; Chen, J.; Zhang, R.R.; Wu, H.P.; Gao, J.; Guo, F.; Liu, W.; et al. Tet and tdg mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell 2014, 14, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Ito, K.; Ala, U.; Kats, L.; Webster, K.; Sun, S.M.; Jongen-Lavrencic, M.; Manova-Todorova, K.; Teruya-Feldstein, J.; Avigan, D.E.; et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 2013, 13, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Yu, Y.; Suzuki, M.; Campbell, N.; Mazdo, J.; Vasanthakumar, A.; Bhagat, T.D.; Nischal, S.; Christopeit, M.; Parekh, S.; et al. Genome-wide hydroxymethylation tested using the help-gt assay shows redistribution in cancer. Nucl. Acids Res. 2013, 41, e157. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, P.; Carrillo-de Santa Pau, E.; Cox, T.; Sainz, B., Jr.; Dusetti, N.; Greenhalf, W.; Rinaldi, L.; Costello, E.; Ghaneh, P.; Malats, N.; et al. Gata6 regulates emt and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut 2016, 66, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; McHale, C.M.; Skibola, C.F.; Smith, A.H.; Smith, M.T.; Zhang, L. An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Environ. Health Perspect. 2011, 119, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.T.; Helin, K. Histone demethylases in development and disease. Trends Cell Biol. 2010, 20, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Snapshot: Histone-modifying enzymes. Cell 2007, 128, 802. [Google Scholar] [CrossRef] [PubMed]
- Vakoc, C.R.; Sachdeva, M.M.; Wang, H.; Blobel, G.A. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol. Cell. Biol. 2006, 26, 9185–9195. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; Tee, W.W.; Reinberg, D. A double take on bivalent promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Olguin, P.; Recillas-Targa, F. Chromatin structure of pluripotent stem cells and induced pluripotent stem cells. Brief. Funct. Genom. 2011, 10, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, J.; Estecio, M.R.; Lu, Y.; Long, H.; Malouf, G.G.; Graber, D.; Huo, Y.; Ramagli, L.; Liang, S.; Kornblau, S.M.; et al. The epigenome of aml stem and progenitor cells. Epigenetics 2013, 8, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Lee, H.; Yoon, J.G.; Madan, A.; Wayner, E.; Tonning, S.; Hothi, P.; Schroeder, B.; Ulasov, I.; Foltz, G.; et al. Global analysis of H3K4me3 and H3K27me3 profiles in glioblastoma stem cells and identification of SLC17A7 as a bivalent tumor suppressor gene. Oncotarget 2015, 6, 5369–5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toll, A.D.; Dasgupta, A.; Potoczek, M.; Yeo, C.J.; Kleer, C.G.; Brody, J.R.; Witkiewicz, A.K. Implications of enhancer of zeste homologue 2 expression in pancreatic ductal adenocarcinoma. Hum. Pathol. 2010, 41, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xie, D.; Yin Li, W.; Man Cheung, C.; Yao, H.; Chan, C.Y.; Chan, C.Y.; Xu, F.P.; Liu, Y.H.; Sung, J.J.; et al. RNAi targeting EZH2 inhibits tumor growth and liver metastasis of pancreatic cancer in vivo. Cancer Lett. 2010, 297, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Van Vlerken, L.E.; Kiefer, C.M.; Morehouse, C.; Li, Y.; Groves, C.; Wilson, S.D.; Yao, Y.; Hollingsworth, R.E.; Hurt, E.M. Ezh2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl. Med. 2013, 2, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. Utx and JMJD3 are histone H3K27 demethylases involved in hox gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Bayliss, P.E.; Rinn, J.L.; Whetstine, J.R.; Wang, J.K.; Chen, S.; Iwase, S.; Alpatov, R.; Issaeva, I.; Canaani, E.; et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 2007, 449, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Welstead, G.G.; Creyghton, M.P.; Bilodeau, S.; Cheng, A.W.; Markoulaki, S.; Young, R.A.; Jaenisch, R. X-linked H3K27me3 demethylase Utx is required for embryonic development in a sex-specific manner. Proc. Natl. Acad. Sci. USA 2012, 109, 13004–13009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.W.; Hong, T.; Hong, S.; Guo, H.; Yu, H.; Kim, D.; Guszczynski, T.; Dressler, G.R.; Copeland, T.D.; Kalkum, M.; et al. Ptip associates with mll3- and mll4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 2007, 282, 20395–20406. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Gao, X.; Morgan, M.A.; Herz, H.M.; Smith, E.R.; Shilatifard, A. The MLL3/MLL4 branches of the compass family function as major histone H3K4 monomethylases at enhancers. Mol. Cell Biol. 2013, 33, 4745–4754. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by polycomb and compass families. Science 2016, 352. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to BET inhibitors. Cancer Cell 2018, 33, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Shpargel, K.B.; Sengoku, T.; Yokoyama, S.; Magnuson, T. Utx and uty demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012, 8, e1002964. [Google Scholar] [CrossRef] [PubMed]
- Shpargel, K.B.; Starmer, J.; Yee, D.; Pohlers, M.; Magnuson, T. Kdm6 demethylase independent loss of histone H3 lysine 27 trimethylation during early embryonic development. PLoS Genet. 2014, 10, e1004507. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Sphyris, N.; Johnson, K.S.; Reisenauer, K.N.; Nesbit, T.A.; Joseph, R.; Vijay, G.V.; Sarkar, T.R.; Bhangre, N.A.; Song, J.J.; et al. The H3K27me3-demethylase KDM6A is suppressed in breast cancer stem-like cells, and enables the resolution of bivalency during the mesenchymal-epithelial transition. Oncotarget 2017, 8, 65548–65565. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 2017, 170, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Rack, B.; Schindlbeck, C.; Juckstock, J.; Andergassen, U.; Hepp, P.; Zwingers, T.; Friedl, T.W.; Lorenz, R.; Tesch, H.; Fasching, P.A.; et al. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. Emt and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. Gencode: The reference human genome annotation for the encode project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Kapranov, P.; Drenkow, J.; Dike, S.; Brubaker, S.; Patel, S.; Long, J.; Stern, D.; Tammana, H.; Helt, G.; et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 2005, 308, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.S.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.; Weissman, S.; et al. Global identification of human transcribed sequences with genome tiling arrays. Science 2004, 306, 2242–2246. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Kloc, M.; Wilk, K.; Vargas, D.; Shirato, Y.; Bilinski, S.; Etkin, L.D. Potential structural role of non-coding and coding RNAs in the organization of the cytoskeleton at the vegetal cortex of xenopus oocytes. Development 2005, 132, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Deniz, E.; Erman, B. Long noncoding RNA (lincRNA), a new paradigm in gene expression control. Funct. Integr. Genom. 2017, 17, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Wang, K. Long noncoding RNAs: Pivotal regulators in acute myeloid leukemia. Exp. Hematol. Oncol. 2015, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Sana, J.; Faltejskova, P.; Svoboda, M.; Slaby, O. Novel classes of non-coding RNAs and cancer. J. Transl. Med. 2012, 10, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. Metazoan microRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, J.; Cairns, M.J. Identifying miRNAs, targets and functions. Brief. Bioinform. 2014, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Odenthal, M.; Fries, J.W. Exosomes as miRNA carriers: Formation-function-future. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Kai, K.; Dittmar, R.L.; Sen, S. Secretory microRNAs as biomarkers of cancer. Semin. Cell Dev. Biol. 2018, 78, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Nouraee, N.; Mowla, S.J. MiRNA therapeutics in cardiovascular diseases: Promises and problems. Front. Genet. 2015, 6, 232. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Zhao, B.; Zhao, J.; Li, S. Potential roles of exosomal microRNAs as diagnostic biomarkers and therapeutic application in alzheimer’s disease. Neural Plast. 2017, 2017, 7027380. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.Y.; Choi, Y.H.; Hwang, S.; Jeong, J.Y.; An, H.J. Clinical impact of microRNAs associated with cancer stem cells as a prognostic factor in ovarian carcinoma. J. Cancer 2017, 8, 3538–3547. [Google Scholar] [CrossRef] [PubMed]
