An Easy and Reliable Strategy for Making Type I Interferon Signature Analysis Comparable among Research Centers

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Sample Collection, RNA Isolation and cDNA Preparation
2.3. IFN Signature Analysis
2.4. RNAseq Analysis
2.5. Statistical Analyses
3. Results
3.1. Variability Assessment in IFN-Stimulated Genes Expression in Healthy Controls (Dataset A) Analyzed by qPCR
3.2. IFN-Stimulated Genes Expression Evaluated by qPCR or RNAseq Analysis Are Comparable
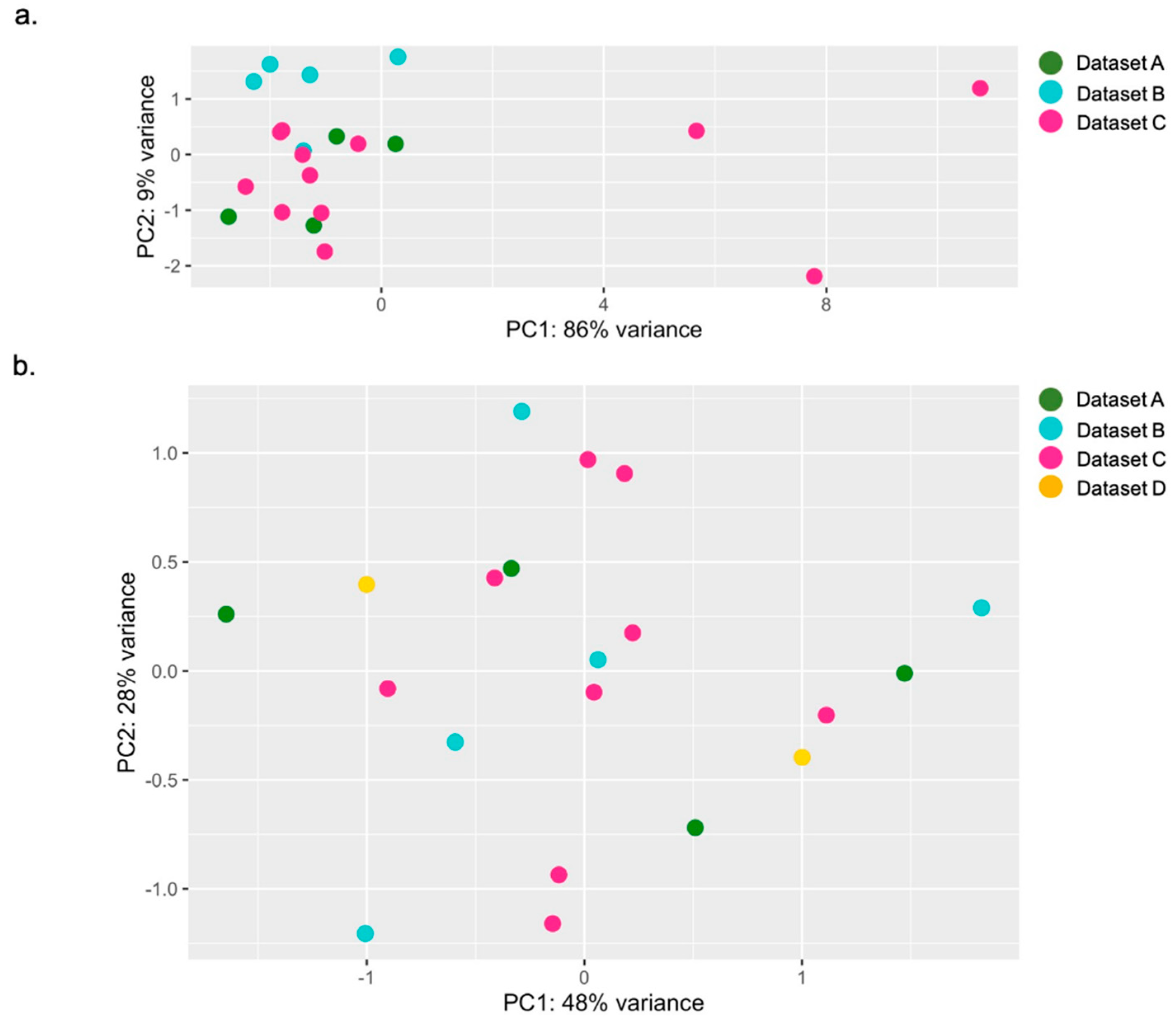
3.3. Preliminary Analysis for Sample Selection and Variability Assessment in IFN-Stimulated Genes Expression Analyzed by RNAseq
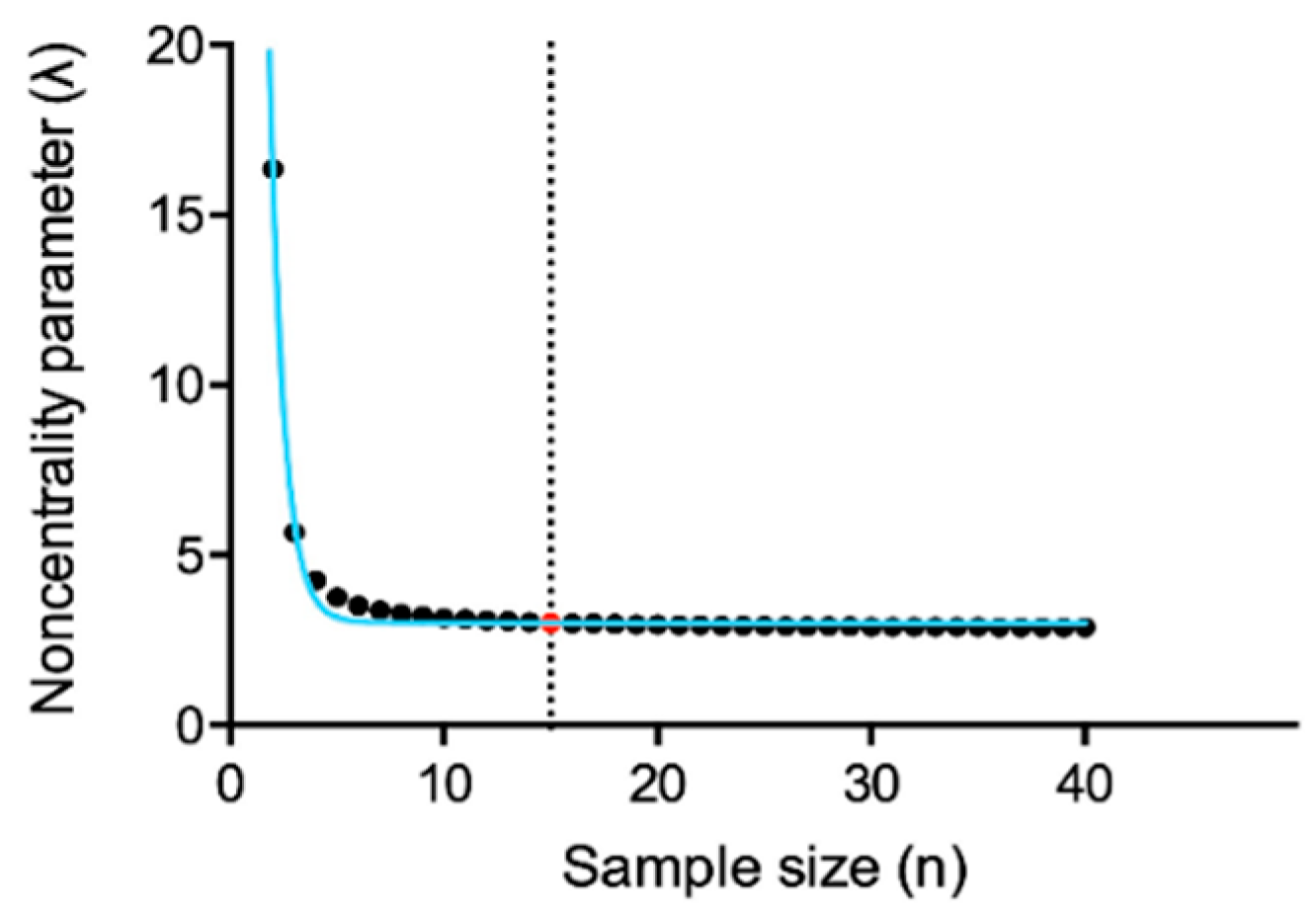
3.4. Pooling Twenty Subjects Could Be Considered an Optimal Strategy to Minimize Gene Expression Variability among Healthy Controls for on Wet IFN Signature Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [PubMed]
- Crow, Y.J. Type I interferonopathies: A novel set of inborn errors of immunity. Ann. N. Y. Acad. Sci. 2011, 1238, 91–98. [Google Scholar] [CrossRef]
- Crow, Y.J.; Black, D.N.; Ali, M.; Bond, J.; Jackson, A.P.; Lefson, M.; Michaud, J.; Roberts, E.; Stephenson, J.B.; Woods, C.G.; et al. Cree encephalitis is allelic with Aicardi-Goutiéres syndrome: Implications for the pathogenesis of disorders of interferon α metabolism. J. Med. Genet. 2003, 40, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, G.I.; Forte, G.M.; Szynkiewicz, M.; Chase, D.S.; Aeby, A.; Abdel-Hamid, M.S.; Ackroyd, S.; Allcock, R.; Bailey, K.M.; Balottin, U.; et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A case-control study. Lancet Neurol. 2013, 12, 1159–1169. [Google Scholar] [CrossRef]
