Non-Invasive Screening Tools for Down’s Syndrome: A Review

Abstract
:1. Introduction
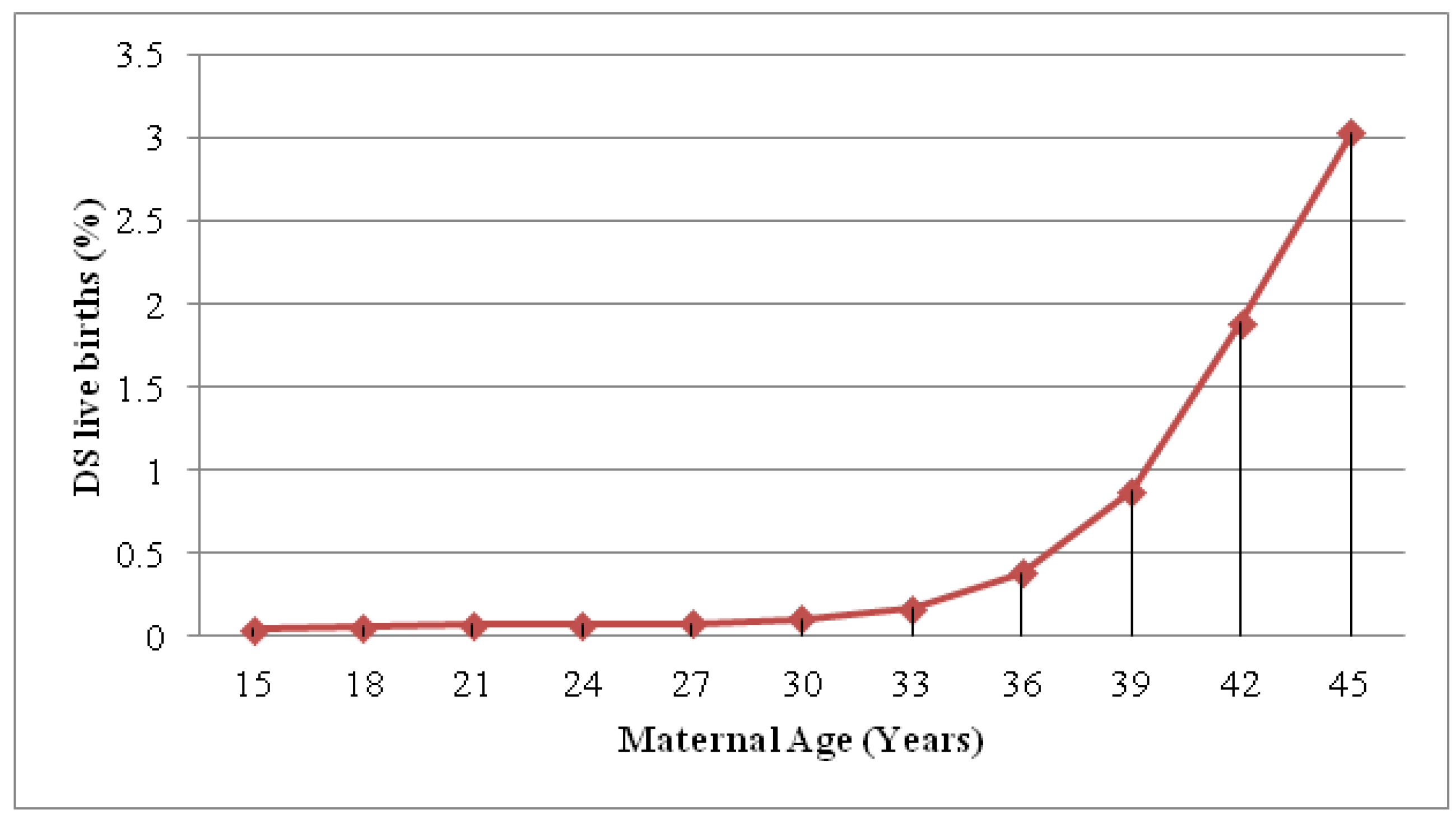
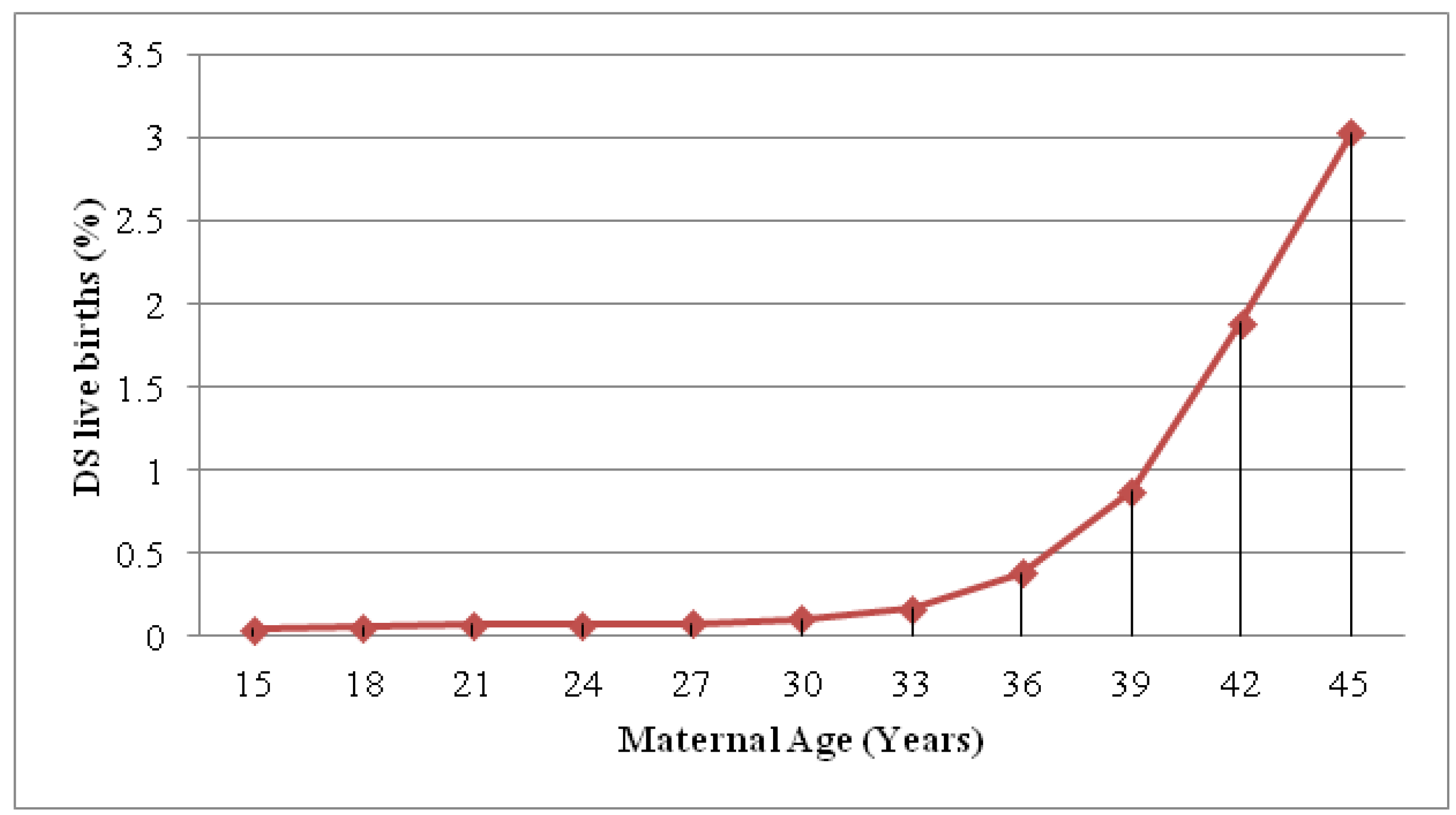
2. Definitions

3. Screening: Past to Present
3.1. Historical Overview
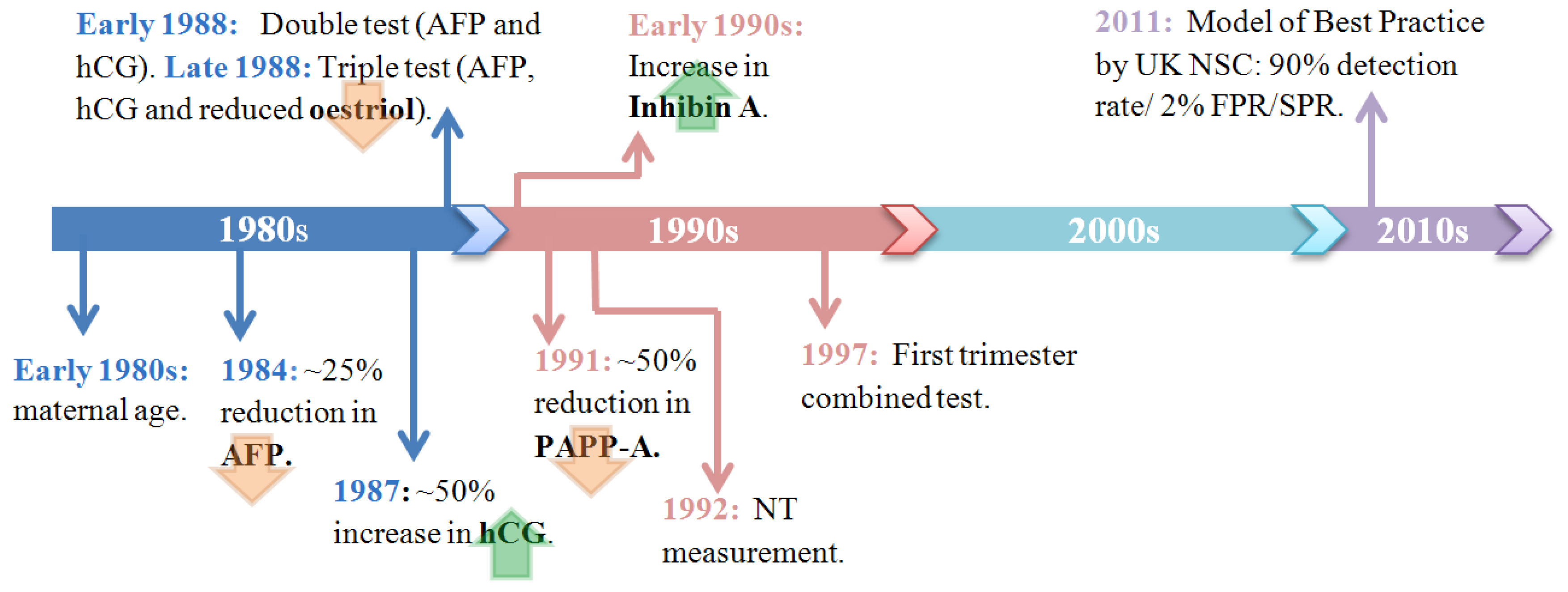
3.2. Current Methods
4. Further Developments
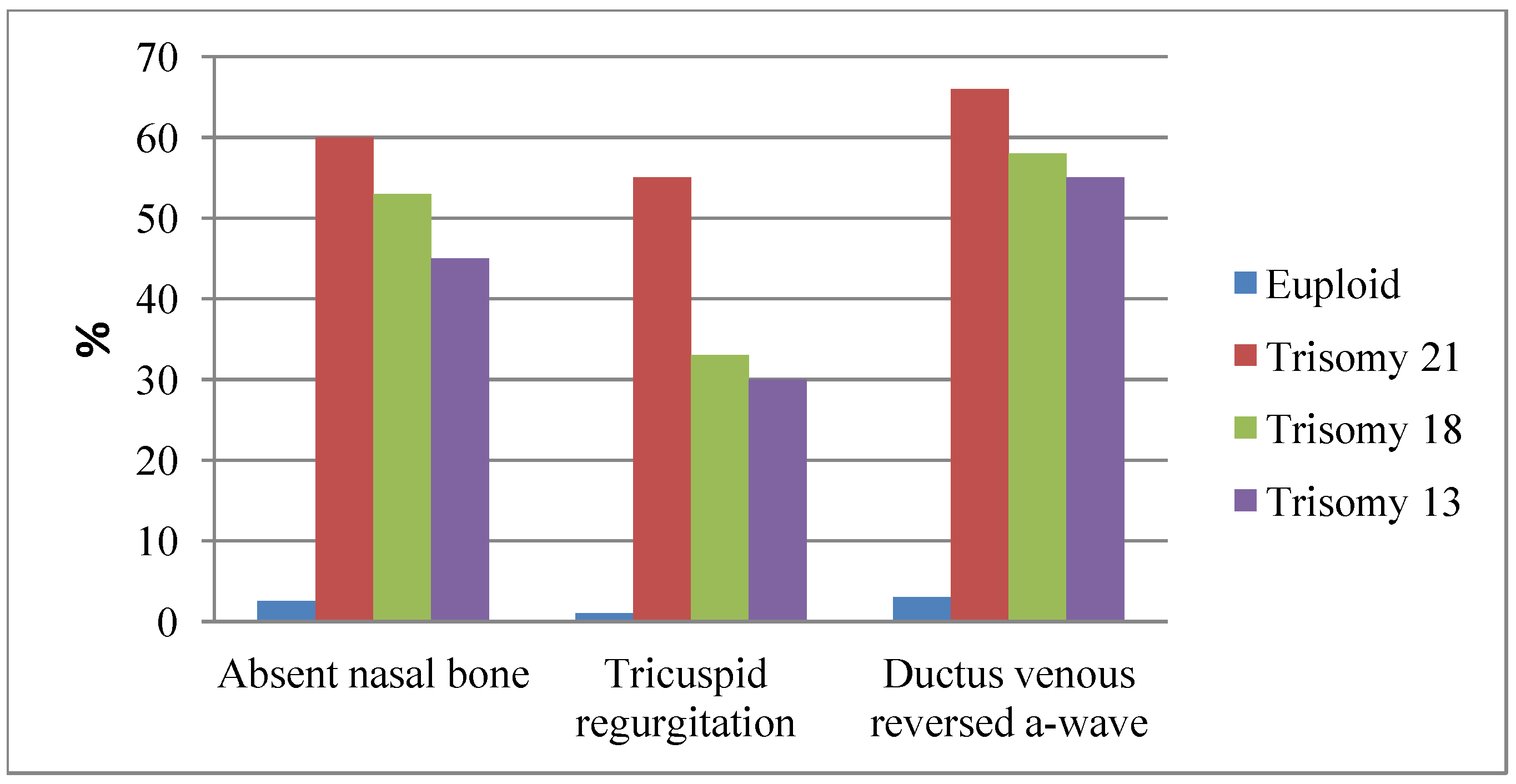
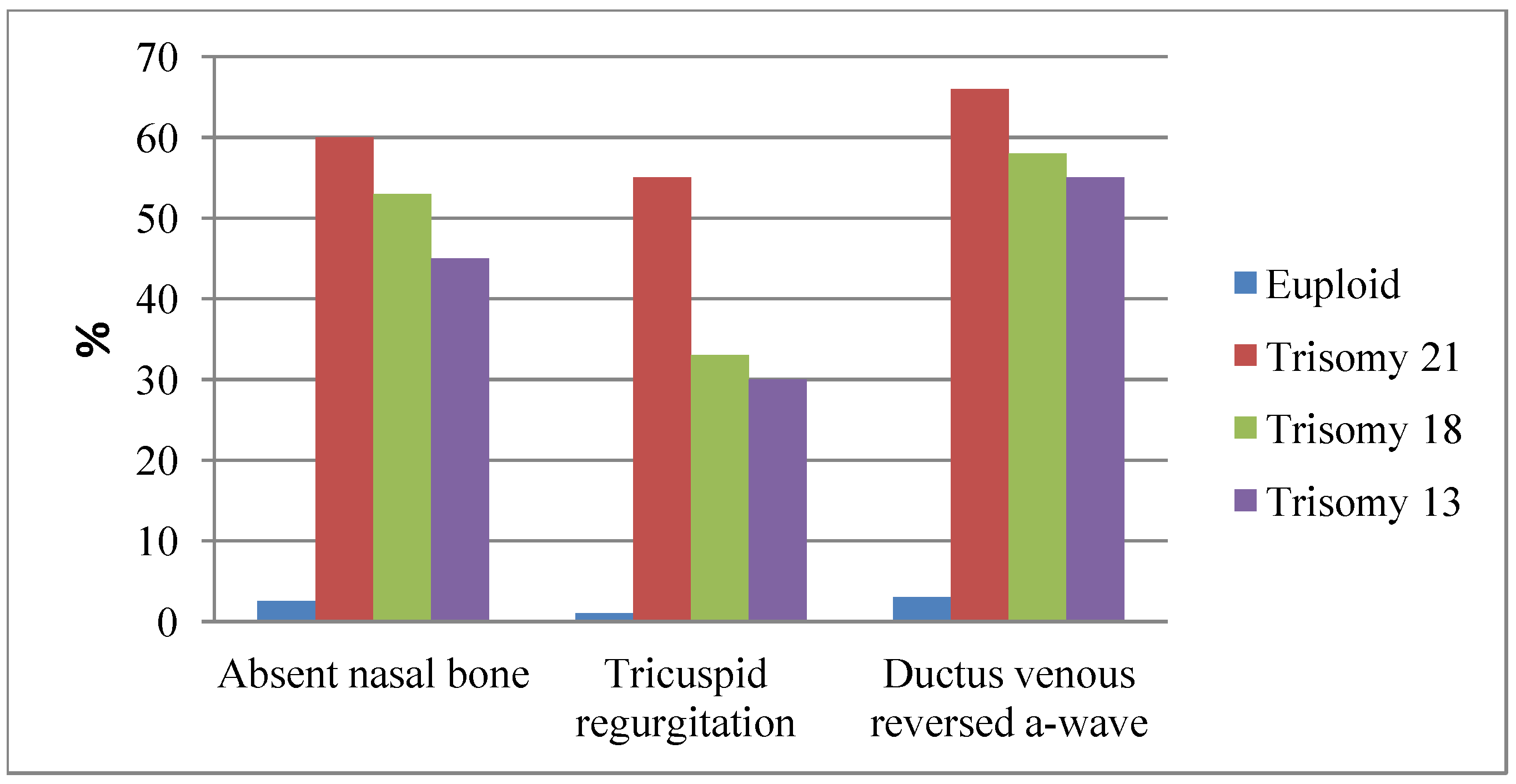
4.1. Sonographic Markers of DS
4.2. New Serum Biomarkers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Epigenetic Markers | |||
|---|---|---|---|
| Study | Marker | Assay | Results |
| Cowens et al. [54] | Placental growth factor (PlGF) | DELFIA Xpress immunoassay platform. | Increase during early first trimester in affected DS pregnancies (1 MoM in unaffected pregnancies, 1.3 MoM in DS pregnancies, p < 0.0001). |
| Wang et al. [64] | ADAM12 | Auto DELFIA/DELFIA ADAM12 Research kit (PerkinElmer Life and Analytical Sciences, Finland). | Reduction during early first-trimester in affected DS pregnancies (1 MoM in unaffected pregnancies, 1.26 MoM in DS pregnancies, p < 0.05). |
| Akinlade et al. [55] | CA15-3C A19-9 | Quantified by the Kryptor Analyzer. | No difference between euploid and DS pregnancies. Significantly elevated in DS pregnancies. (0.98 MoM in euploid, 1.16 MoM in trisomy 21, p = 0.024). |
| Kamyab et al. [56] | DSCAM DYRK1A | Multiplex assay with cytogenetic analysis and QF-PCR. | The mean gene dosage rate was significantly increased for both genes in DS pregnancies compared to euploid pregnancies (p < 0.001). |
| Epigenetic Markers | |||
| Lim et al. [57] | PDE9A | Quantitative methylation specific-PCR. | M-PDE9A (maternal) did not differ between pregnancies, but levels of U-PDE9A (fetal) were significantly higher in DS pregnancies. |
| Du et al. [58] | DSCR4 | Methylation specific primers and digital PCR. | Hypomethylated in placental tissue and methylated in maternal cells. Can detect and quantify unmethylated DSCR4 in the first-trimester maternal plasma, successfully detect DS by RCD against a reference gene (e.g., ZFY). |