- Mukohyama, J.; Shimono, Y.; Minami, H.; Kakeji, Y.; Suzuki, A. Roles of microRNAs and RNA-binding proteins in the regulation of colorectal cancer stem cells. Cancers 2017, 9, 143. [Google Scholar] [CrossRef] [PubMed]
- Salvador, M.A.; Birnbaum, D.; Charafe-Jauffret, E.; Ginestier, C. Breast cancer stem cells programs: Enter the (non)-code. Brief. Funct. Genom. 2016, 15, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Moriya, C.; Igarashi, H.; Saitoh, A.; Yamamoto, H.; Adachi, Y.; Imai, K. Cancer stem cells in human gastrointestinal cancer. Cancer Sci. 2016, 107, 1556–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioffi, M.; Trabulo, S.M.; Sanchez-Ripoll, Y.; Miranda-Lorenzo, I.; Lonardo, E.; Dorado, J.; Reis Vieira, C.; Ramirez, J.C.; Hidalgo, M.; Aicher, A.; et al. The miR-17-92 cluster counteracts quiescence and chemoresistance in a distinct subpopulation of pancreatic cancer stem cells. Gut 2015, 64, 1936–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A. Role of stem cell derived exosomes in tumor biology. Int. J. Cancer 2018, 142, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Bajdak, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The ZEB1/miR-200 feedback loop controls notch signalling in cancer cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Ru, P.; Steele, R.; Newhall, P.; Phillips, N.J.; Toth, K.; Ray, R.B. MiRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol. Cancer Ther. 2012, 11, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, H.; Liu, J.; Tu, X.; Zang, Y.; Zhu, J.; Chen, J.; Dong, L.; Zhang, J. miR-30 inhibits TGF-β1-induced epithelial-to-mesenchymal transition in hepatocyte by targeting snail1. Biochem. Biophys. Res. Commun. 2012, 417, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Gandellini, P.; Giannoni, E.; Casamichele, A.; Taddei, M.L.; Callari, M.; Piovan, C.; Valdagni, R.; Pierotti, M.A.; Zaffaroni, N.; Chiarugi, P. miR-205 hinders the malignant interplay between prostate cancer cells and associated fibroblasts. Antioxid. Redox Signal. 2014, 20, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Fu, X.; Chen, X.; Zeng, S.; Tian, Y.; Jove, R.; Xu, R.; Huang, W. miR-194 is a marker of hepatic epithelial cells and suppresses metastasis of liver cancer cells in mice. Hepatology 2010, 52, 2148–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetter, G.; Saumet, A.; Moes, M.; Vallar, L.; Le Bechec, A.; Laurini, C.; Sabbah, M.; Arar, K.; Theillet, C.; Lecellier, C.H.; et al. miR-661 expression in SNAI1-induced epithelial to mesenchymal transition contributes to breast cancer cell invasion by targeting Nectin-1 and StarD10 messengers. Oncogene 2010, 29, 4436–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Fan, J.; Ding, X.; Peng, W.; Yu, X.; Chen, Y.; Nie, J. TGF-{β}-induced miR-491-5p expression promotes par-3 degradation in rat proximal tubular epithelial cells. J. Biol. Chem. 2010, 285, 40019–40027. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Satoh, K.; Miura, S.; Hirota, M.; Kanno, A.; Masamune, A.; Kikuta, K.; Kume, K.; Unno, J.; Egawa, S.; et al. miR-197 induces epithelial-mesenchymal transition in pancreatic cancer cells by targeting p120 catenin. J. Cell Physiol. 2013, 228, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Nalls, D.; Tang, S.N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34A for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Hao, X.; Zhang, M.; Tang, W.; Yang, M.; Li, L.; Xiang, D.; Desano, J.T.; Bommer, G.T.; Fan, D.; et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS ONE 2009, 4, e6816. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.G.; Yu, S.N.; Lu, Z.H.; Ma, Y.H.; Gu, Y.M.; Chen, J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting kras. Carcinogenesis 2010, 31, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Lu, Z.; Liu, C.; Meng, Y.; Ma, Y.; Zhao, W.; Liu, J.; Yu, J.; Chen, J. miRNA-96 suppresses kras and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res. 2010, 70, 6015–6025. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.R.; Frampton, A.E.; Jacob, J.; Pellegrino, L.; Krell, J.; Giamas, G.; Tsim, N.; Vlavianos, P.; Cohen, P.; Ahmad, R.; et al. MicroRNAs targeting oncogenes are down-regulated in pancreatic malignant transformation from benign tumors. PLoS ONE 2012, 7, e32068. [Google Scholar] [CrossRef] [PubMed]
