
Synergistic Mutations of LRP6 and WNT10A in Familial Tooth Agenesis

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Enrollment of FTA Families
2.2. Mutational Analyses
2.3. Prediction of Structural Alterations Caused by LRP6 Mutations
2.4. Literature Review and Statistical Analyses
3. Results
3.1. Family 1
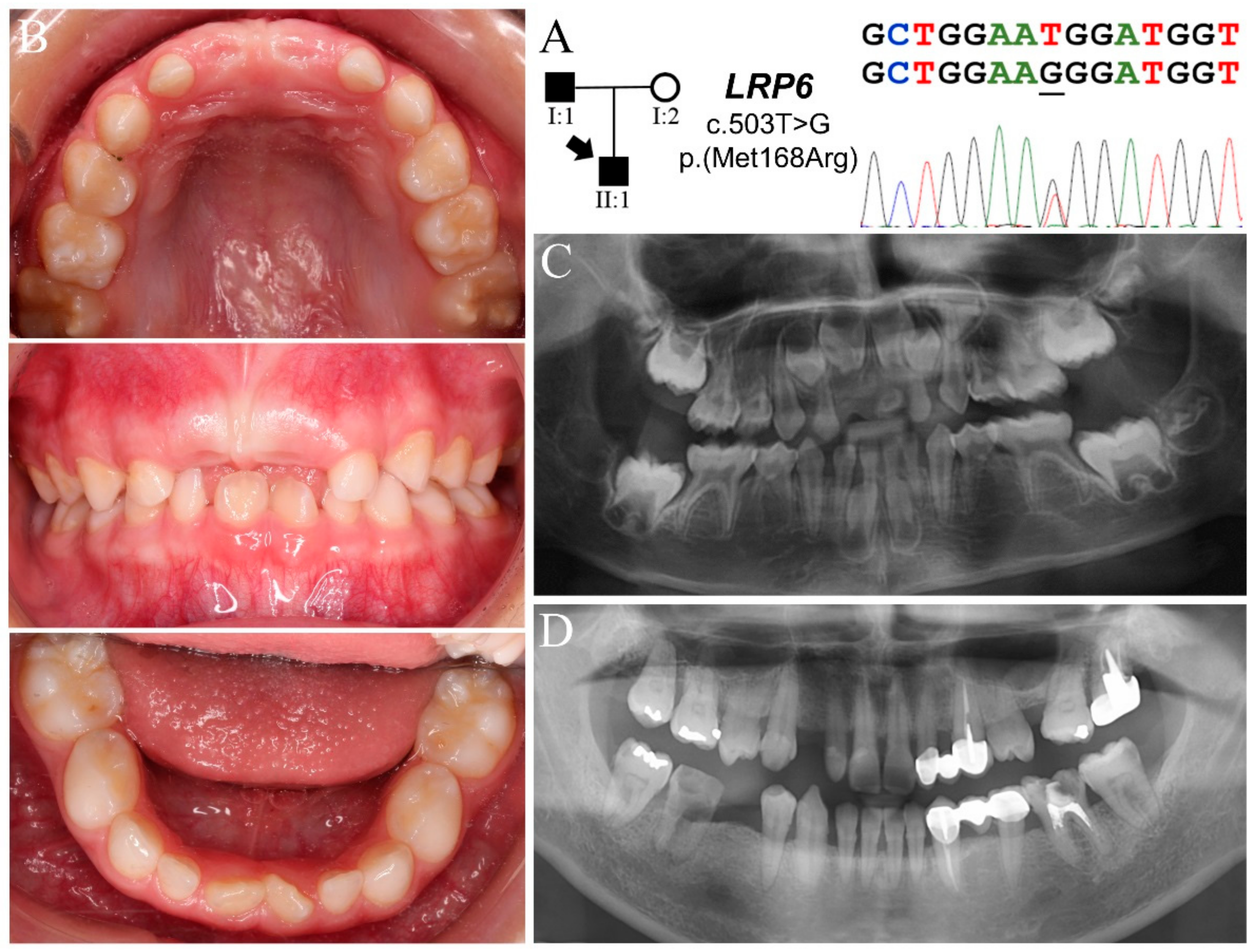
3.2. Family 2
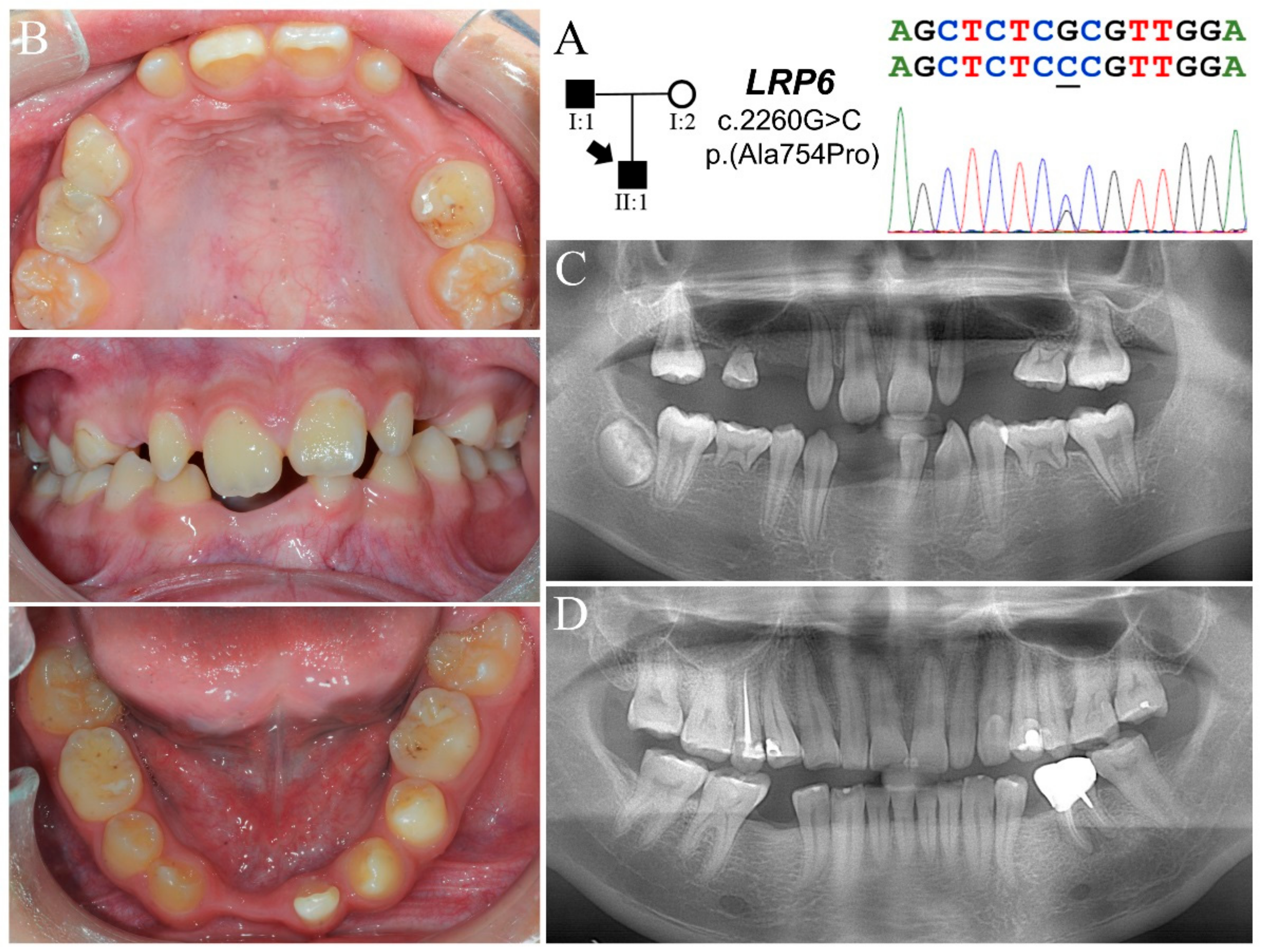
3.3. Family 3
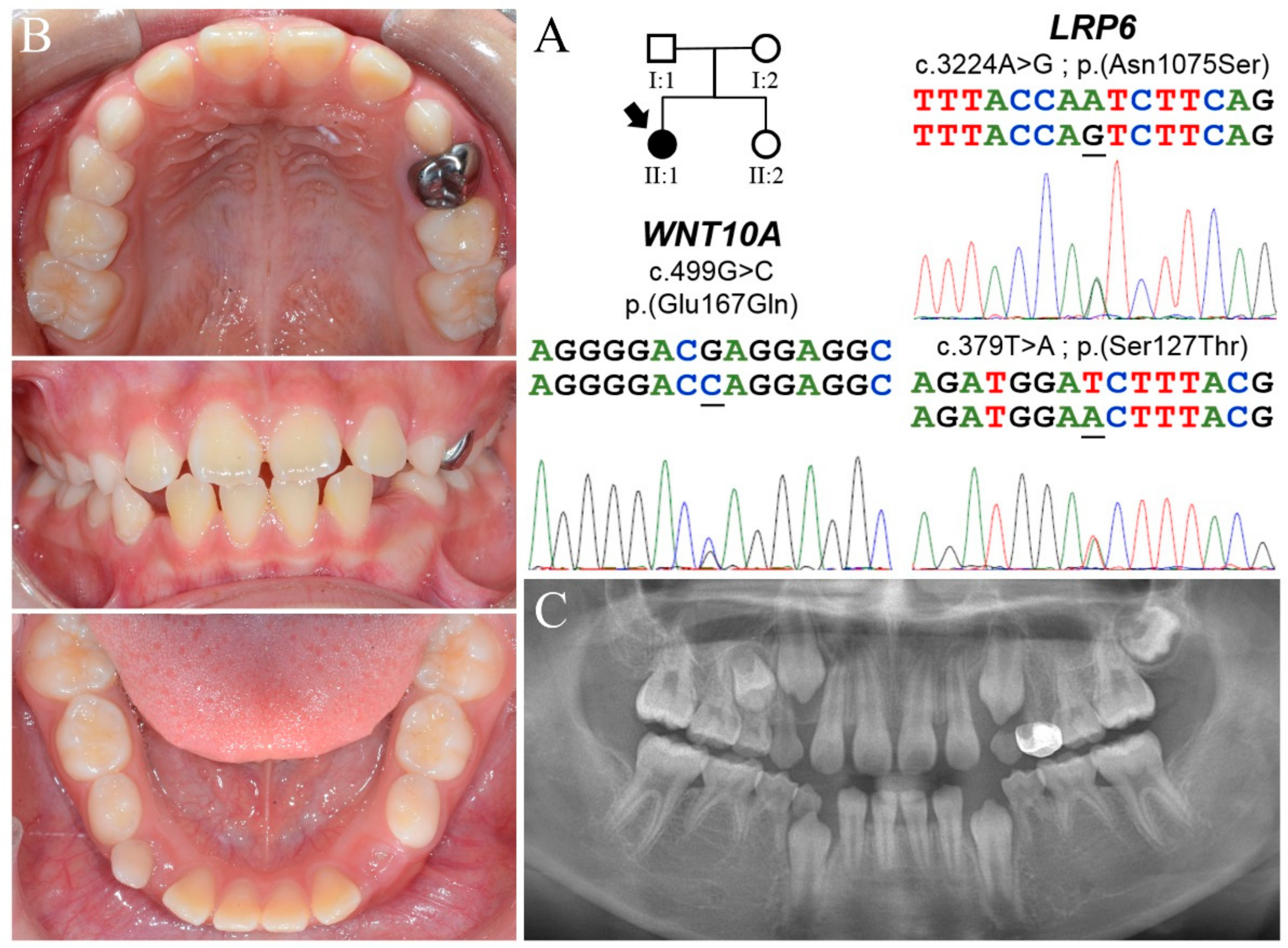
3.4. Family 4
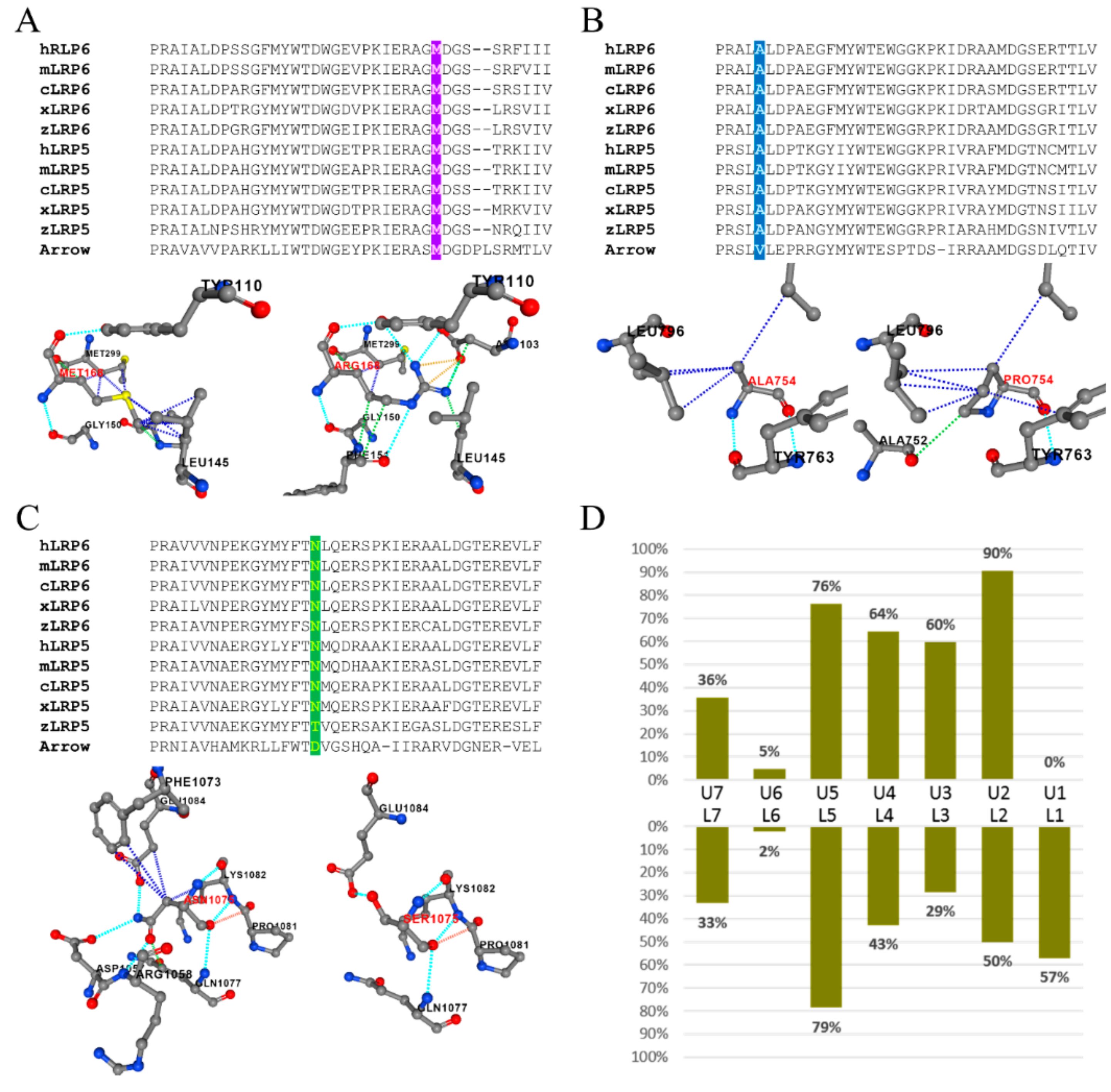
3.5. Predicted Structural Alterations and Pathogenicity of LRP6 Missense Mutations
3.6. Pattern of Missing Teeth Associated with LRP6 Loss-of-Function Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balic, A.; Thesleff, I. Tissue Interactions Regulating Tooth Development and Renewal. Curr. Top. Dev. Biol. 2015, 115, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Juuri, E.; Balic, A. The Biology Underlying Abnormalities of Tooth Number in Humans. J. Dent. Res. 2017, 96, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Nieminen, P. Genetic basis of tooth agenesis. J. Exp. Zool. B Mol. Dev. Evol. 2009, 312, 320–342. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Letra, A. The Changing Landscape in the Genetic Etiology of Human Tooth Agenesis. Genes 2018, 9, 255. [Google Scholar] [CrossRef] [Green Version]
- Vastardis, H.; Karimbux, N.; Guthua, S.W.; Seidman, J.G.; Seidman, C.E. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat. Genet. 1996, 13, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Stockton, D.W.; Das, P.; Goldenberg, M.; D’Souza, R.N.; Patel, P.I. Mutation of PAX9 is associated with oligodontia. Nat. Genet. 2000, 24, 18–19. [Google Scholar] [CrossRef]
