Nitrogenous Derivatives of Phosphorus and the Origins of Life: Plausible Prebiotic Phosphorylating Agents in Water


Abstract
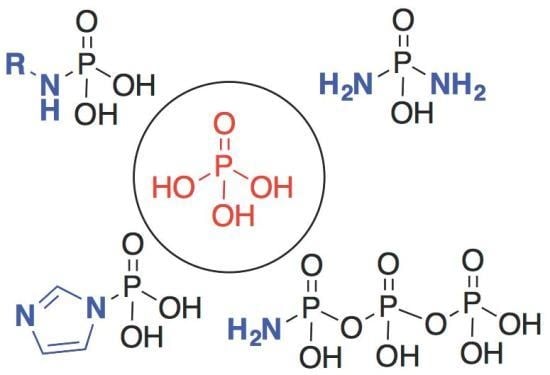
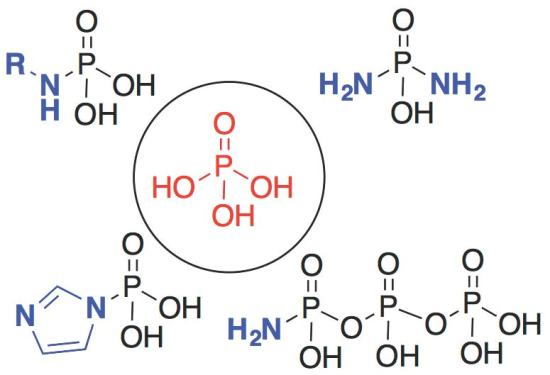
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
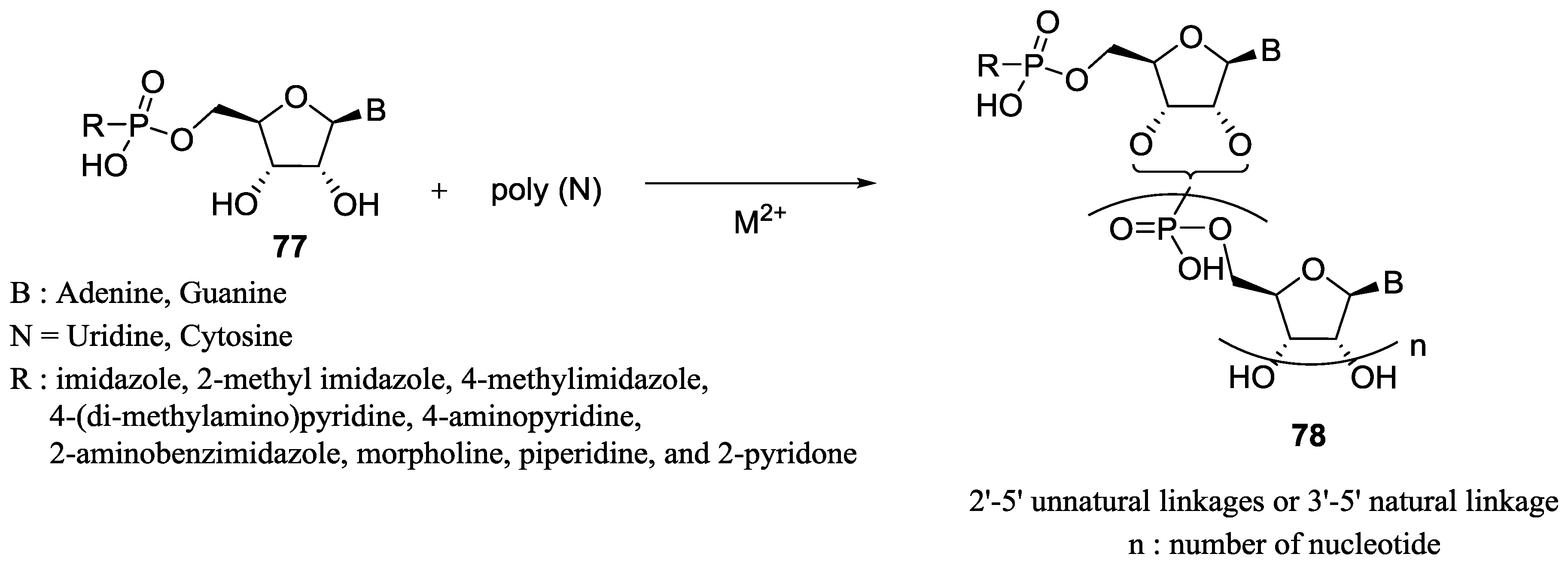{kind=link}