| Chim et al. [65] | SERPINB5 (coding for Maspin) | Bisulphite genomic sequencing and RT-Quantitative methylation-specific PCR. | Hypomethylated in placental tissue and methylated in maternal cells. SERPINB5 was the first fetal-specific hypomethylated gene to be identified in maternal plasma. |
4.3. Digital PCR and Next Generation Sequencing (NGS)

| Study | Method | DR (%) | FPR (%) |
|---|---|---|---|
| Chiu et al. [90] | Shotgun (2-plex protocol) | 100 | 2.1 |
| Chiu et al. [90] | Shotgun (8-plex protocol) | 79.1 | 1.2 |
| Ehrich et al. [1] | Shotgun | 100 | 0.2 |
| Bianchi et al. [91] | Shotgun | 100 | 0 |
| Jensen et al. [92] | Shotgun | 100 | 0.9 |
| Sparks et al. [93] | Targeted | 100 | 0.8 |
| Ashoor et al. [88] | Targeted | 100 | 0 |
| Norton et al. [94] | Targeted | 100 | 0.1 |
| Liang et al. [95] | Targeted | 100 | 0 |
| PCR-based sequencing | Single end reads per run | Run Time | |
|---|---|---|---|
| HiSeq™2000 (Illumina, Inc.) | Sequencing-by-synthesis | 3 billion | 5–14 days |
| HiSeq™2500 (rapid run) (Illumina, Inc.) | Sequencing-by-synthesis | ~300 million (10 Gb) | 7 h |
| SOLiD4™ (Life Technologies™/Applied Biosystems ™) | Sequencing-by-ligation | ~0.7 billion | 5–10 days |
| HeliScope® Single Molecule Sequencer (Helicos™Biosciences) | Single-molecule-sequencing-by-synthesis | ~840 million (28 Gb) | 8 days |
| Benchtop: MiSeq™ (Illumina, Inc.) | Sequence-by-synthesis | ~12 million (3.4 Gb) | 16.5 h |
| Benchtop: Ion Torrent™ (Life Technologies™) | Semiconductor sequencing technology | ~5 million (1 Gb) | 4.4 h |
| Company | Test | Released | Trisomies Tested | Genetic Testing Method | Accuracy | Sensitivity | Cost |
|---|---|---|---|---|---|---|---|
| Sequenom | MaterniT21 Plus | February 2012 | 13, 18, 21, sex chromo-somes | MPSS | >99% | 92–99% | $2,762 |
| Verinata | Verifi Prenatal Test | March 2012 | 13, 18, 21, sex chromo-somes | MPSS | 100% | 87–99% | $1,500 |
| Aria Diagnostics | Harmony Prenatal Test | May 2012 | 13, 18, 21 | Chromosome-selective sequencing | >99% | 80–99% | $795 |
| Natera | Panorama | March 2013 | 13, 18, 21 | Single nucleotide polymorphism | 100% | 92–99% | $1,495 |
5. Conclusions
Conflict of Interest
References