- Lammi, L.; Arte, S.; Somer, M.; Jarvinen, H.; Lahermo, P.; Thesleff, I.; Pirinen, S.; Nieminen, P. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am. J. Hum. Genet. 2004, 74, 1043–1050. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Han, D.; Qu, H.; Gong, Y.; Wu, H.; Zhang, X.; Zhong, N.; Feng, H. EDA gene mutations underlie non-syndromic oligodontia. J. Dent. Res. 2009, 88, 126–131. [Google Scholar] [CrossRef] [Green Version]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P.; et al. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massink, M.P.; Créton, M.A.; Spanevello, F.; Fennis, W.M.; Cune, M.S.; Savelberg, S.M.; Nijman, I.J.; Maurice, M.M.; van den Boogaard, M.J.; van Haaften, G. Loss-of-Function Mutations in the WNT Co-receptor LRP6 Cause Autosomal-Dominant Oligodontia. Am. J. Hum. Genet. 2015, 97, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Ockeloen, C.W.; Khandelwal, K.D.; Dreesen, K.; Ludwig, K.U.; Sullivan, R.; van Rooij, I.; Thonissen, M.; Swinnen, S.; Phan, M.; Conte, F.; et al. Novel mutations in LRP6 highlight the role of WNT signaling in tooth agenesis. Genet. Med. 2016, 18, 1158–1162. [Google Scholar] [CrossRef] [Green Version]
- Fournier, B.P.; Bruneau, M.H.; Toupenay, S.; Kerner, S.; Berdal, A.; Cormier-Daire, V.; Hadj-Rabia, S.; Coudert, A.E.; de La Dure-Molla, M. Patterns of Dental Agenesis Highlight the Nature of the Causative Mutated Genes. J. Dent. Res. 2018, 97, 1306–1316. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, H.; Camhi, H.; Seymen, F.; Koruyucu, M.; Kasimoglu, Y.; Kim, J.W.; Kim-Berman, H.; Yuson, N.M.R.; Benke, P.J.; et al. Analyses of oligodontia phenotypes and genetic etiologies. Int. J. Oral Sci. 2021, 13, 32. [Google Scholar] [CrossRef]