- Yao, Y.; Higgs, B.W.; Morehouse, C.; de Los Reyes, M.; Trigona, W.; Brohawn, P.; White, W.; Zhang, J.; White, B.; Coyle, A.J.; et al. Development of Potential Pharmacodynamic and Diagnostic Markers for Anti-IFN-α Monoclonal Antibody Trials in Systemic Lupus Erythematosus. Hum. Genom. Proteom. 2009, 2009. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S.; et al. Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef]
- Merrill, J.T.; Furie, R.; Werth, V.P.; Khamashta, M.; Drappa, J.; Wang, L.; Illei, G.; Tummala, R. Anifrolumab effects on rash and arthritis: Impact of the type I interferon gene signature in the phase IIb MUSE study in patients with systemic lupus erythematosus. Lupus Sci. Med. 2018, 5, e000284. [Google Scholar] [CrossRef]
- Rice, G.I.; Melki, I.; Frémond, M.L.; Briggs, T.A.; Rodero, M.P.; Kitabayashi, N.; Oojageer, A.; Bader-Meunier, B.; Belot, A.; Bodemer, C.; et al. Assessment of Type I Interferon Signaling in Pediatric Inflammatory Disease. J. Clin. Immunol. 2017, 37, 123–132. [Google Scholar] [CrossRef]
- Kim, H.; Sanchez, G.A.; Goldbach-Mansky, R. Insights from Mendelian Interferonopathies: Comparison of CANDLE, SAVI with AGS, Monogenic Lupus. J. Mol. Med. 2016, 94, 1111–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Wu, H.; Grossman, J.M.; Hanvivadhanakul, P.; FitzGerald, J.D.; Park, G.S.; Dong, X.; Chen, W.; Kim, M.H.; Weng, H.H.; et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2951–2962. [Google Scholar] [CrossRef] [PubMed]
- Higgs, B.W.; Liu, Z.; White, B.; Zhu, W.; White, W.I.; Morehouse, C.; Brohawn, P.; Kiener, P.A.; Richman, L.; Fiorentino, D.; et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann. Rheum. Dis. 2011, 70, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.A.; Pinkus, J.L.; Pinkus, G.S.; Burleson, T.; Sanoudou, D.; Tawil, R.; Barohn, R.J.; Saperstein, D.S.; Briemberg, H.R.; Ericsson, M.; et al. Interferon-α/β-mediated innate immune mechanisms in dermatomyositis. Ann. Neurol. 2005, 57, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Salajegheh, M.; Kong, S.W.; Pinkus, J.L.; Walsh, R.J.; Liao, A.; Nazareno, R.; Amato, A.A.; Krastins, B.; Morehouse, C.; Higgs, B.W.; et al. Interferon-stimulated gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy. Ann. Neurol. 2010, 67, 53–63. [Google Scholar] [CrossRef]