- Talotta, F.; Cimmino, A.; Matarazzo, M.R.; Casalino, L.; De Vita, G.; D’Esposito, M.; Di Lauro, R.; Verde, P. An autoregulatory loop mediated by miR-21 and PDCD4 controls the AP-1 activity in ras transformation. Oncogene 2009, 28, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Hong, S.M.; Karikari, C.A.; Hruban, R.H.; Goggins, M.G.; Maitra, A. Aberrant microRNA-155 expression is an early event in the multistep progression of pancreatic adenocarcinoma. Pancreatology 2010, 10, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, A.; Hong, S.M.; Hruban, R.H.; Goggins, M. MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin. Cancer Res. 2012, 18, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, Z.; Li, K.; Gong, L.; Yang, Q.; Huang, X.; Hong, C.; Ding, M.; Yang, H. Linc-DYNC2H1-4 promotes emt and csc phenotypes by acting as a sponge of miR-145 in pancreatic cancer cells. Cell Death Dis. 2017, 8, e2924. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.K.; Mondal, G.; Kumar, V.; Kattel, K.; Mahato, R.I. Chemosensitization and inhibition of pancreatic cancer stem cell proliferation by overexpression of microRNA-205. Cancer Lett. 2017, 402, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, M.; Raza, U.; Saatci, O.; Eyupoglu, E.; Yurdusev, E.; Sahin, O. miR-200C: A versatile watchdog in cancer progression, emt, and drug resistance. J. Mol. Med. 2016, 94, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Eguchi, H.; Nagano, H.; Konno, M.; Tomimaru, Y.; Wada, H.; Hama, N.; Kawamoto, K.; Kobayashi, S.; Nishida, N.; et al. MicroRNA-1246 expression associated with CCNG2-mediated chemoresistance and stemness in pancreatic cancer. Br. J. Cancer 2014, 111, 1572–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Chitkara, D.; Kumar, V.; Behrman, S.W.; Mahato, R.I. MiRNA profiling in pancreatic cancer and restoration of chemosensitivity. Cancer Lett. 2013, 334, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Wang, L.; Yu, B.; Zhuang, Q.Y.; Wang, Y.P. Expression signatures of long noncoding RNAs in adolescent idiopathic scoliosis. Biomed. Res. Int. 2015, 2015, 276049. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Yu, X.; Lai, J.; Yang, L.; Chen, S.; Li, Y. Overexpression of the long non-coding RNA PVT1 is correlated with leukemic cell proliferation in acute promyelocytic leukemia. J. Hematol. Oncol. 2015, 8, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.D.; Chen, W.M.; Qi, F.Z.; Xia, R.; Sun, M.; Xu, T.P.; Yin, L.; Zhang, E.B.; De, W.; Shu, Y.Q. Long non-coding RNA anril is upregulated in hepatocellular carcinoma and regulates cell proliferation by epigenetic silencing of KLF2. J Hematol. Oncol. 2015, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Qu, X.; Li, W.; Zhong, X.; Li, P.; Yang, S.; Chen, X.; Shao, M.; Zhang, L. The long non-coding RNA, GAS5, enhances gefitinib-induced cell death in innate egfr tyrosine kinase inhibitor-resistant lung adenocarcinoma cells with wide-type egfr via downregulation of the IGF-1r expression. J. Hematol. Oncol. 2015, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.P.; Huang, M.D.; Xia, R.; Liu, X.X.; Sun, M.; Yin, L.; Chen, W.M.; Han, L.; Zhang, E.B.; Kong, R.; et al. Decreased expression of the long non-coding RNA fendrr is associated with poor prognosis in gastric cancer and fendrr regulates gastric cancer cell metastasis by affecting FIBRONECTIN1 expression. J. Hematol. Oncol. 2014, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Tian, J.; Tang, X.; Ma, J.; Wang, S. Long non-coding RNAs in the regulation of myeloid cells. J. Hematol. Oncol. 