- Brown, S.D.; Twells, R.C.; Hey, P.J.; Cox, R.D.; Levy, E.R.; Soderman, A.R.; Metzker, M.L.; Caskey, C.T.; Todd, J.A.; Hess, J.F. Isolation and characterization of LRP6, a novel member of the low density lipoprotein receptor gene family. Biochem. Biophys. Res. Commun. 1998, 248, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Semenov, M.; Kato, Y.; Spokony, R.; Liu, C.; Katsuyama, Y.; Hess, F.; Saint-Jeannet, J.P.; He, X. LDL-receptor-related proteins in Wnt signal transduction. Nature 2000, 407, 530–535. [Google Scholar] [CrossRef]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Davidson, G. LRPs in WNT Signalling. Handb. Exp. Pharmacol. 2021, 269, 45–73. [Google Scholar] [CrossRef]
- Jeong, W.; Jho, E.H. Regulation of the Low-Density Lipoprotein Receptor-Related Protein LRP6 and Its Association with Disease: Wnt/β-Catenin Signaling and Beyond. Front. Cell Dev. Biol. 2021, 9, 714330. [Google Scholar] [CrossRef] [PubMed]
- Mani, A.; Radhakrishnan, J.; Wang, H.; Mani, A.; Mani, M.A.; Nelson-Williams, C.; Carew, K.S.; Mane, S.; Najmabadi, H.; Wu, D.; et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science 2007, 315, 1278–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, M.P.; McAlister, W.H.; Zhang, F.; Bijanki, V.N.; Nenninger, A.; Gottesman, G.S.; Lin, E.L.; Huskey, M.; Duan, S.; Dahir, K.; et al. New explanation for autosomal dominant high bone mass: Mutation of low-density lipoprotein receptor-related protein 6. Bone 2019, 127, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Fathe, K.; McCartney, D.; Zhu, H.; Yang, W.; Ross, M.E.; Shaw, G.M.; Finnell, R.H. Rare LRP6 variants identified in spina bifida patients. Hum. Mutat. 2015, 36, 342–349. [Google Scholar] [CrossRef] [Green Version]