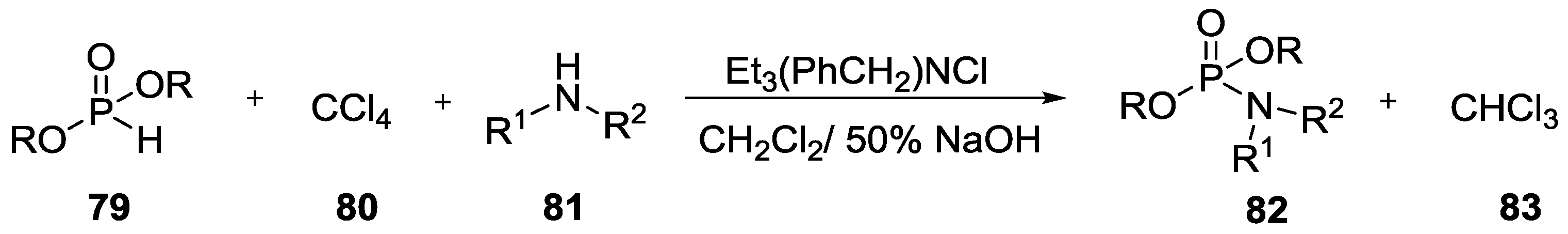{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
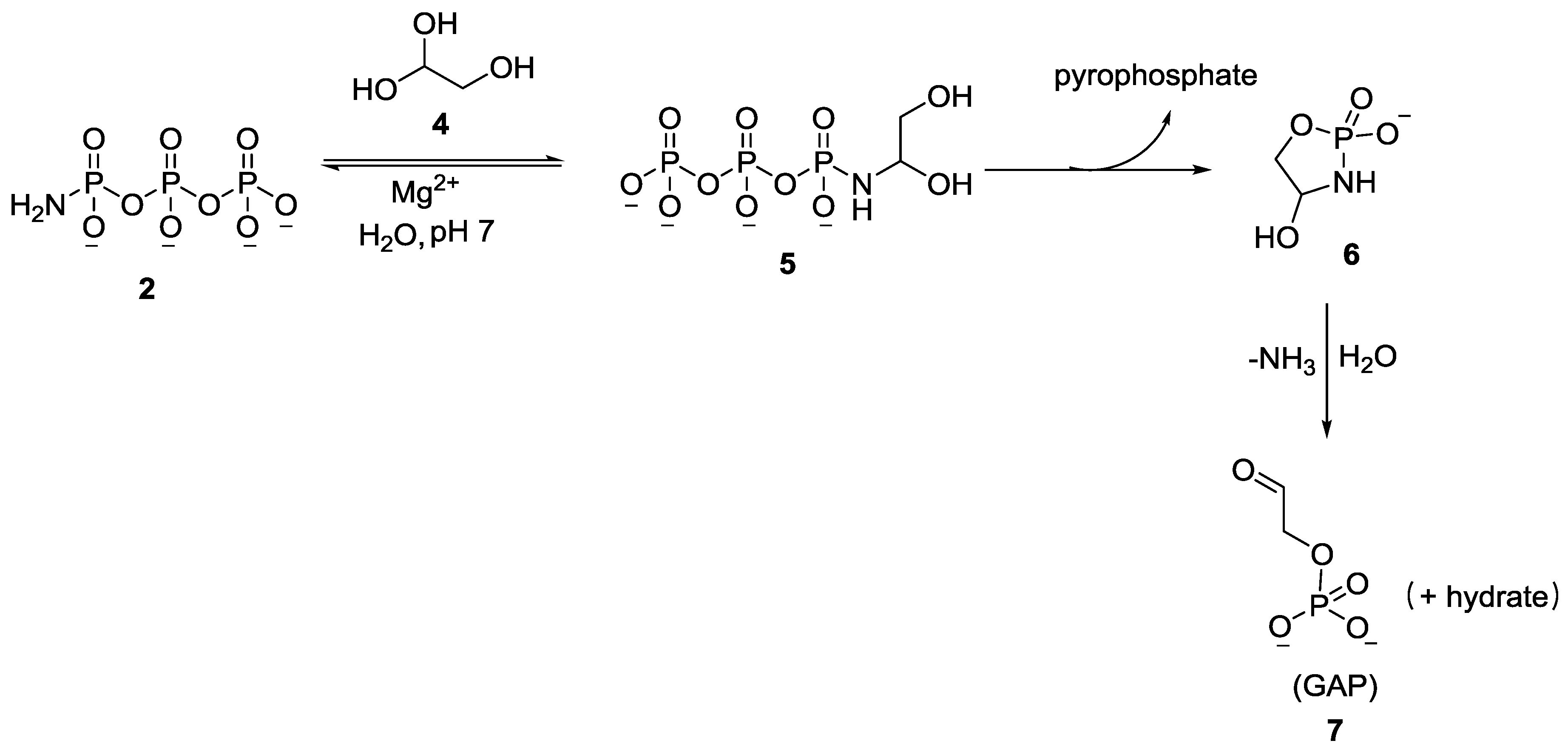