2016, 9, 99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Peng, G. Non-coding RNAs: An emerging player in DNA damage response. Mutat. Res. Rev. Mutat. Res. 2015, 763, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.Y.; Liao, Y.W.; Chen, P.Y.; Hsieh, P.L.; Fang, C.Y.; Wu, C.Y.; Yen, M.L.; Peng, B.Y.; Wang, D.P.; Cheng, H.C.; et al. Targeting lncRNA hotair suppresses cancer stemness and metastasis in oral carcinomas stem cells through modulation of emt. Oncotarget 2017, 8, 98542–98552. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Lv, Y.; Jin, F.; Liu, Y.; Ma, Y.; Xiong, Y.; Liu, L.; Zhang, S.; Sun, Y.; Tipoe, G.L.; et al. LncRNA hanr promotes tumorigenesis and increase of chemoresistance in hepatocellular carcinoma. Cell Physiol. Biochem. 2017, 43, 1926–1938. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Hu, H.; Han, T.; Yuan, C.; Wang, L.; Jin, Z.; Guo, Z.; Wang, L. Long noncoding RNA MALAT-1 enhances stem cell-like phenotypes in pancreatic cancer cells. Int. J. Mol. Sci. 2015, 16, 6677–6693. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Maeda, S.; Liu, C.; Karin, M.; Edgington, T.S. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene 2007, 26, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yang, M.; Tian, J.; Wang, X.; Li, Z. Malat-1: A long non-coding RNA and its important 3′ end functional motif in colorectal cancer metastasis. Int. J. Oncol. 2011, 39, 169–175. [Google Scholar] [PubMed]
- Wu, M.; Lin, Z.; Li, X.; Xin, X.; An, J.; Zheng, Q.; Yang, Y.; Lu, D. Hulc cooperates with MALAT1 to aggravate liver cancer stem cells growth through telomere repeat-binding factor 2. Sci. Rep. 2016, 6, 36045. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.T.; Shi, D.B.; Wang, Y.W.; Li, X.X.; Xu, Y.; Tripathi, P.; Gu, W.L.; Cai, G.X.; Cai, S.J. High expression of lncRNA MALAT1 suggests a biomarker of poor prognosis in colorectal cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 3174–3181. [Google Scholar] [PubMed]
- Han, Y.; Zhou, L.; Wu, T.; Huang, Y.; Cheng, Z.; Li, X.; Sun, T.; Zhou, Y.; Du, Z. Downregulation of lncRNA-MALAT1 affects proliferation and the expression of stemness markers in glioma stem cell line SHG139s. Cell Mol. Neurobiol. 2016, 36, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Chen, C.; Zhou, Q.; Wang, Y.; Zhao, Y.; Zhao, X.; Li, W.; Zheng, S.; Ye, H.; Wang, L.; et al. lncRNA hottip modulates cancer stem cell properties in human pancreatic cancer by regulating HOXA9. Cancer Lett. 2017, 410, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.Z.; Guo, Y.J.; Tao, Q.F.; Liu, F.; Pan, W.; Wang, T.T.; Zhou, C.C.; et al. A long noncoding RNA activated by tgf-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes emt and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Yang, X.; Song, W.; Sun, W.; Li, X.; Wang, J.; Zhong, Y.; Shang, R.; Ruan, B.; Zhang, Z.; et al. Downregulation of lncRNA-ATB correlates with clinical progression and unfavorable prognosis in pancreatic cancer. Tumour Biol. 2016, 37, 3933–3938. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.Q.; Jiang, Y.; Zhu, F.; Sun, D.L.; He, X.Z. Long noncoding RNA PVT1 promotes emt and cell proliferation and migration through downregulating p21 in pancreatic cancer cells. Technol. Cancer Res. Treat. 2017, 16, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Sun, H.; Kong, H.; Chen, Z.; Chen, B.; Zhou, M. The lncRNA-TUG1/EZH2 axis promotes pancreatic cancer cell proliferation, migration and emt phenotype formation through sponging miR-382. Cell Physiol. Biochem. 2017, 42, 2145–2158. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Zhao, J.; Vieth, E.; Nephew, K.P.; Matei, D. EZH2 inhibition promotes epithelial-to-mesenchymal transition in ovarian cancer cells. Oncotarget 2016, 7, 84453–84467. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Yu, J.; Dhanasekaran, S.M.; Kim, J.H.; Mani, R.S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; et al. Repression of e-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef] [PubMed]