- van den Boogaard, M.J.; Créton, M.; Bronkhorst, Y.; van der Hout, A.; Hennekam, E.; Lindhout, D.; Cune, M.; Ploos van Amstel, H.K. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J. Med. Genet. 2012, 49, 327–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, Y.A.; Kamal, L.; Rayyan, A.A.; Dweik, D.; Pierce, S.; Lee, M.K.; King, M.C.; Walsh, T.; Kanaan, M. Mutation of KREMEN1, a modulator of Wnt signaling, is responsible for ectodermal dysplasia including oligodontia in Palestinian families. Eur. J. Hum. Genet. 2016, 24, 1430–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Wong, S.W.; Han, D.; Cai, T. Genetic analysis: Wnt and other pathways in nonsyndromic tooth agenesis. Oral Dis. 2019, 25, 646–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lu, H.; Zhang, N.; Zhu, Z.; Wang, S.; Li, M. PremPS: Predicting the impact of missense mutations on protein stability. PLoS Comput. Biol. 2020, 16, e1008543. [Google Scholar] [CrossRef]
- Ross, J.; Fennis, W.; de Leeuw, N.; Cune, M.; Willemze, A.; Rosenberg, A.; Ploos van Amstel, H.K.; Créton, M.; van den Boogaard, M.J. Concurrent manifestation of oligodontia and thrombocytopenia caused by a contiguous gene deletion in 12p13.2: A three-generation clinical report. Mol. Genet. Genom. Med. 2019, 7, e679. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Yang, J.H.; Chiang, C.W.K.; Hsiung, C.N.; Wu, P.E.; Chang, L.C.; Chu, H.W.; Chang, J.; Song, I.W.; Yang, S.L.; et al. Population structure of Han Chinese in the modern Taiwanese population based on 10,000 participants in the Taiwan Biobank project. Hum. Mol. Genet. 2016, 25, 5321–5331. [Google Scholar] [CrossRef] [Green Version]