- Ehrich, M.; Deciu, C.; Zwiefelhofer, T.; Tynan, J.A.; Cagasan, L.; Tim, R.; Lu, V.; McCullough, R.; McCarthy, E.; Nygren, A.O. Nonivasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: A study in a clinical setting. Am. J. Obstet. Gynecol. 2011, 204, 201–211. [Google Scholar]
- Egan, J.F.; Benn, P.A.; Zelop, C.M.; Bolnick, A.; Gianferrari, E.; Borgida, A.F. Down syndrome births in the United States from 1989 to 2001. Am. J. Obstet. Gynecol. 2004, 191, 1044–1048. [Google Scholar] [CrossRef]
- Morris, J.K.; Mutton, D.E.; Alberman, E. Revised estimates of the maternal age specific live birth prevalence of Down’s syndrome. J. Med. Screen 2002, 9, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Crane, E.; Morris, J.K. Changes in maternal age in England and Wales—Implications for Down syndrome. Down Syndr. Res. Prac. 2006, 10, 41–43. [Google Scholar] [CrossRef]
- McEwan, A.; Godfrey, A.; Wilkins, J. Screening for Down syndrome. Obstet. Gynaecol. Reprod. Med. 2012, 22, 70–75. [Google Scholar] [CrossRef]
- Rozenberg, P.; Bussières, L.; Chevret, S.; Bernard, J.P.; Malagrida, L.; Cuckle, H.; Chabry, C.; Durand-Zaleski, I.; Bidat, L.; Lacroix, I. Screening for Down syndrome using first-trimester combined screening followed by second trimester ultrasound examination in an unselected population. Gynecol. Obstet. Fertil. 2007, 35, 303–311. [Google Scholar] [CrossRef]
- Morris, J.K.; Wald, N.J.; Watt, H.C. Fetal loss in Down syndrome pregnancies. Prenat. Diagn. 1999, 19, 142–145. [Google Scholar] [CrossRef]
- Chiu, R.W.K.; Cantor, C.R.; Lo, Y.M.D. Non-invasive prenatal diagnosis by single molecule counting technologies. Trends Genet. 2009, 25, 324–331. [Google Scholar] [CrossRef]
- Ferguson-Smith, M.A. Placental mRNA in maternal plasma: Prospects for fetal screening. Proc. Natl. Acad. Sci. USA 2003, 100, 4360–4362. [Google Scholar] [CrossRef]
- Wald, N.J. Guidance on terminology. J. Med. Screen 2006, 13, 53. [Google Scholar] [CrossRef]
- Buckley, F.; Buckley, S. Wrongful deaths and rightful lives—Screening for Down syndrome. Down Syndr. Res. Prac. 2008, 12, 79–86. [Google Scholar] [CrossRef]