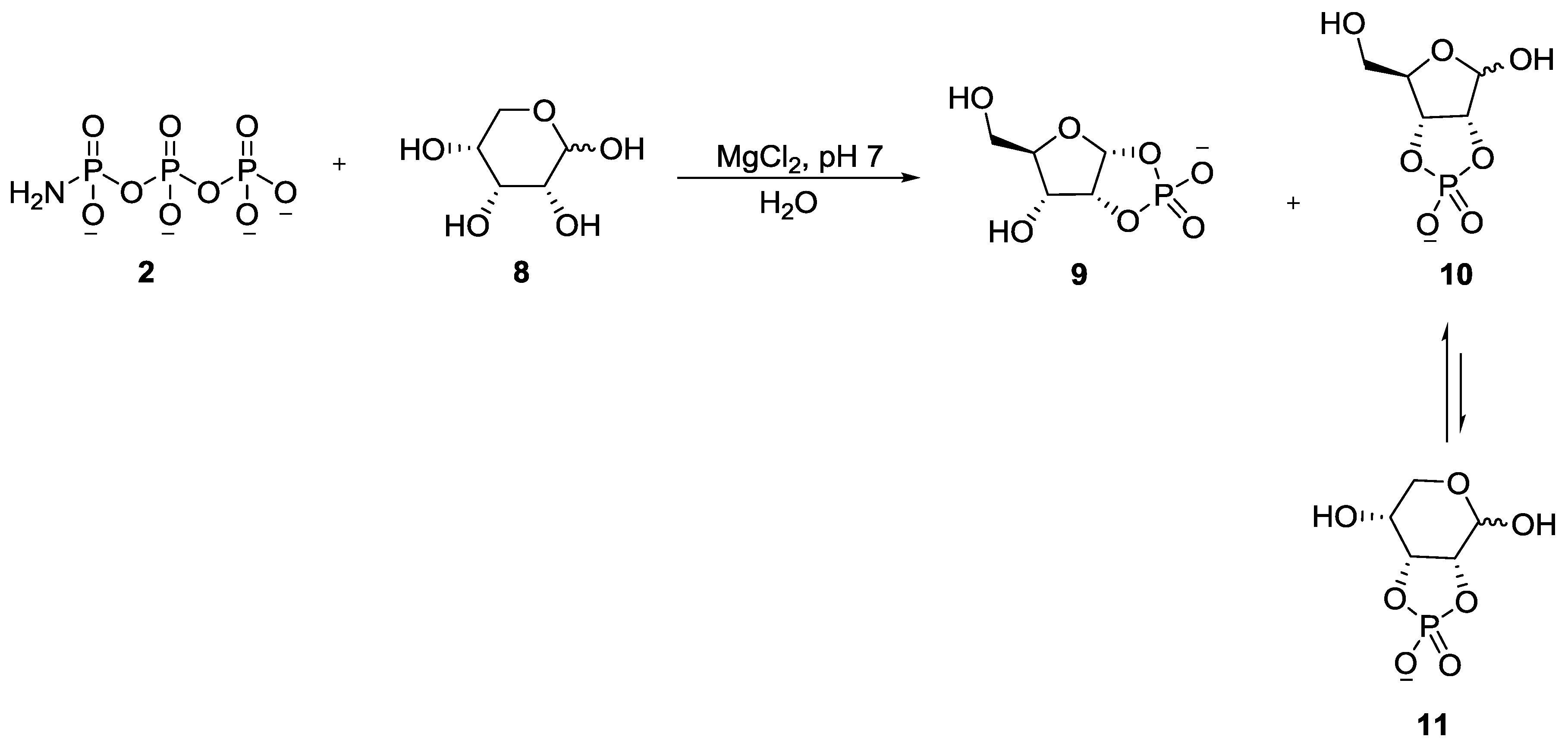
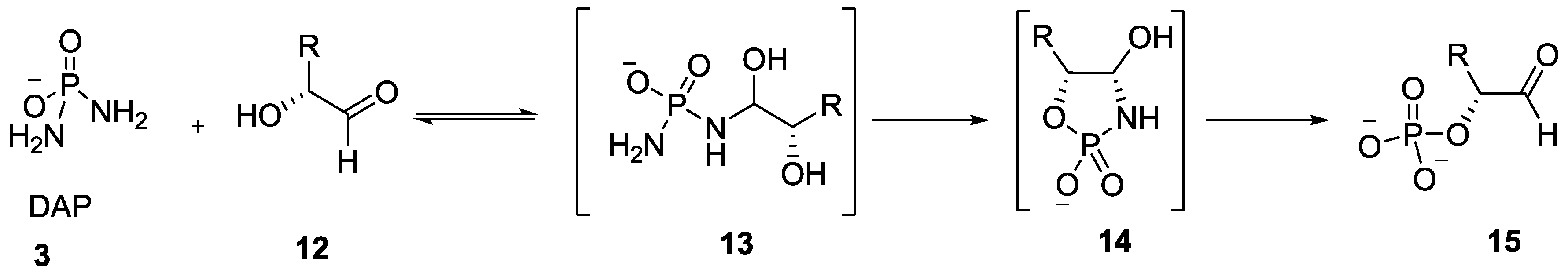
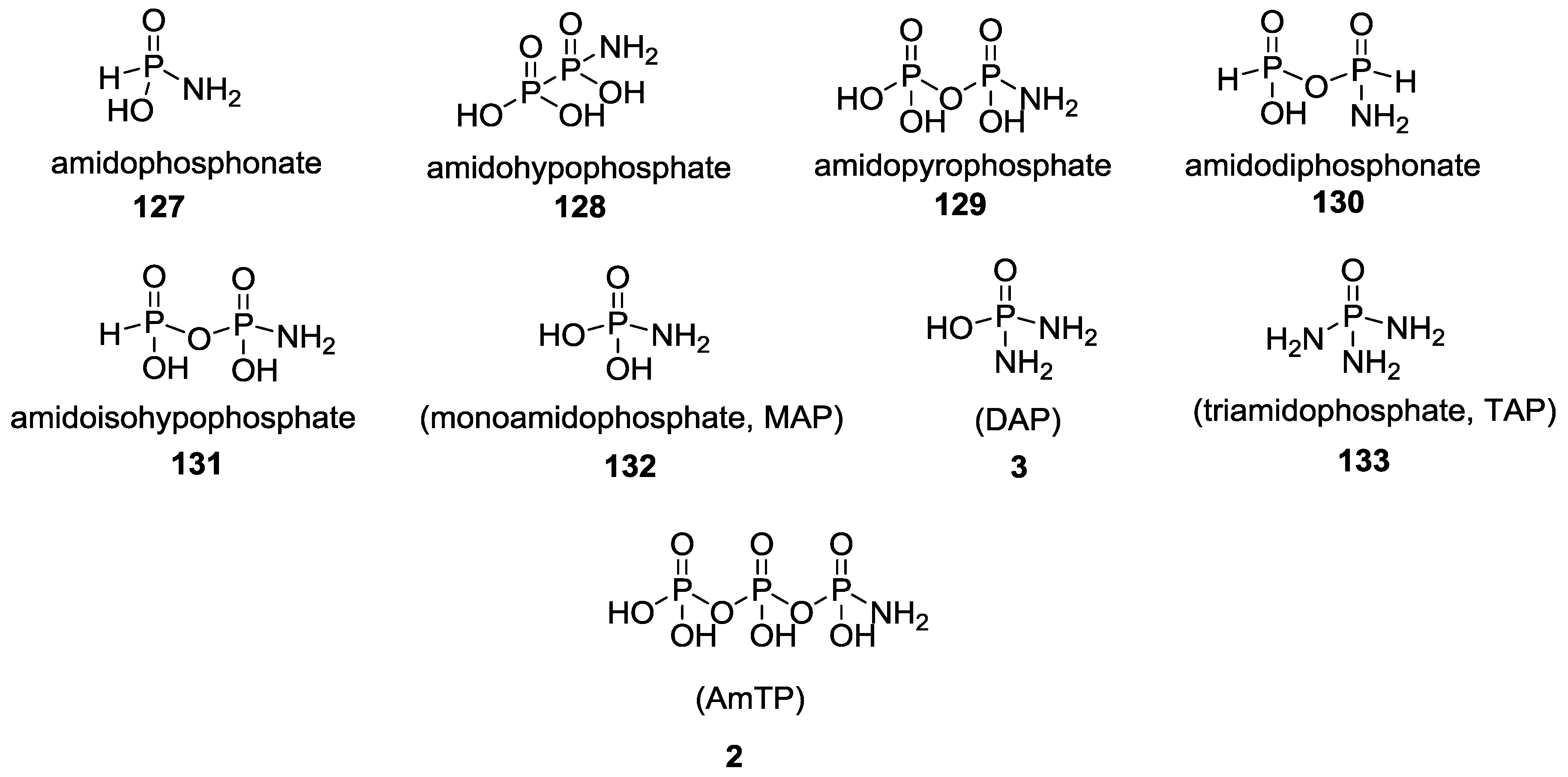{kind=link}
1. Introduction