- Kantaputra, P.; Kaewgahya, M.; Kantaputra, W. WNT10A mutations also associated with agenesis of the maxillary permanent canines, a separate entity. Am. J. Med. Genet. A 2014, 164, 360–363. [Google Scholar] [CrossRef]
- Song, S.; Zhao, R.; He, H.; Zhang, J.; Feng, H.; Lin, L. WNT10A variants are associated with non-syndromic tooth agenesis in the general population. Hum. Genet. 2014, 133, 117–124. [Google Scholar] [CrossRef]
- Lindeboom, R.G.; Supek, F.; Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Bubeck, D.; MacDonald, B.T.; Liang, W.X.; Mao, J.H.; Malinauskas, T.; Llorca, O.; Aricescu, A.R.; Siebold, C.; He, X.; et al. Structural and functional studies of LRP6 ectodomain reveal a platform for Wnt signaling. Dev. Cell 2011, 21, 848–861. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Biechele, T.; Wei, Z.; Morrone, S.; Moon, R.T.; Wang, L.; Xu, W. Crystal structures of the extracellular domain of LRP6 and its complex with DKK1. Nat. Struct. Mol. Biol. 2011, 18, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, S.K.; Choi, M.; Reid, B.M.; Hu, Y.; Lee, Y.L.; Herzog, C.R.; Kim-Berman, H.; Lee, M.; Benke, P.J.; et al. Taurodontism, variations in tooth number, and misshapened crowns in Wnt10a null mice and human kindreds. Mol. Genet. Genom. Med. 2015, 3, 40–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Pérez, J.M.; Candela, H.; Micol, J.L. Understanding synergy in genetic interactions. Trends Genet. 2009, 25, 368–376. [Google Scholar] [CrossRef]