- Haddow, J.E.; Palomaki, G.E.; Knight, G.J.; Williams, J.; Pulkkinen, A.; Canick, J.A.; Saller, D.N.; Bowers, G.B. Prenatal screening for Down’s syndrome with use of maternal serum markers. N. Engl. J. Med. 1992, 327, 588–593. [Google Scholar] [CrossRef]
- Benacerraf, B.R.; Gelman, R.; Friholetto, F.D. Sonographic identification of second-trimester fetuses with Down’s syndrome. N. Engl. J. Med. 1987, 317, 1371–1376. [Google Scholar] [CrossRef]
- Merkatz, I.; Nitowsky, H.; Marci, J.; Johnson, W. An association between low maternal serum alpha-fetoprotein and fetal chromosomal abnormalities. Am. J. Obstet. Gynecol. 1984, 148, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Cuckle, H.; Wald, N.; Lindenbaum, R. Maternal serum alpha-fetoprotein measurement: A screening test for Down syndrome. Lancet 1984, 1, 926–929. [Google Scholar] [CrossRef]
- Furhmann, W.; Wendt, P.; Weitzel, H. Maternal serum-AFP as screening test for Down syndrome. Lancet 1984, 2. [Google Scholar] [CrossRef]
- Gillespie, G.; Uversky, V. Structure and function of alpha-fetoprotein: A biophysical overview. Biochem. Biophys. Acta 2000, 1480, 41–56. [Google Scholar] [PubMed]
- DiMaio, M.; Baumgarten, A.; Greenstein, R.; Mahoney, M. Screening for fetal Down’s syndrome in pregnancy by measuring maternal serum alpha-fetoprotein levels. N. Engl. J. Med. 1987, 6, 342–346. [Google Scholar]
- Cole, L. New discoveries on the biology and detection of human chorionic gonadotrophin. Reprod. Biol. Endocrinol. 2009, 7. [Google Scholar] [CrossRef]
- Bogart, M.; Pandian, M.; Jones, O. Abnormal maternal serum chorionic gonadotropin levels in pregnancies with fetal chromosome abnormalities. Prenat. Diagn. 1987, 7, 623–630. [Google Scholar] [CrossRef]
- Agarwal, R. Prenatal diagnosis of chromosomal anomalies: Pictorial essay. Indian J. Radiol. Imaging 2003, 13, 173–188. [Google Scholar]
- Canick, J.; Kellner, L. First trimester screening for aneuploidy: Serum biochemical markers. Semin. Perinatol. 1999, 5, 359–368. [Google Scholar] [CrossRef]
- Crossley, J.; Aitken, D.; Connor, J. Second-trimester unconjugated oestriol levels in maternal serum from chromosomally abnormal pregnancies using an optimized assay. Prenat. Diagn. 1993, 13, 271–280. [Google Scholar] [CrossRef]