- Arnes, L.; Liu, Z.; Wang, J.; Carlo Maurer, H.; Sagalovskiy, I.; Sanchez-Martin, M.; Bommakanti, N.; Garofalo, D.C.; Balderes, D.A.; Sussel, L.; et al. Comprehensive characterisation of compartment-specific long non-coding RNAs associated with pancreatic ductal adenocarcinoma. Gut 2018. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Huang, X.; Tan, W.; Yu, D.; Du, Z.; Chang, J.; Wei, L.; Han, Y.; Wang, C.; Che, X.; et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat. Genet. 2016, 48, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Gorgulu, K.; Dantes, Z.; Wormann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juttermann, R.; Li, E.; Jaenisch, R. Toxicity of 5-AZA-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. USA 1994, 91, 11797–11801. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, K.; Datta, J.; Majumder, S.; Bai, S.; Kutay, H.; Motiwala, T.; Jacob, S.T. 5-AZA-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the ken box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell Biol. 2005, 25, 4727–4741. [Google Scholar] [CrossRef] [PubMed]
- Abele, R.; Clavel, M.; Dodion, P.; Bruntsch, U.; Gundersen, S.; Smyth, J.; Renard, J.; van Glabbeke, M.; Pinedo, H.M. The eortc early clinical trials cooperative group experience with 5-AZA-2′-deoxycytidine (NSC 127716) in patients with colo-rectal, head and neck, renal carcinomas and malignant melanomas. Eur. J. Cancer Clin. Oncol. 1987, 23, 1921–1924. [Google Scholar] [CrossRef]
- Clavel, M.; Monfardini, S.; Fossa, S.; Smyth, J.; Renard, J.; Kaye, S.B. 5-AZA-2′-deoxycytidine (NSC 127716) in non-seminomatous testicular cancer. Phase ii from the eortc early clinical trials cooperative group and genito-urinary group. Ann. Oncol. 1992, 3, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Stadler, W.M.; Margolin, K.; Ferber, S.; McCulloch, W.; Thompson, J.A. A phase ii study of depsipeptide in refractory metastatic renal cell cancer. Clin. Genitourin. Cancer 2006, 5, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Ohwada, S.; Saitoh, F.; Adachi, M.; Morishita, Y.; Hozumi, M. Induction of ley antigen by 5-AZA-2′-deoxycytidine in association with differentiation and apoptosis in human pancreatic cancer cells. Anticancer Res. 1996, 16, 735–740. [Google Scholar] [PubMed]
- Lefebvre, B.; Belaich, S.; Longue, J.; Vandewalle, B.; Oberholzer, J.; Gmyr, V.; Pattou, F.; Kerr-Conte, J. 5′-AZA induces NGN3 expression and endocrine differentiation in the PANC-1 human ductal cell line. Biochem. Biophys. Res. Commun. 2010, 391, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cardenas, H.; Fang, F.; Condello, S.; Taverna, P.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014, 74, 4922–4936. [Google Scholar] [CrossRef] [PubMed]
- Schech, A.; Kazi, A.; Yu, S.; Shah, P.; Sabnis, G. Histone deacetylase inhibitor entinostat inhibits tumor-initiating cells in triple-negative breast cancer cells. Mol. Cancer Ther. 2015, 14, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Morales, P.; Gomez-Martinez, A.; Carrato, A.; Martinez-Lacaci, I.; Barbera, V.M.; Soto, J.L.; Carrasco-Garcia, E.; Menendez-Gutierrez, M.P.; Castro-Galache, M.D.; Ferragut, J.A.; et al. Histone deacetylase inhibitors induced caspase-independent apoptosis in human pancreatic adenocarcinoma cell lines. Mol. Cancer Ther. 2005, 4, 1222–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, W.; Lee, D.H.; Zheng, Y.; Wuensche, P.; Alvarez, R.; Wen, D.L.; Aribi, A.M.; Thean, S.M.; Doan, N.B.; Said, J.W.; et al. Growth inhibition of pancreatic cancer cells by histone deacetylase inhibitor belinostat through suppression of multiple pathways including HIF, NFkB, and mTOR signaling in vitro and in vivo. Mol. Carcinog. 2014, 53, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.T.; Mock, K.; Ruh, M.; Schuler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debeb, B.G.; Lacerda, L.; Xu, W.; Larson, R.; Solley, T.; Atkinson, R.; Sulman, E.P.; Ueno, N.T.; Krishnamurthy, S.; Reuben, J.M.; et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through wnt/beta-catenin signaling. Stem Cells 2012, 30, 2366–2377. [Google Scholar] [CrossRef] [PubMed]