- Deltas, C. Digenic inheritance and genetic modifiers. Clin. Genet. 2018, 93, 429–438. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Green, R.C. Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 2014, 370, 2418–2425. [Google Scholar] [CrossRef] [Green Version]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, A.L.; Manolio, T.A.; Mensah, G.A.; Peterson, J.F.; Roden, D.M.; Tamburro, C.; Williams, M.S.; Green, E.D. Genomic medicine for undiagnosed diseases. Lancet 2019, 394, 533–540. [Google Scholar] [CrossRef]
- Prasad, M.K.; Geoffroy, V.; Vicaire, S.; Jost, B.; Dumas, M.; Le Gras, S.; Switala, M.; Gasse, B.; Laugel-Haushalter, V.; Paschaki, M.; et al. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J. Med. Genet. 2016, 53, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Fan, Z.; Wong, S.W.; Sun, K.; Zhang, L.; Liu, H.; Feng, H.; Liu, Y.; Han, D. Lrp6 Dynamic Expression in Tooth Development and Mutations in Oligodontia. J. Dent. Res. 2021, 100, 415–422. [Google Scholar] [CrossRef]
- Brance, M.L.; Brun, L.R.; Cóccaro, N.M.; Aravena, A.; Duan, S.; Mumm, S.; Whyte, M.P. High bone mass from mutation of low-density lipoprotein receptor-related protein 6 (LRP6). Bone 2020, 141, 115550. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Y.; Zheng, Y.; Zhao, X.; Lin, S.; Zhang, Q.; Zhang, X. A novel missense mutation of LRP6 identified by whole-exome sequencing in a Chinese family with non-syndromic tooth agenesis. Orthod. Craniofac. Res. 2021, 24, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Keskin, G.; Karaer, K.; Uçar Gündoğar, Z. Targeted next-generation sequencing (NGS) analysis of mutations in nonsyndromic tooth agenesis candidate genes: Analysis of a Turkish cohort. J. Orofac. Orthop. 2021. [Google Scholar] [CrossRef]