- Ryall, R.; Staples, A.; Robertson, E.; Pollard, A. Improved performance in a prenatal screening programme for Down’s syndrome incorporating serum-free hCG subunit analyses. Prenat. Diagn. 1992, 12, 251–261. [Google Scholar] [CrossRef]
- Reynolds, T. The triple test as a screening technique for Down syndrome: Reliability and relevance. Int. J. Wom. Health 2010, 2010, 83–88. [Google Scholar] [CrossRef]
- Aitken, D.A.; Wallace, E.M.; Crossley, J.A.; Swanston, I.A.; Pareren, Y.V.; Maarle, M.V.; Groome, N.P.; Macri, J.N.; Connor, M.J. Dimeric inhibin A as a marker for Down’s syndrome in early pregnancy. N. Engl. J. Med. 1996, 334, 1231–1236. [Google Scholar] [CrossRef]
- Brambati, B.; Macintosh, M.C.; Teisner, B.; Maguiness, S.; Shrimanker, K.; Lanzani, A.; Bonacchi, I.; Tului, L.; Chard, T.; Grudzinskas, J.G. Low maternal serum levels of pregnancy associated plasma protein a (PAPP-A) in the first trimester in association with abnormal fetal karyotype. Br. J. Obstet. Gynaecol. 1993, 100, 324–326. [Google Scholar] [CrossRef]
- Knight, G.; Palomaki, G.; Haddow, J.E. Pregnancy associated plasma protein A as a marker for Down syndrome in the second trimester of pregnancy. Prenat. Diagn. 1993, 13, 222–223. [Google Scholar] [CrossRef]
- Reis, F.; D’Antona, D.; Petraglia, F. Predictive value of hormone measurements in maternal and fetal complications of pregnancy. Endo Rev. 2002, 23, 230–257. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Azar, G.; Byrne, D.; Mansur, C.; Marks, K. Fetal nuchal translucency: Ultrasound screening for chromosomal defects in first trimester of pregnancy. BMJ 1992, 304, 867–869. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Brizot, M.L.; Snijders, R.J. Fetal nuchal translucency: Ultrasound screening for chromosomal defects in first trimester of pregnancy. J. Obstet. Gynaecol. 1994, 101, 782–786. [Google Scholar] [CrossRef]
- Committee, T.N.S. National Down’s Syndrome Screening Program (DoSYSP). Available online: http://fetalanomaly.screening.nhs.uk/standardsandpolicies (accessed on 20 May 2013).