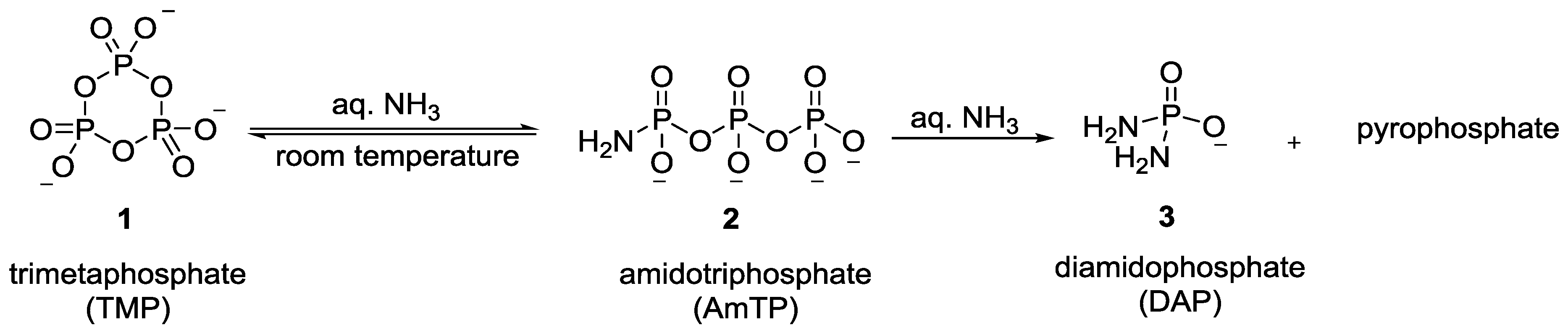
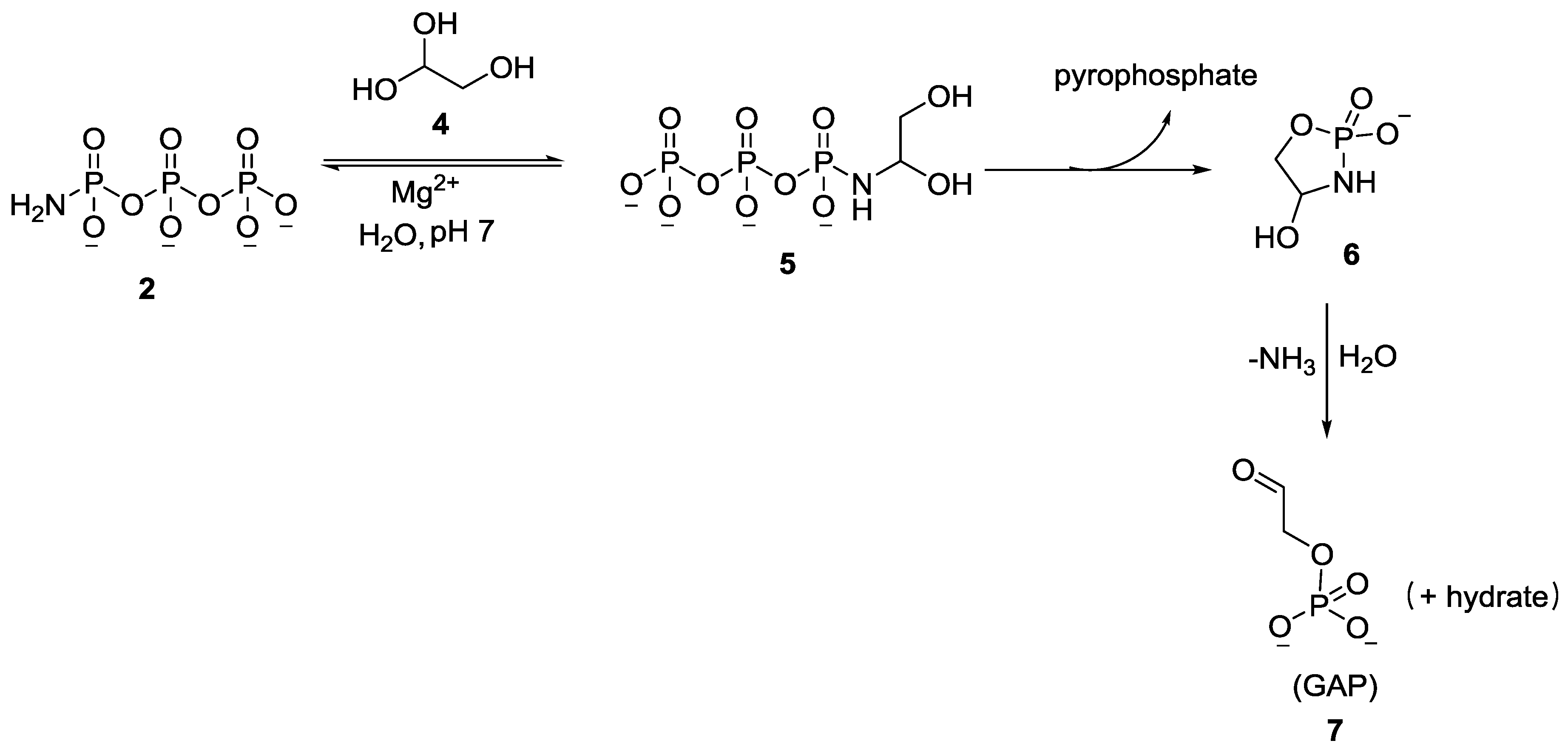
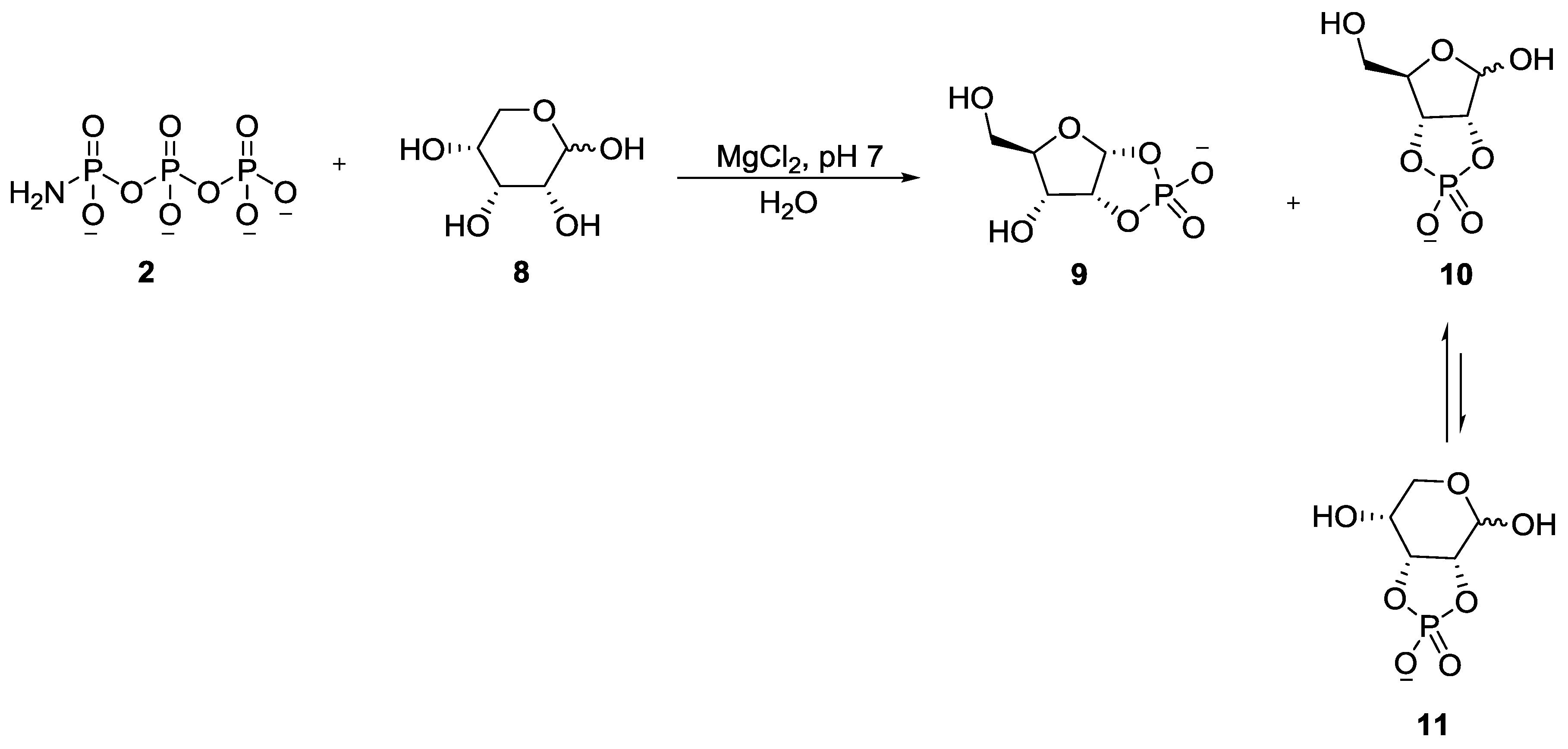
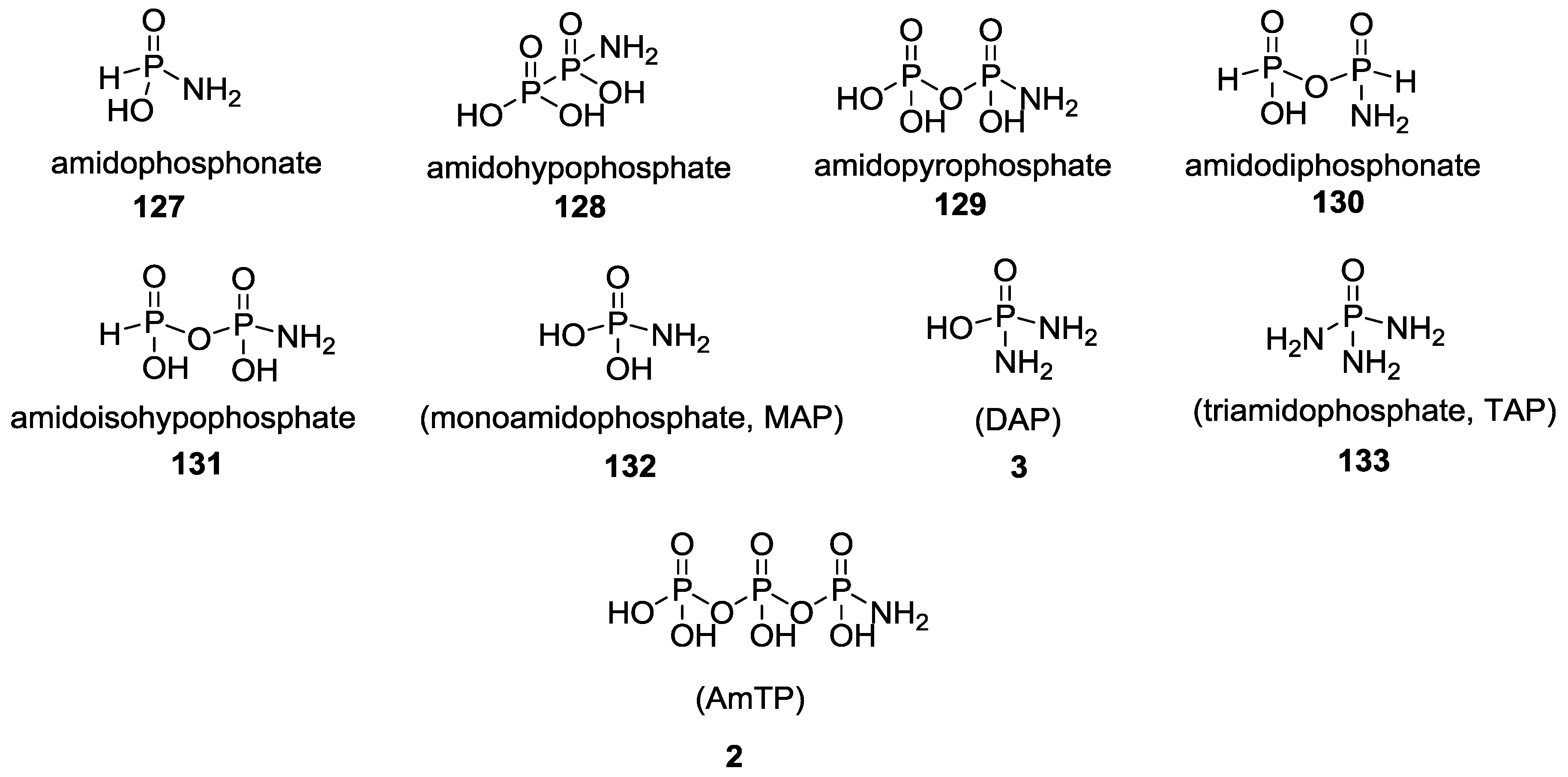