- Goto, H.; Kimura, M.; Machida, J.; Ota, A.; Nakashima, M.; Tsuchida, N.; Adachi, J.; Aoki, Y.; Tatematsu, T.; Takahashi, K.; et al. A novel LRP6 variant in a Japanese family with oligodontia. Hum. Genome Var. 2021, 8, 30. [Google Scholar] [CrossRef]
- Huang, Y.X.; Gao, C.Y.; Zheng, C.Y.; Chen, X.; Yan, Y.S.; Sun, Y.Q.; Dong, X.Y.; Yang, K.; Zhang, D.L. Investigation of a Novel LRP6 Variant Causing Autosomal-Dominant Tooth Agenesis. Front. Genet. 2021, 12, 688241. [Google Scholar] [CrossRef]
- Basha, M.; Demeer, B.; Revencu, N.; Helaers, R.; Theys, S.; Bou Saba, S.; Boute, O.; Devauchelle, B.; Francois, G.; Bayet, B.; et al. Whole exome sequencing identifies mutations in 10% of patients with familial non-syndromic cleft lip and/or palate in genes mutated in well-known syndromes. J. Med. Genet. 2018, 55, 449–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinckan, N.; Du, R.; Petty, L.E.; Coban-Akdemir, Z.; Jhangiani, S.N.; Paine, I.; Baugh, E.H.; Erdem, A.P.; Kayserili, H.; Doddapaneni, H.; et al. Whole-Exome Sequencing Identifies Novel Variants for Tooth Agenesis. J. Dent. Res. 2018, 97, 49–59. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Subject | 8 | 7 | 6 | 5 | 4 | 3 | 2 | 1 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | No | LRP6 Mutation | WNT10A Mutation | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 1 | I:1 | Max | X | E | X | X | E | E | X | X | 6 | c.3754C>T p.(Gln1252*) | c.338G>A p.(Arg113His) | ||||||||