- Chitty, L.S.; Pandya, P.P. Ultrasound screening for fetal abnormalities in the first trimester. Prenat. Diagn. 1997, 17, 1269–1281. [Google Scholar] [CrossRef]
- Pandya, P.P.; Altman, D.G.; Brizot, M.L.; Pettersen, H.; Nicolaides, K.H. Repeatability of measurement of fetal nuchal translucency thickness. Ultrasound Obstet. Gynecol. 1995, 5, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Wald, N.; Hackshaw, A.K. Combining ultrasound and biochemistry in first trimester screening for Down’s syndrome. Prenat. Diagn. 1997, 17, 821–829. [Google Scholar] [CrossRef]
- Spencer, K.; Nicolaides, K.H. Screening for trisomy 21 in twins using first trimester ultrasound and maternal serum biochemistry in a one-stop clinic: A review of three years experience. Br. J. Obstet. Gynaecol. 2003, 110, 276–280. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Spencer, K.; Avqidou, K.; Faiola, S.; Falcon, O. Multicenter study of first-trimester screening for trisomy 21 in 75,821 pregnancies: Results and estimation of the potential impact of individual risk-orientated two-stage first-trimester screening. Ultrasound Obstet. Gynecol. 2005, 25, 221–226. [Google Scholar] [CrossRef]
- Jaques, A.M.; Halliday, J.L.; Francis, I.; Bonacquisto, L.; Forbes, R.; Cronin, A.; Sheffield, L.J. Follow up and evaluation of the victorian first-trimester combined screening programme for Down syndrome and trisomy 18. BJOG 2007, 114, 812–818. [Google Scholar] [CrossRef]
- Valinen, Y.; Rapakko, K.; Kokkonen, H.; Laitinen, P.; Tekay, A.; Ahola, T.; Ryynanen, M. Clinical first-trimester routine screening for Down syndrome in singleton pregnancies in northern Finland. Am. J. Obstet. Gynecol. 2007, 196, 278. [Google Scholar] [CrossRef]
- Wald, N.J.; Huttly, W.J.; Murphy, K.W.; Ali, K.; Bestwick, J.P.; Rodeck, C.H. Antenatal screening for Down’s syndrome using the integrated test at two London hospitals. J. Med. Screen 2009, 16, 7–10. [Google Scholar] [CrossRef]
- Benn, P.; Borell, A.; Chiu, R.; Cuckle, H.; Dugoff, L.; Faas, B.; Gross, S.; Johnson, J.; Maymon, R.; Norton, M.; et al. Position statement from the aneuploidy screening committee on behalf of the board of the international society for prenatal diagnosis, April 2013. Prenat. Diagn. 2013, 32, 1–2. [Google Scholar]
- NHS Fetal Anomaly Screening Programme. Screening for Down’s syndrome: UK NSC Policy Recommendations 2011–2014 Model of Best Practice. Available online: http://www.fetalanomaly. screening.nhs.uk/getdata.php?id=11393 (accessed on 13 May 2013).
- Fang, Y.M.V.; Benn, P.; Campbell, W.; Bolnick, J.; Prabulos, A.M.; Egan, J.F.X. Down syndrome screening in the united states in 2001 and 2007: A survey of maternal-fetal medicine specialists. Am. J. Obstet. Gynecol. 2009, 201, e91–e95. [Google Scholar]
- Benn, P.; Borrell, A.; Crossley, J.; Cuckle, H.; Dugoff, L.; Gross, S.; Johnson, J.-A.; Maymon, R.; Odibo, A.; Schielen, P.; et al. Aneuploidy screening: A position statement from a committee on behalf of the board of the international society for prenatal diagnosis, January 2011. Prenat. Diagn. 2011, 31, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Maslen, C.L.; Babcock, D.; Robinson, S.W.; Bean, L.J.; Dooley, K.J.; Willour, V.L.; Sherman, S.L. Creld1 mutations contribute to the occurrence of cardiac atrioventricular septal defects in Down syndrome. Am. J. Med. Genet. 2006, 140, 2501–2505. [Google Scholar] [PubMed]
- Nyberg, D.; Souter, V. Sonographic markers of fetal trisomies: Second trimester. J. Ultrasound Med. 2001, 20, 655–674. [Google Scholar] [PubMed]
- Stoll, C.; Dott, B.; Alembik, Y.; Roth, M. Evaluation of routine prenatal ultrasound examination in detecting fetal chromosomal abnormalities in a low risk population. Hum. Genet. 1993, 91, 37–41. [Google Scholar] [PubMed]
- Hill, L. The sonographic detection of trisomies 13, 18 and 21. Clin. Obstet. Gynecol. 1996, 39, 831–850. [Google Scholar] [CrossRef] [PubMed]
- Kagan, K.O.; Etchegaray, A.; Zhou, Y.; Wright, D.; Nicolaides, K.H. Prospective validation of first-trimester combined screening for trisomy 21. Ultrasound Obstet. Gynecol. 2009, 34, 14–18. [Google Scholar] [PubMed]
- Nicolaides, K. Screening for fetal aneuploidies at 11 to 13 weeks. Prenat. Diagn. 2011, 31, 7–15. [Google Scholar] [CrossRef]
- Smith-Bindman, R.; Hosmer, W.; Feldstein, V.A.; Deeks, J.J.; Goldberg, J.D. Second-trimester ultrasound to detect fetuses with Down syndrome: A meta-analysis. JAMA 2001, 285, 1044–1055. [Google Scholar] [CrossRef]
- Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Tsui, D.W.; Chan, K.C.; Chim, S.S.; Chan, L.W.; Leung, T.Y.; Lau, T.K.; Lo, Y.M.; Chiu, R.W. Quantitative aberrations of hypermethylated RASSF1A gene sequences in maternal plasma in pre-eclampsia. Prenat. Diagn. 2007, 27, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Cowans, N.; Stamatopoulou, A.; Torring, N.; Spencer, K. Early first trimester maternal serum placental growth factor in trisomy 21 pregnancies. Ultrasound Obstet. Gynecol. 2011, 37, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Akinlade, F.; Cowans, N.; Kisanga, M.; Spencer, K. Maternal serum CA 19-9 and CA 15-3 levels in pregnancies affected by trisomy 21. Prenat. Diagn. 2012, 32, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Kamyab, A.R.; Shahrokhi, F.; Shamsian, E.; Nosaied, M.H.; Dibajnia, P.; Hashemi, M.; Mahdian, R. Determination of sensitivity and specificity of a novel gene dosage assay for prenatal screening of trisomy 21 syndrome. Clin. Biochem. 2012, 45, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Kim, S.Y.; Park, S.Y.; Lee, S.Y.; Kim, M.J.; Han, Y.J.; Lee, S.W.; Chung, J.H.; Kim, M.Y.; Yang, J.H.; et al. Non-invasive epigenetic detection of fetal trisomy 21 in first trimester maternal plasma. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, J.; Wang, H.; Yan, X.; Yang, Y.; Yang, L.; Luo, X.; Chen, Y.; Duan, T.; Ma, D. Hypomethylated dscr4 is a placenta-derived epigenetic marker for trisomy 21. Prenat. Diagn. 2011, 31, 207–214. [Google Scholar] [CrossRef]