- Uruha, A.; Nishikawa, A.; Tsuburaya, R.S.; Hamanaka, K.; Kuwana, M.; Watanabe, Y.; Suzuki, S.; Suzuki, N.; Nishino, I. Sarcoplasmic MxA expression: A valuable marker of dermatomyositis. Neurology 2017, 88, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R.; Serrano, R.; Pons-Estel, G.J.; Ceberio-Hualde, L.; Shoenfeld, Y.; de Ramón, E.; Buonaiuto, V.; Jacobsen, S.; Zeher, M.M.; Tarr, T.; et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: A multicentre prospective study of 1000 patients. Ann. Rheum. Dis. 2015, 74, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Palli, E.; Kravvariti, E.; Tektonidou, M.G. Type I Interferon Signature in Primary Antiphospholipid Syndrome: Clinical and Laboratory Associations. Front. Immunol. 2019, 10, 487. [Google Scholar] [CrossRef] [Green Version]
- Bernales, I.; Fullaondo, A.; Marín-Vidalled, M.J.; Ucar, E.; Martínez-Taboada, V.; López-Hoyos, M.; Zubiaga, A.M. Innate immune response gene expression profiles characterize primary antiphospholipid syndrome. Genes Immun. 2008, 9, 38–46. [Google Scholar] [CrossRef]
- Grenn, R.C.; Yalavarthi, S.; Gandhi, A.A.; Kazzaz, N.M.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Cabral, A.R.; McCune, W.J.; Bockenstedt, P.L.; Knight, J.S. Endothelial progenitor dysfunction associates with a type I interferon signature in primary antiphospholipid syndrome. Ann. Rheum. Dis. 2017, 76, 450–457. [Google Scholar] [CrossRef]