| Man | X | X | X | X | X | ||||||||||||||||
| II:1 | Max | ? | X | X | X | X | X | X | ? | 8 | |||||||||||
| Man | ? | X | X | ? | |||||||||||||||||
| Family 2 | I:1 | Max | X | X | P | X | X | 4 | c.503T>G p.(Met168Arg) | - | |||||||||||
| Man | X | X | X | X | |||||||||||||||||
| II:1 | Max | ? | X | X | X | X | X | ? | 13 | c.637G>A p.(Gly213Ser) | |||||||||||
| Man | ? | X | X | X | X | X | X | X | X | ? | |||||||||||
| Family 3 | I:1 | Max | X | P | P | X | 2 | c.2260G>C p.(Ala754Pro) | - | ||||||||||||
| Man | X | X | X | X | |||||||||||||||||
| II:1 | Max | ? | X | X | X | X | P | P | X | X | X | X | ? | 14 | |||||||
| Man | ? | X | X | X | X | X | X | ? | |||||||||||||
| Family 4 | II:1 | Max | ? | X | X | X | X | ? | 10 | c.3224A>G p.(Asn1075Ser) | c.499G>C p.(Glu167Gln) | ||||||||||
| Man | ? | X | X | X | X | X | X | ? |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, K.-Y.; Wang, Y.-L.; Chou, Y.-R.; Chen, J.-T.; Wang, Y.-P.; Simmer, J.P.; Hu, J.C.-C.; Wang, S.-K. Synergistic Mutations of LRP6 and WNT10A in Familial Tooth Agenesis. J. Pers. Med. 2021, 11, 1217. https://doi.org/10.3390/jpm11111217
Chu K-Y, Wang Y-L, Chou Y-R, Chen J-T, Wang Y-P, Simmer JP, Hu JC-C, Wang S-K. Synergistic Mutations of LRP6 and WNT10A in Familial Tooth Agenesis. Journal of Personalized Medicine. 2021; 11(11):1217. https://doi.org/10.3390/jpm11111217
Chicago/Turabian StyleChu, Kuan-Yu, Yin-Lin Wang, Yu-Ren Chou, Jung-Tsu Chen, Yi-Ping Wang, James P. Simmer, Jan C.-C. Hu, and Shih-Kai Wang. 2021. "Synergistic Mutations of LRP6 and WNT10A in Familial Tooth Agenesis" Journal of Personalized Medicine 11, no. 11: 1217. https://doi.org/10.3390/jpm11111217