2. The Development of Amidophosphate as a Potential Prebiotic Phosphorylating Reagent
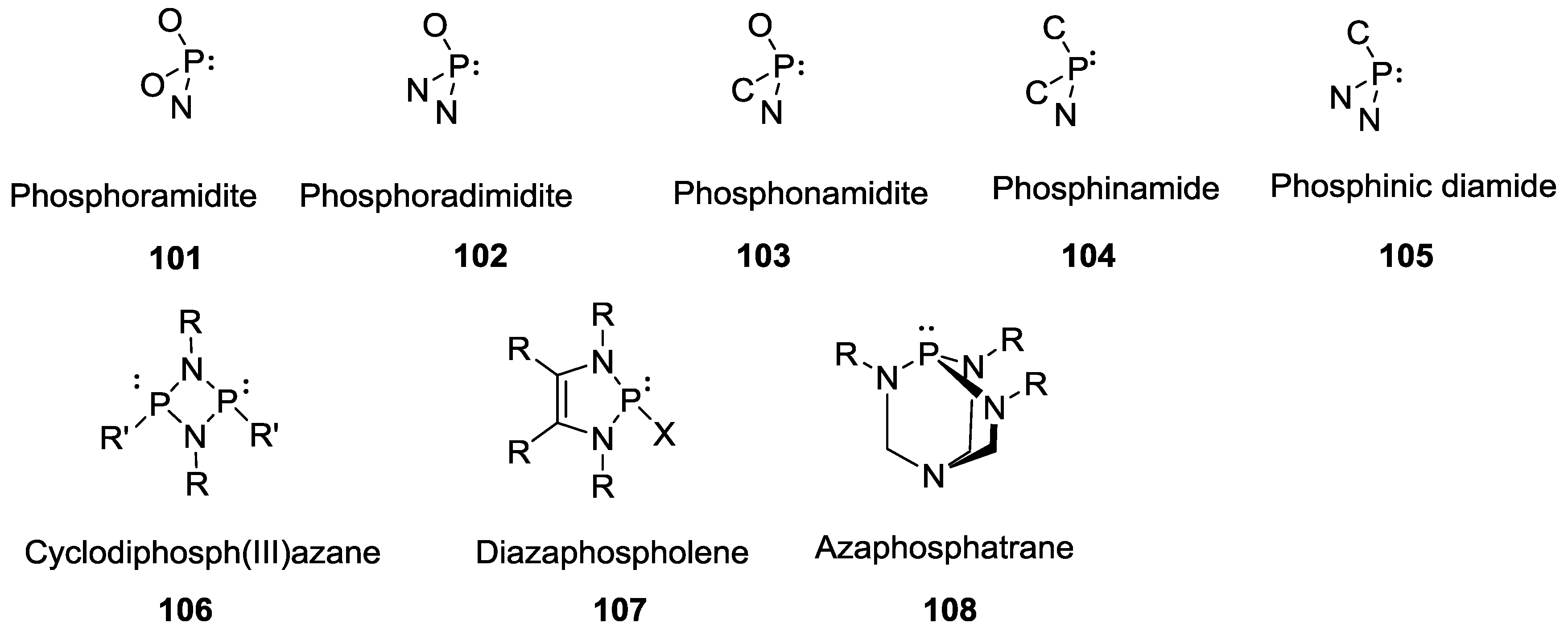
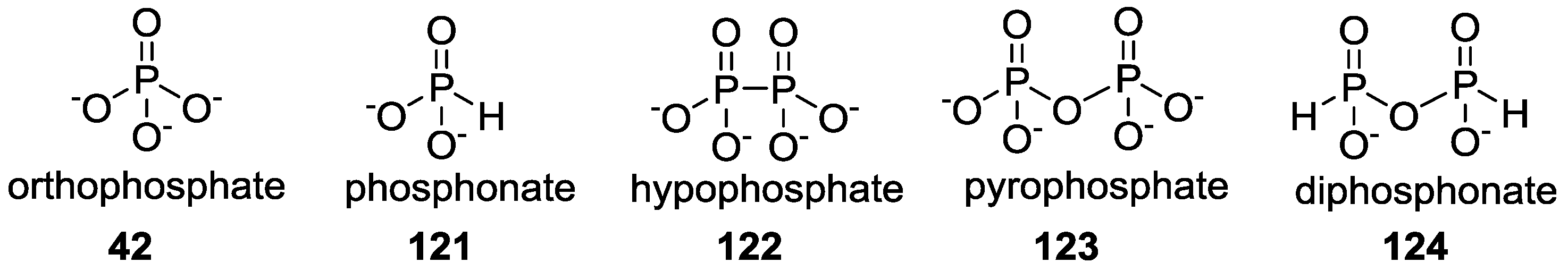