- van den Hoogen, L.L.; Fritsch-Stork, R.D.; Versnel, M.A.; Derksen, R.H.; van Roon, J.A.; Radstake, T.R. Monocyte type I interferon signature in antiphospholipid syndrome is related to proinflammatory monocyte subsets, hydroxychloroquine and statin use. Ann. Rheum. Dis. 2016, 75, e81. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Meng, H.; Coit, P.; Yalavarthi, S.; Sule, G.; Gandhi, A.A.; Grenn, R.C.; Mazza, L.F.; Ali, R.A.; Renauer, P.; et al. Activated signature of antiphospholipid syndrome neutrophils reveals potential therapeutic target. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fryer, J.F.; Baylis, S.A.; Gottlieb, A.L.; Ferguson, M.; Vincini, G.A.; Bevan, V.M.; Carman, W.F.; Minor, P.D. Development of working reference materials for clinical virology. J. Clin. Virol. 2008, 43, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; de Jesus, A.A.; Brooks, S.R.; Liu, Y.; Huang, Y.; VanTries, R.; Montealegre Sanchez, G.A.; Rotman, Y.; Gadina, M.; Goldbach-Mansky, R. Development of a Validated Interferon Score Using NanoString Technology. J. Interferon Cytokine Res. 2018, 38, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Faul, F.; Erdfelder, E.; Lang, A.G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodewes, I.L.A.; Björk, A.; Versnel, M.A.; Wahren-Herlenius, M. Innate immunity and interferons in the pathogenesis of Sjögren’s syndrome. Rheumatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Nezos, A.; Gravani, F.; Tassidou, A.; Kapsogeorgou, E.K.; Voulgarelis, M.; Koutsilieris, M.; Crow, M.K.; Mavragani, C.P. Type I and II interferon signatures in Sjogren’s syndrome pathogenesis: Contributions in distinct clinical phenotypes and Sjogren’s related lymphomagenesis. J. Autoimmun. 2015, 63, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Cantaert, T.; van Baarsen, L.G.; Wijbrandts, C.A.; Thurlings, R.M.; van de Sande, M.G.; Bos, C.; van der Pouw Kraan, T.C.; van der Pouw, T.K.; Verweij, C.L.; Tak, P.P.; et al. Type I interferons have no major influence on humoral autoimmunity in rheumatoid arthritis. Rheumatology 2010, 49, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-Delgado, J.E.; Bastián-Hernandez, Y.; Macias-Segura, N.; Santiago-Algarra, D.; Castillo-Ortiz, J.D.; Alemán-Navarro, A.L.; Martínez-Tejada, P.; Enciso-Moreno, L.; Garcia-De Lira, Y.; Olguín-Calderón, D.; et al. Type I Interferon Gene Response Is Increased in Early and Established Rheumatoid Arthritis and Correlates with Autoantibody Production. Front. Immunol. 2017, 8, 285. [Google Scholar] [CrossRef] [Green Version]
- Ekholm, L.; Vosslamber, S.; Tjärnlund, A.; de Jong, T.D.; Betteridge, Z.; McHugh, N.; Plestilova, L.; Klein, M.; Padyukov, L.; Voskuyl, A.E.; et al. Autoantibody Specificities and Type I Interferon Pathway Activation in Idiopathic Inflammatory Myopathies. Scand. J. Immunol. 2016, 84, 100–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brkic, Z.; van Bon, L.; Cossu, M.; van Helden-Meeuwsen, C.G.; Vonk, M.C.; Knaapen, H.; van den Berg, W.; Dalm, V.A.; Van Daele, P.L.; Severino, A.; et al. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann. Rheum. Dis. 2016, 75, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Quartuccio, L.; Mavragani, C.P.; Nezos, A.; Gandolfo, S.; Tzioufas, A.G.; De Vita, S. Type I interferon signature may influence the effect of belimumab on immunoglobulin levels, including rheumatoid factor in Sjögren’s syndrome. Clin. Exp. Rheumatol. 2017, 35, 719–720. [Google Scholar]
- de Jong, T.D.; Vosslamber, S.; Blits, M.; Wolbink, G.; Nurmohamed, M.T.; van der Laken, C.J.; Jansen, G.; Voskuyl, A.E.; Verweij, C.L. Effect of prednisone on type I interferon signature in rheumatoid arthritis: Consequences for response prediction to rituximab. Arthritis Res. Ther. 2015, 17, 78. [Google Scholar] [CrossRef]