- Maddocks, D.G.; Alberry, M.S.; Attilakos, G.; Madgett, T.E.; Choi, K.; Soothill, P.W.; Avent, N.D. The SAFE project: Towards non-invasive prenatal diagnosis. Biochem. Soc. Trans. 2009, 37, 460–465. [Google Scholar] [CrossRef]
- Chitty, L.S.; van der Schoot, C.E.; Hahn, S.; Avent, N.D. SAFE-the special non-invasive advances in fetal and neonatal evaluation network: Aims and achievements. Prenat. Diagn. 2008, 28, 83–88. [Google Scholar] [CrossRef]
- Shipp, T.; Benacerraf, B. Second trimester ultrasound screening for chromosomal abnormalities. Prenat. Diagn. 2002, 22, 296–307. [Google Scholar] [CrossRef]
- Cicero, S.; Curcio, P.; Papageorghiou, A.; Sonek, J.; Nicolaides, K. Absence of nasal bone in fetuses with trisomy 21 at 11–14 weeks of gestation: An observational study. Lancet 2001, 358, 1665–1667. [Google Scholar] [CrossRef]
- Donaldson, K.; Turner, S.; Morrison, L.; Liitte, P.; Nilsson, C.; Cuckle, H. Maternal serum placental growth factor and α-fetoprotein testing in first trimester screening for Down syndrome. Prenat. Diagn. 2013, 33, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lu, S.; Zhu, Y.; Li, H. Adam12 is an effective marker in the second trimester of pregnancy for prenatal screening of Down syndrome. Prenat. Diagn. 2010, 30, 561–564. [Google Scholar] [PubMed]
- Chim, S.S.; Tong, Y.K.; Chiu, R.W.; Lau, T.K.; Leung, T.Y.; Chan, L.Y.S.; Oudejans, C.B.M.; Ding, C.; Lo, Y.M. Detection of the placental epignetic signature of the maspin gene in maternal plasma. Proc. Natl. Acad. Sci. USA 2005, 11, 14753–14758. [Google Scholar]
- Kang, Y.; Dong, X.; Zhou, Q.; Zhang, Y.; Cheng, Y.; Hu, R.; Su, C.; Jin, H.; Liu, X.; Ma, D.; et al. Identification of novel candidate maternal serum protein markers for Down syndrome by integrated proteomic and bioinformatic analysis. Prenat. Diagn. 2012, 32, 284–292. [Google Scholar] [CrossRef]
- Avent, N.D. Maternal plasma biomarkers for Down syndrome: Present and future. Drugs Today 2013, 49, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Miharu, N.; Ohashi, Y.; Samura, O.; Kinutani, M.; Hara, T.; Ohama, K. Fetal gender determination in early pregnancy through qualitative and quantitative analysis of fetal DNA in maternal serum. Hum. Genet. 2002, 110, 75–79. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Avent, N.D.; Costa, J.M.; van der Schoot, C.E. Noninvasive prenatal diagnosis of fetal rhesus D: Ready for prime(r) time. Obstet. Gynecol. 2005, 106, 841–844. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Hjelm, M.; Fidler, C.; Sargent, I.L.; Murphy, M.F.; Chamberlain, P.F.; Poon, P.M.K.; Redman, C.W.G.; Wainscoat, J.S. Prenatal diagnosis of fetal rhd status by molecular analysis of maternal plasma. N. Engl. J. Med. 1998, 339, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Satio, H.; Sekizawa, A.; Morimoto, T. Prenatal DNA diagnosis of a single-gene disorder from maternal plasma. Lancet 2000, 356. [Google Scholar] [CrossRef]
- Sehnert, A.J.; Rhees, B.; Comstock, D.; de Feo, E.; Heilek, G.; Burke, J.; Rava, R.P. Optimal detection of fetal chromosomal abnormalities by massively parallel DNA sequencing of cell-free fetal DNA from maternal blood. Clin. Chem. 2011, 57, 1042–1049. [Google Scholar] [CrossRef]
- Dan, S.; Chen, F.; Choy, K.W.; Jiang, F.; Lin, J.; Xuan, Z.; Wang, W.; Chen, S.; Li, X.; Jiang, H.; et al. Prenatal detection of aneuploidy and imbalanced chromosomal parallel sequencing. PLoS One 2011, 7. [Google Scholar] [CrossRef]
- Papageorgiou, E.A.; Patsalis, P.C. Non-invasive prenatal diagnosis of aneuploidies: New technologies and clinical applications. Genom. Med. 2012, 4. [Google Scholar] [CrossRef]
- Hulten, M.; Dhanjal, S.; Pertl, B. Rapid and simple prenatal diagnosis of common chromosome disorders: Advantages and disadvantages of the molecular methods fish and QF-PCR. Reproduction 2003, 126, 279–297. [Google Scholar] [CrossRef]
- Shaffer, L.; Bui, T. Molecular cytogenetic and rapid aneuploidy detection methods in prenatal diagnosis. Am. J. Med. Genet. 2007, 145, 87–98. [Google Scholar] [CrossRef]
- Zimmermann, B.; Holzgreve, W.; Wenzel, F.; Hahn, S. Novel real-time quantitative PCR test for trisomy 21. Clin. Chem. 2002, 48, 362–363. [Google Scholar] [PubMed]
- Vogelstein, B.; Kinzler, K. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9261. [Google Scholar] [CrossRef]
- White, R.; Blainey, P.; Fan, H.; Quake, S. Digital PCR provides sensitive and absolute calibration for high throughput sequencing. BMC 2009, 10, 116. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; El-Sayed, Y.Y.; Chueh, J.; Quake, S.R. Microfluidic digital PCR enables rapid prenatal diagnosis of fetal aneuploidy. Am. J. Med. Genet. 2009, 200, e541–e547. [Google Scholar]
- Zimmermann, B.G.; Grill, S.; Holzgreve, W.; Zhong, X.Y.; Jackson, L.G.; Hahn, S. Digital PCR: A powerful new tool for noninvasive prenatal diagnosis? Prenat. Diagn. 2008, 28, 1087–1093. [Google Scholar] [CrossRef]
- Lun, F.M.; Chiu, R.W.; Chan, K.C.; Leung, T.Y.; Lau, T.K.; Lo, Y.M. Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin. Chem. 2008, 54, 1664–1672. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Lun, F.M.; Chan, K.C.; Tsui, N.B.Y.; Chong, K.C.; Lau, T.K.; Leung, T.Y.; Zee, T.Y.; Cantor, C.R.; Chiu, R.W. Digital PCR for the molecular detection of fetal chromosomal aneuploidy. Proc. Natl. Acad. Sci. USA 2007, 104, 13116–13121. [Google Scholar] [CrossRef]