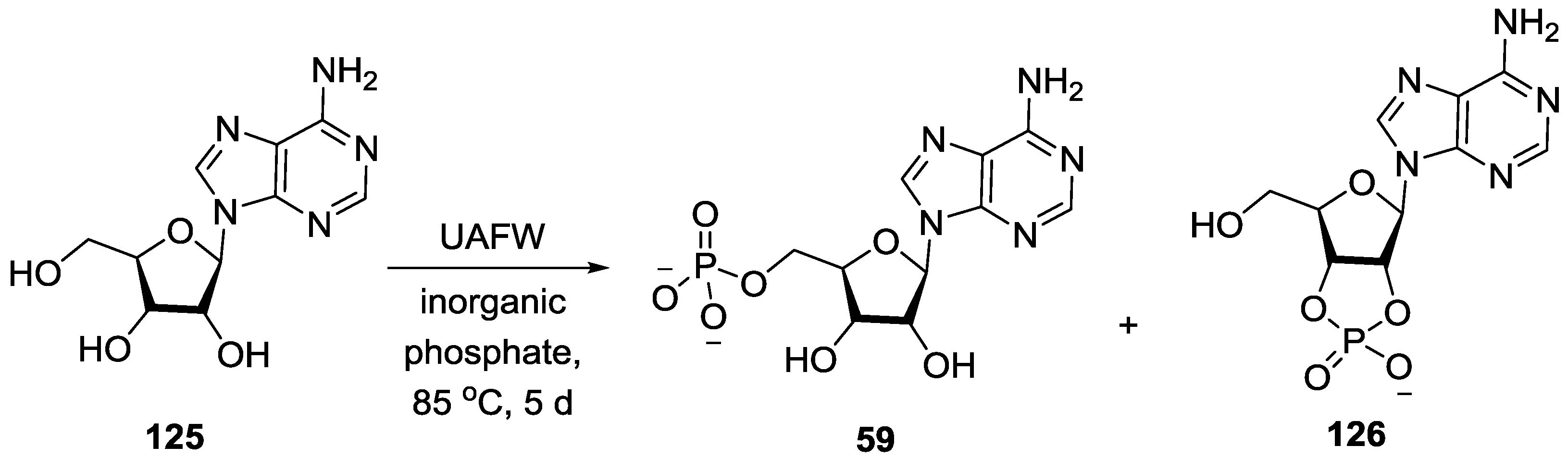
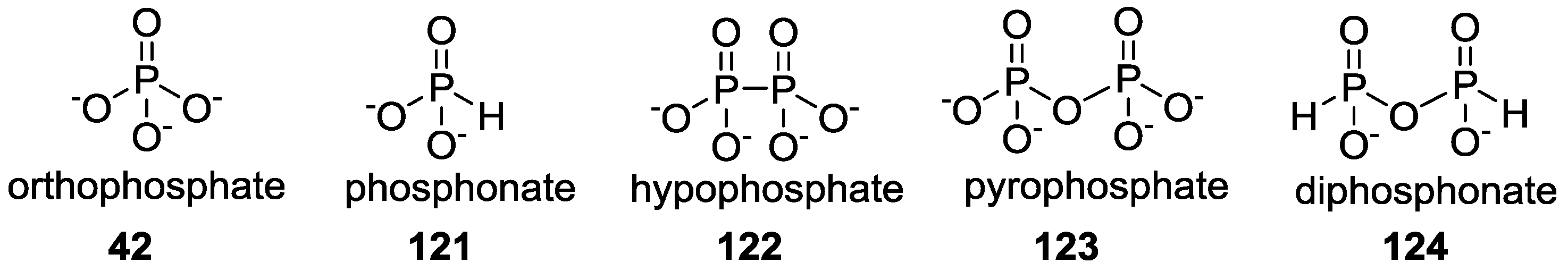
3. Other P–N Derivatives and Their Chemistries