- Wright, H.L.; Thomas, H.B.; Moots, R.J.; Edwards, S.W. Interferon gene expression signature in rheumatoid arthritis neutrophils correlates with a good response to TNFi therapy. Rheumatology 2015, 54, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Raterman, H.G.; Vosslamber, S.; de Ridder, S.; Nurmohamed, M.T.; Lems, W.F.; Boers, M.; van de Wiel, M.; Dijkmans, B.A.; Verweij, C.L.; Voskuyl, A.E. The interferon type I signature towards prediction of non-response to rituximab in rheumatoid arthritis patients. Arthritis Res. Ther. 2012, 14, R95. [Google Scholar] [CrossRef] [PubMed]
- Everaert, C.; Luypaert, M.; Maag, J.L.V.; Cheng, Q.X.; Dinger, M.E.; Hellemans, J.; Mestdagh, P. Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data. Sci. Rep. 2017, 7, 1559. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Subjects | n Total (F/M) | Method (n) | Purpose |
|---|---|---|---|---|
| A—Data of from our center; Accession: # | Healthy donors | 11 (5/6) | qPCR (10) RNAseq (3) | To test the variability of expression of the six ISGs in a healthy donor small group available at out center |
| RNAseq (1) | To increase the healthy donor group size, to improve the power of the variability measurement | |||
| B—Accession: E-MTAB-5735 | Healthy donors | 5 (2/3) | RNAseq (5) | To increase the healthy donor group size, to improve the power of the variability measurement |
| C—Accession: GSE112057 | Healthy donors | 12 (6/6) | RNAseq (9) | To increase the healthy donor group size, to improve the power of the variability measurement |
| D—Accession: GSE90081 | Healthy donors | 12 (12/0) | RNAseq (2) | To increase the healthy donor group size, to improve the power of the variability measurement |
| E—Patients and patient’s relatives recruited at our center | Patients | 20 (9/11) | qPCR (20) RNAseq (20) | To compare IFN signature results between qPCR and RNAseq analyses |
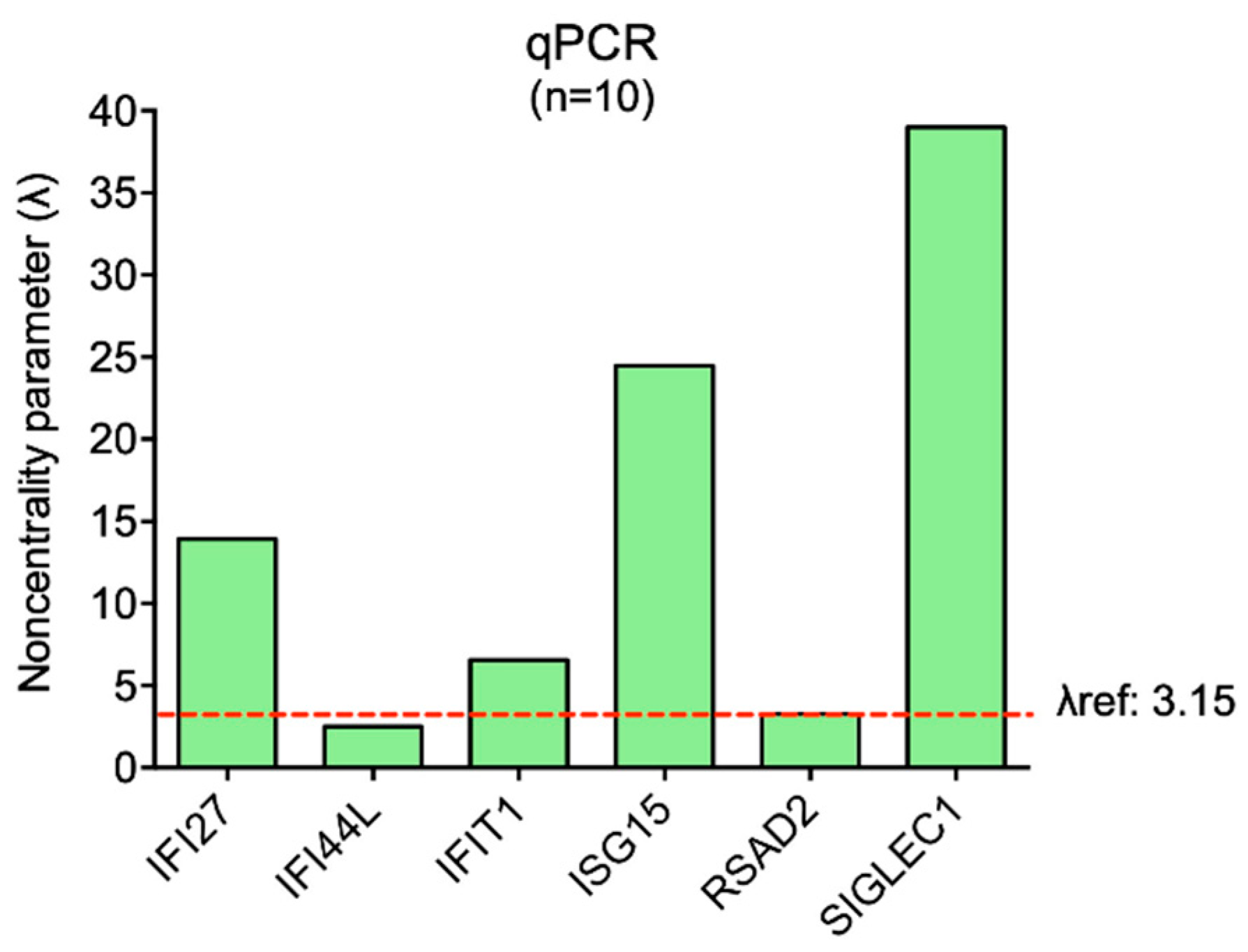
| IFI27 | IFI44L | IFIT1 | ISG15 | RSAD2 | SIGLEC1 | |
|---|---|---|---|---|---|---|
| Mean | 4.57 | 0.72 | 2.18 | 4.61 | 1.24 | 5.14 |
| SD | 1.04 | 0.91 | 1.05 | 0.60 | 1.20 | 0.42 |