- Debeb, B.G.; Lacerda, L.; Larson, R.; Wolfe, A.R.; Krishnamurthy, S.; Reuben, J.M.; Ueno, N.T.; Gilcrease, M.; Woodward, W.A. Histone deacetylase inhibitor-induced cancer stem cells exhibit high pentose phosphate pathway metabolism. Oncotarget 2016, 7, 28329–28339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs-microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Kasinski, A.L.; Slack, F.J. Aberrant regulation and function of microRNAs in cancer. Curr. Biol. 2014, 24, R762–R776. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, D.; Campbell, N.R.; Karikari, C.; Chivukula, R.; Kent, O.A.; Mendell, J.T.; Maitra, A. Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol. Cancer Ther. 2011, 10, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Setua, S.; Khan, S.; Yallapu, M.M.; Behrman, S.W.; Sikander, M.; Khan, S.S.; Jaggi, M.; Chauhan, S.C. Restitution of tumor suppressor microRNA-145 using magnetic nanoformulation for pancreatic cancer therapy. J. Gastrointest. Surg. 2017, 21, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, L.; Ischenko, I.; Bao, Q.; Schwarz, B.; Niess, H.; Wang, Y.; Renner, A.; Mysliwietz, J.; Jauch, K.W.; et al. Antisense inhibition of microRNA-21 and microRNA-221 in tumor-initiating stem-like cells modulates tumorigenesis, metastasis, and chemotherapy resistance in pancreatic cancer. Target. Oncol. 2015, 10, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Chitkara, D.; Behrman, S.W.; Mahato, R.I. Efficacy of gemcitabine conjugated and miRNA-205 complexed micelles for treatment of advanced pancreatic cancer. Biomaterials 2014, 35, 7077–7087. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y.; Li, J.; Zhang, Z.; Huang, C.; Lian, G.; Yang, K.; Chen, S.; Lin, Y.; Wang, L.; et al. Co-delivery of microRNA-21 antisense oligonucleotides and gemcitabine using nanomedicine for pancreatic cancer therapy. Cancer Sci. 2017, 108, 1493–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Mondal, G.; Slavik, P.; Rachagani, S.; Batra, S.K.; Mahato, R.I. Codelivery of small molecule hedgehog inhibitor and miRNA for treating pancreatic cancer. Mol. Pharm. 2015, 12, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Smaldone, M.C.; Davies, B.J. BC-819, a plasmid comprising the H19 gene regulatory sequences and diphtheria toxin a, for the potential targeted therapy of cancers. Curr. Opin. Mol. Ther. 2010, 12, 607–616. [Google Scholar] [PubMed]
- Hanna, N.; Ohana, P.; Konikoff, F.M.; Leichtmann, G.; Hubert, A.; Appelbaum, L.; Kopelman, Y.; Czerniak, A.; Hochberg, A. Phase 1/2A, dose-escalation, safety, pharmacokinetic and preliminary efficacy study of intratumoral administration of BC-819 in patients with unresectable pancreatic cancer. Cancer Gene. Ther. 2012, 19, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Bonasio, R.; Saldana-Meyer, R.; Yoshida, T.; Son, J.; Nishino, K.; Umezawa, A.; Reinberg, D. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol. Cell 2014, 53, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Sunamura, N.; Ohira, T.; Kataoka, M.; Inaoka, D.; Tanabe, H.; Nakayama, Y.; Oshimura, M.; Kugoh, H. Regulation of functional KCNQ1OT1 lncRNA by β-catenin. Sci. Rep. 2016, 6, 20690. [Google Scholar] [CrossRef] [PubMed]
- Paris, O.; Ferraro, L.; Grober, O.M.; Ravo, M.; De Filippo, M.R.; Giurato, G.; Nassa, G.; Tarallo, R.; Cantarella, C.; Rizzo, F.; et al. Direct regulation of microRNA biogenesis and expression by estrogen receptor β in hormone-responsive breast cancer. Oncogene 2012, 31, 4196–4206. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Chen, Y. Small molecule targeting miR-34A for cancer therapy. Mol. Cell Oncol. 2015, 2, e977160. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zagorac, S.; Garcia-Bermejo, L.; Sainz, B., Jr. The Epigenetic Landscape of Pancreatic Cancer Stem Cells. Epigenomes 2018, 2, 10. https://doi.org/10.3390/epigenomes2020010
Zagorac S, Garcia-Bermejo L, Sainz B Jr. The Epigenetic Landscape of Pancreatic Cancer Stem Cells. Epigenomes. 2018; 2(2):10. https://doi.org/10.3390/epigenomes2020010
Chicago/Turabian StyleZagorac, Sladjana, Laura Garcia-Bermejo, and Bruno Sainz, Jr. 2018. "The Epigenetic Landscape of Pancreatic Cancer Stem Cells" Epigenomes 2, no. 2: 10. https://doi.org/10.3390/epigenomes2020010