- Lo, Y.M.; Chan, K.C.; Chiu, R.W. Nonivasive fetal trisomy 21 detection using chromosom selective sequencing: A variation of the molecular counting theme. Expert Rev. Mol. Diagn. 2012, 12, 329–331. [Google Scholar] [CrossRef]
- Evans, M.I.; Wright, D.A.; Pergament, E.; Cuckle, H.S.; Nicolaides, K.H. Digital PCR for noninvasive detection of aneuploidy: Power analysis equations for feasibility. Fetal Diagn. Ther. 2012, 31, 244–247. [Google Scholar] [CrossRef]
- Sonek, J.; Molina, F.; Hiett, A.K.; Glover, M.; McKenna, D.; Nicolaides, K. Paper abstracts of the ISPD 16th international conference on prenatal diagnosis and therapy. Prenat. Diagn. 2012, 32, 1–128. [Google Scholar] [PubMed]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef] [PubMed]
- Ashoor, G.; Syngelaki, A.; Wagner, M.; Birdir, C.; Nicolaides, K. Chromosome-selective sequencing of maternal plasma cell-free DNA for first trimester detection of trisomy 21 and trisomy 18. Am. J. Obstet. Gynecol. 2012, 206, e321–e325. [Google Scholar]
- Avent, N.D. Refining noninvasive prenatal diagnosis with single-molecular next-generation sequencing. Clin. Chem. 2012, 58, 657–658. [Google Scholar] [CrossRef]
- Chiu, R.W.; Akolekar, R.; Zheng, Y.W.; Leung, T.Y.; Sun, H.; Chan, K.C.; Lun, F.M.; Go, A.T.; Lau, E.T.; To, W.W.; et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: Large scale validity study. BMJ 2011, 11. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, D.W.; Platt, L.; Goldberg, J.D.; Abuhamad, A.Z.; Sehnert, A.J.; Rava, R.P. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet. Gynecol. 2012, 119, 890–901. [Google Scholar] [CrossRef]
- Jensen, T.; Zwiefelhofer, T.; Tim, R.; Dzakula, Z.; Kim, S.; Mazloom, A.; Zhu, Z.; Tynan, J.; Lu, T.; McLennan, G. High-throughput massively parallel sequencing for fetal aneuploidy detection from maternal plasma. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Sparks, A.B.; Struble, C.A.; Wang, E.T.; Song, K.; Oliphant, A. Nonivasive prenatal detection and selective analysis of cell-free DNA obtained from maternal blood: Evaluation for trisomy 21 and trisomy 18. Am. J. Obstet. Gynecol. 2012, 206, e311–e319. [Google Scholar]
- Norton, M.E.; Brar, H.; Weiss, J.; Karimi, A.; Laurent, L.C.; Caughey, A.B.; Rodriguez, H.; Williams, J.I.; Mitchell, M.E.; Adair, C.D. Non-invasive chromosomal evaluation (nice) study: Results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am. J. Obstet. Gynecol. 2012, 207, e131–e138. [Google Scholar]
- Liang, D.; Lv, W.; Wang, H.; Xu, L.; Liu, J.; Li, H.; Hu, L.; Peng, Y.; Wu, L. Non-invasive prental testing of fetal whole chromosome aneuploidy by massively parallel sequencing. Prenat. Diagn. 2013, 33, 409–415. [Google Scholar] [CrossRef]
- Boon, E.M.J.; Faas, B.H.W. Benefits and limitations of whole genome versus targeted approaches for noninvasive prenatal testing for fetal aneuploidies. Prenat. Diagn. 2013. [Google Scholar] [CrossRef]
- Chitty, L.S.; Hill, L.; White, H.; Wright, D.; Morris, S. Noninvasive prenatal testing for aneuploidy-ready for prime time? Am. J. Obstet. Gynecol. 2012, 206, 269–275. [Google Scholar] [CrossRef]
- GenomeWeb. Aria Kicks off Clinical Trial for Non-Invasive Down Syndrome Test. Available online: http://www.genomeweb.com/mdx/aria-kicks-clinical-trial-non-invasive-down-syndrome-test (accessed on 12 May 2013).
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012. [Google Scholar] [CrossRef]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pareek, C.S.; Smoczynski, R.; Trefyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef]
- Benn, P.; Borrell, A.; Cuckle, H.; Dugoff, L.; Gross, S.; Johnson, J.A.; Maymon, R.; Odibo, A.; Schielen, P.; Spencer, K. Prenatal detection of Down syndrome using massively parallel sequencing (MPS): A rapid response statement from a committee on behalf of the board of the international society for prenatal diagnosis. Prenat. Diagn. 2012, 32, 1–2. [Google Scholar] [PubMed]
- Hui, L. Non-invasive prenatal testing for fetal aneuploidy: Charting the course from clinical validation to clinical utility. Ultrasound Obstet. Gynecol. 2013, 41, 2–6. [Google Scholar] [CrossRef]
- Jiang, K. Competition intensifies over market for DNA-based prenatal tests. Nat. Med. 2013, 19. [Google Scholar] [CrossRef]
- Perkel, J. Finding the true $1,000 genome. BioTechniques 2013, 54, 71–74. [Google Scholar] [PubMed]
- Nicolaides, K.; Syngelaki, A.; Ashoor, G.; Birdir, C.; Touzet, G. Noninvasive prenatal testing for fetal trisomies in a routinely screened first-trimester population. Am. J. Obstet. Gynecol. 2012, 207, e371–e376. [Google Scholar]
- Anderson, C.; Brown, C. Fetal chromosome abnormalities: Antenatal screening and diagnosis. Am. Fam. Physic. 2009, 79, 117–123. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sillence, K.A.; Madgett, T.E.; Roberts, L.A.; Overton, T.G.; Avent, N.D. Non-Invasive Screening Tools for Down’s Syndrome: A Review. Diagnostics 2013, 3, 291-314. https://doi.org/10.3390/diagnostics3020291
Sillence KA, Madgett TE, Roberts LA, Overton TG, Avent ND. Non-Invasive Screening Tools for Down’s Syndrome: A Review. Diagnostics. 2013; 3(2):291-314. https://doi.org/10.3390/diagnostics3020291
Chicago/Turabian StyleSillence, Kelly A., Tracey E. Madgett, Llinos A. Roberts, Timothy G. Overton, and Neil D. Avent. 2013. "Non-Invasive Screening Tools for Down’s Syndrome: A Review" Diagnostics 3, no. 2: 291-314. https://doi.org/10.3390/diagnostics3020291