| Variability coefficient | 0.23 | 1.26 | 0.48 | 0.13 | 0.96 | 0.08 |
| λ (n = 10) | 13.92 | 2.52 | 6.56 | 24.47 | 3.28 | 38.99 |
| λref: 3.15 |
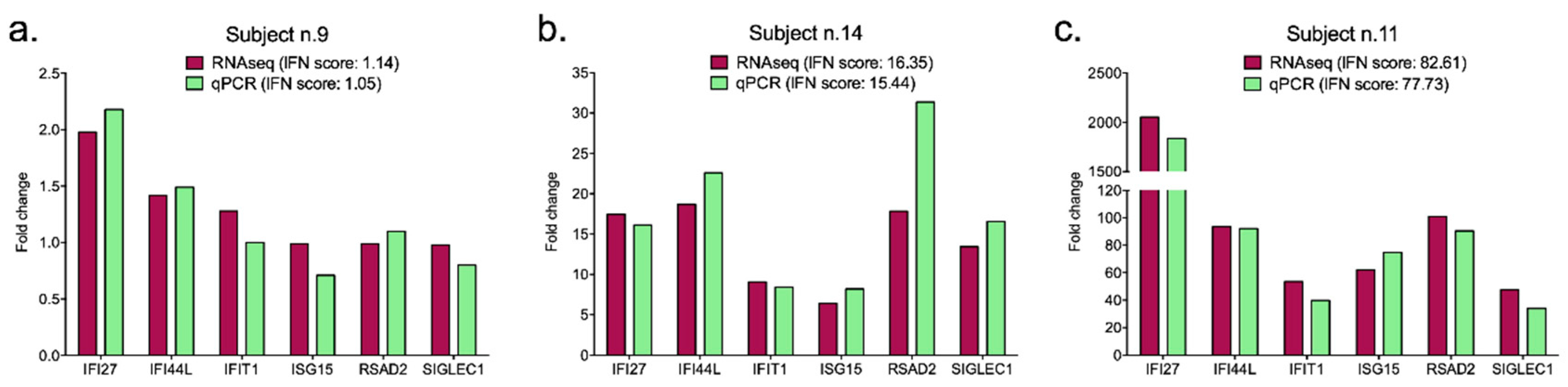
| Subject n. | IFN Score | |
|---|---|---|
| In Silico RNAseq | On Wet qPCR | |
| 1 | 3.26 | 5.01 |
| 2 | 6.79 | 10.05 |
| 3 | 7.12 | 9.85 |
| 4 | 8.58 | 10.74 |
| 5 | 1.55 | 1.37 |
| 6 | 1.11 | 0.97 |
| 7 | 0.55 | 0.67 |
| 8 | 0.22 | 0.19 |
| 9 | 1.14 | 1.05 |
| 10 | 3.68 | 2.60 |
| 11 | 77.73 | 82.61 |
| 12 | 44.03 | 94.25 |
| 13 | 17.44 | 16.48 |
| 14 | 15.44 | 16.35 |
| 15 | 37.43 | 49.98 |
| 16 | 37.44 | 84.99 |
| 17 | 1.07 | 1.18 |
| 18 | 2.39 | 4.70 |
| 19 | 3.01 | 1.71 |
| 20 | 1.82 | 1.82 |
| Mean | 19.83 | 13.59 |
| SD | 31.19 | 20.33 |
| Variability coefficient | 1.57 | 1.50 |
| Datasets | Subjects | RNAseq Details | |||
|---|---|---|---|---|---|
| Authors & Accession | Female (n = 9) | Male (n = 11) | Whole Blood collection/RNA extraction | RNAseq library preparation/platform | Read Length |
| A—Data from our center; Accession:# | 2 | 2 | PAXgene blood RNA tube/PAXgene Blood RNA Kit | Illumina TruSeq stranded mRNA library protocol/Novaseq | Paired-end 100 bp reads |
| B—Rodero MP, et al., 2017; Accession: E-MTAB-5735 | 2 | 3 | PAXgene blood RNA tube/PAXgene Blood RNA Kit | Illumina TruSeq stranded mRNA library protocol/Illumina HiSeq 2000 | Paired-end 75 bp reads |
| C—Mo A., et al., 2018; Accession: GSE112057 | 3 | 6 | Tempus Tube/Tempus Spin isolation RNA kit | Illumina TruSeq stranded mRNA library protocol/Illumina HiSeq Rapid Run | Paired-end 100 bp reads |
| D—Shchetynsky K., et al., 2017; Accession: GSE90081 | 2 | - | PAXgene blood RNA tube/PAXgene Blood miRNA kit | Standard illumina TruSeq RNA protocol, following PolyA enrichment/Illumina HiSeq 2000 | Paired-end 100 bp reads |
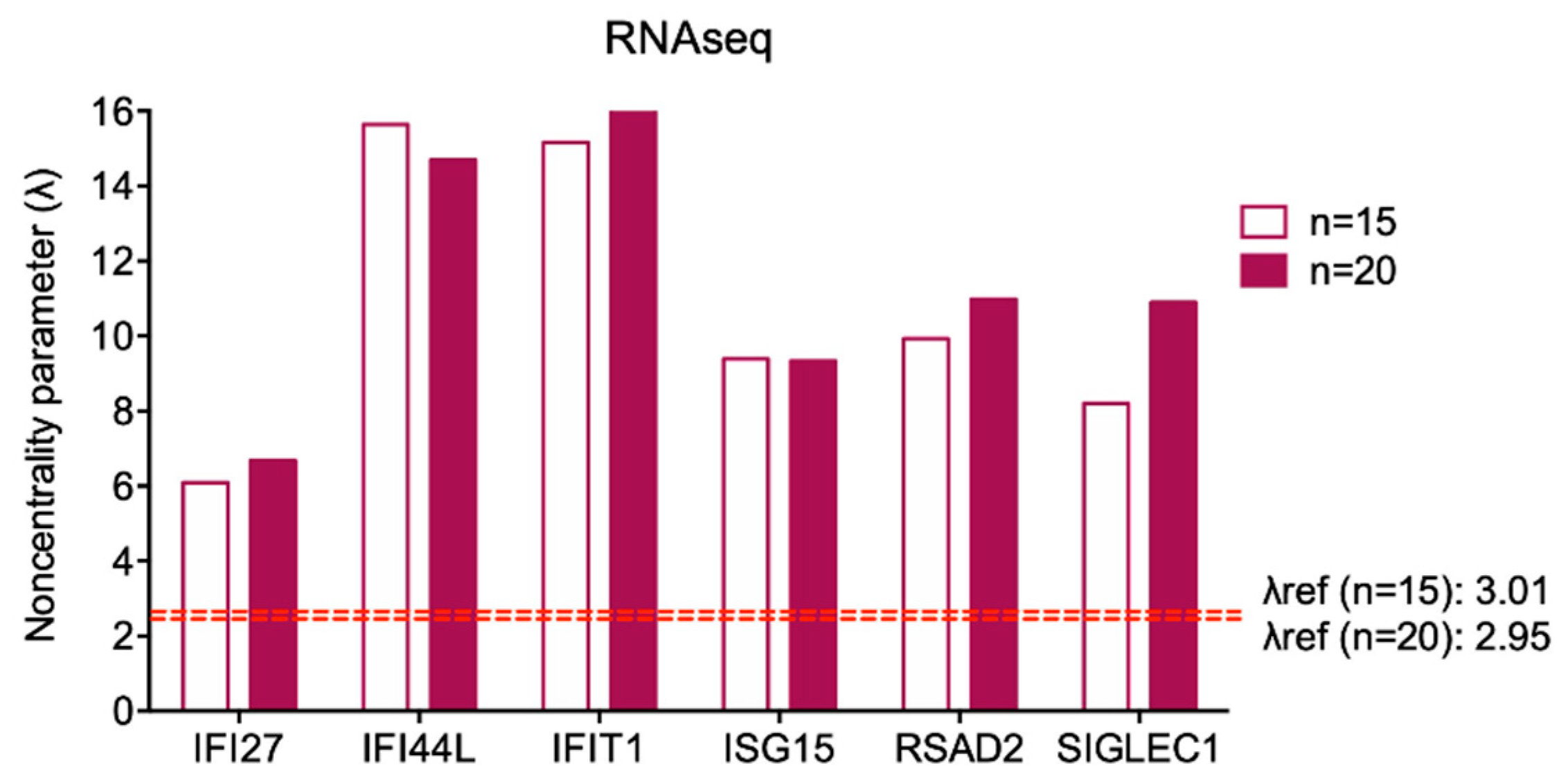
| IFI27 | IFI44L | IFIT1 | ISG15 | RSAD2 | SIGLEC1 | ||
|---|---|---|---|---|---|---|---|
| n = 15 | Mean | 0.35 | 1.14 | 5.06 | 30.17 | 2.21 | 1.17 |
| SD | 0.23 | 0.28 | 1.29 | 12.44 | 0.86 | 0.55 | |
| Variability coefficient | 0.64 | 0.25 | 0.26 | 0.41 | 0.39 | 0.47 | |
| λ | 6.09 | 15.64 | 15.16 | 9.39 | 9.93 | 8.21 | |
| Λref: 3.01 | |||||||
| n = 20 | Mean | 0.33 | 1.26 | 5.17 | 30.09 | 2.47 | 1.22 |
| SD | 0.22 | 0.38 | 1.38 | 14.41 | 1.00 | 0.50 | |
| Variability coefficient | 0.67 | 0.30 | 0.27 | 0.48 | 0.41 | 0.41 | |
| λ | 6.68 | 14.70 | 16.77 | 9.34 | 10.98 | 10.90 | |
| λref: 2.95 |
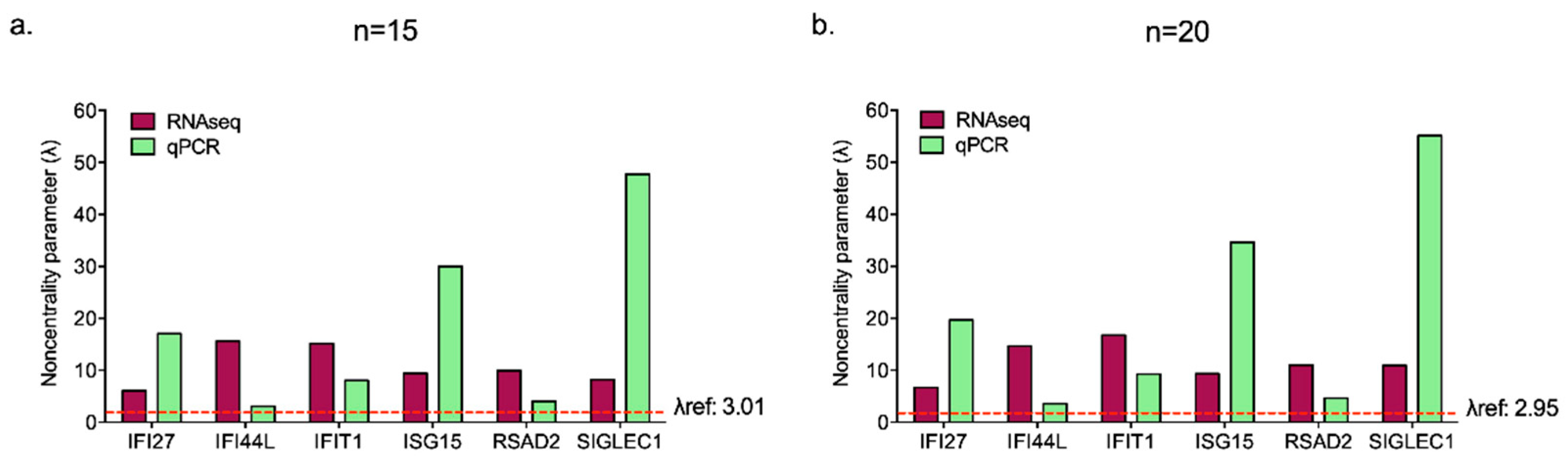
| IFI27 | IFI44L | IFIT1 | ISG15 | RSAD2 | SIGLEC1 | |
|---|---|---|---|---|---|---|
| Mean | 4.57 | 0.72 | 2.18 | 4.61 | 1.24 | 5.14 |
| SD | 1.04 | 0.91 | 1.05 | 0.60 | 1.20 | 0.42 |
| Variability coefficient | 0.23 | 1.26 | 0.48 | 0.13 | 0.96 | 0.08 |
| λ (n = 15) | 17.05 | 3.08 | 8.04 | 29.97 | 4.02 | 47.75 |
| λref: 3.01 | ||||||
| λ (n = 20) | 19.69 | 3.56 | 9.28 | 34.61 | 4.64 | 55.14 |
| λref: 2.95 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pin, A.; Monasta, L.; Taddio, A.; Piscianz, E.; Tommasini, A.; Tesser, A. An Easy and Reliable Strategy for Making Type I Interferon Signature Analysis Comparable among Research Centers. Diagnostics 2019, 9, 113. https://doi.org/10.3390/diagnostics9030113
Pin A, Monasta L, Taddio A, Piscianz E, Tommasini A, Tesser A. An Easy and Reliable Strategy for Making Type I Interferon Signature Analysis Comparable among Research Centers. Diagnostics. 2019; 9(3):113. https://doi.org/10.3390/diagnostics9030113
Chicago/Turabian StylePin, Alessia, Lorenzo Monasta, Andrea Taddio, Elisa Piscianz, Alberto Tommasini, and Alessandra Tesser. 2019. "An Easy and Reliable Strategy for Making Type I Interferon Signature Analysis Comparable among Research Centers" Diagnostics 9, no. 3: 113. https://doi.org/10.3390/diagnostics9030113