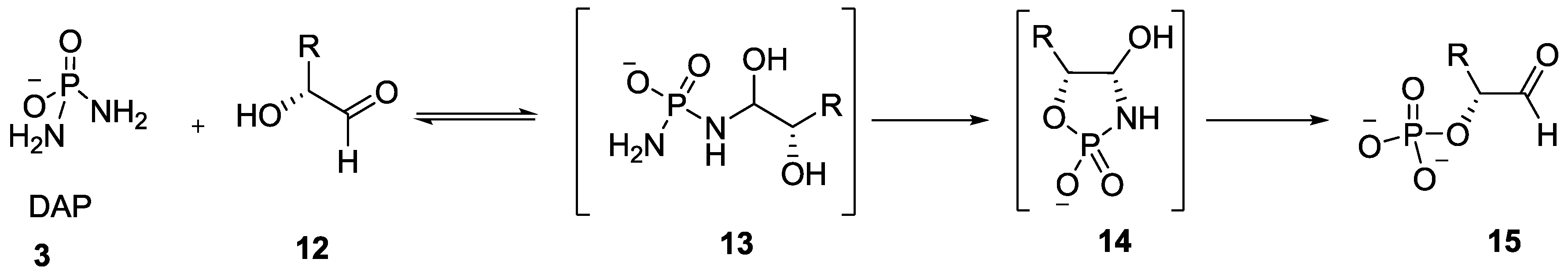
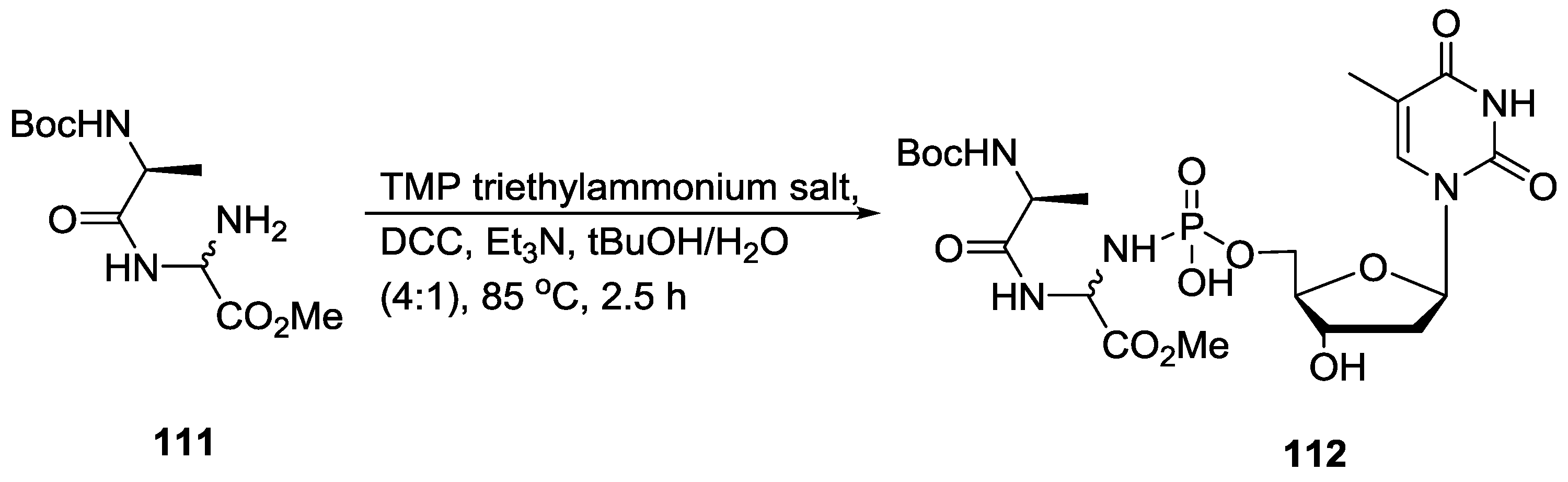
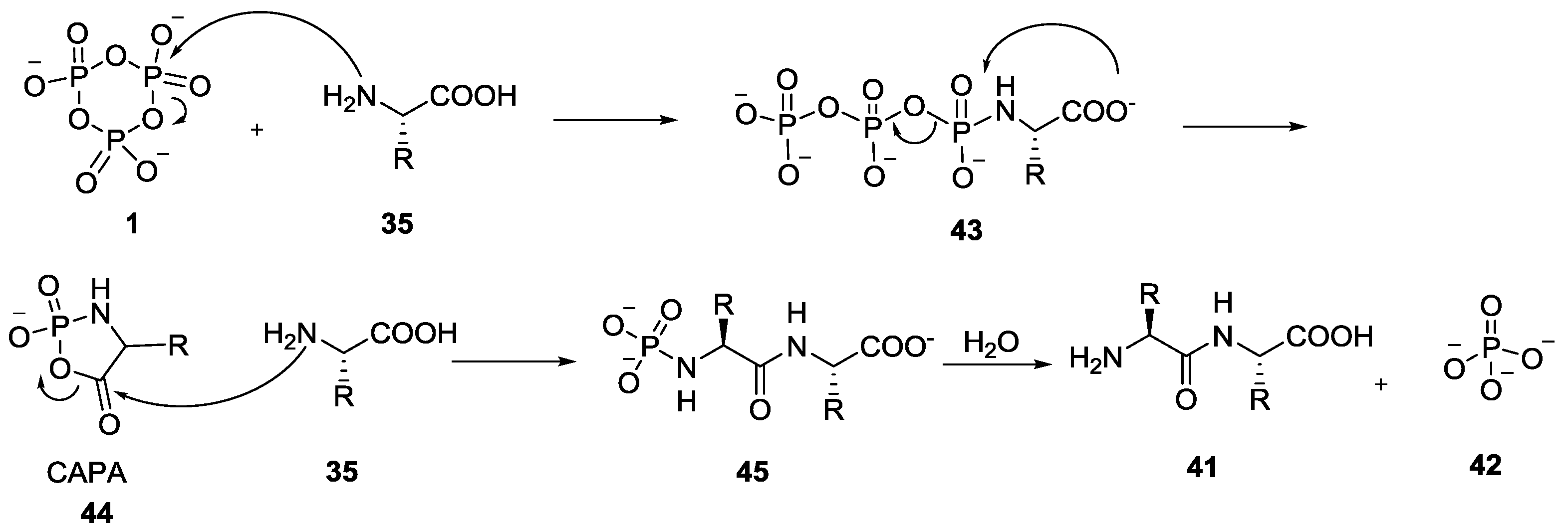
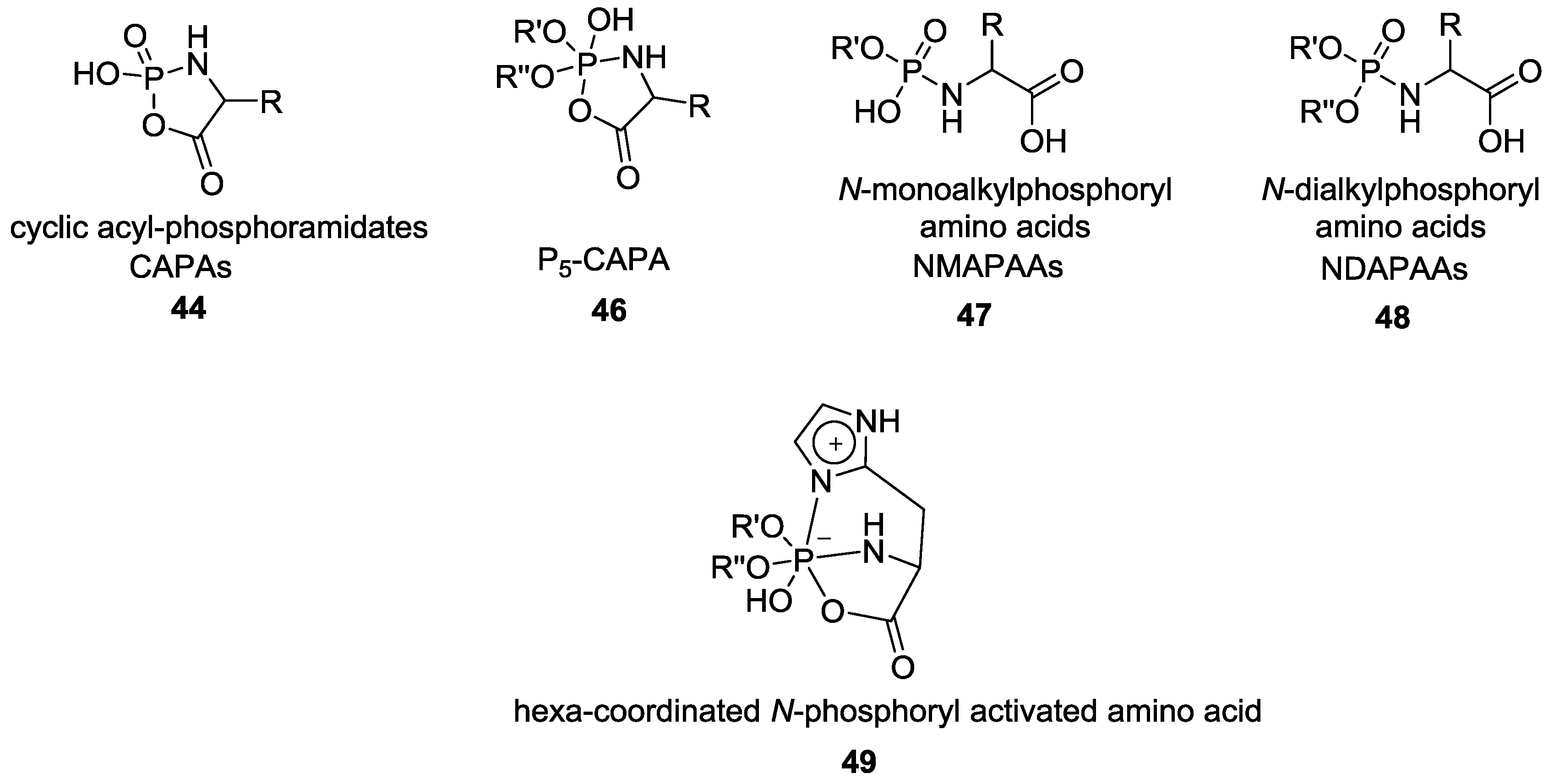
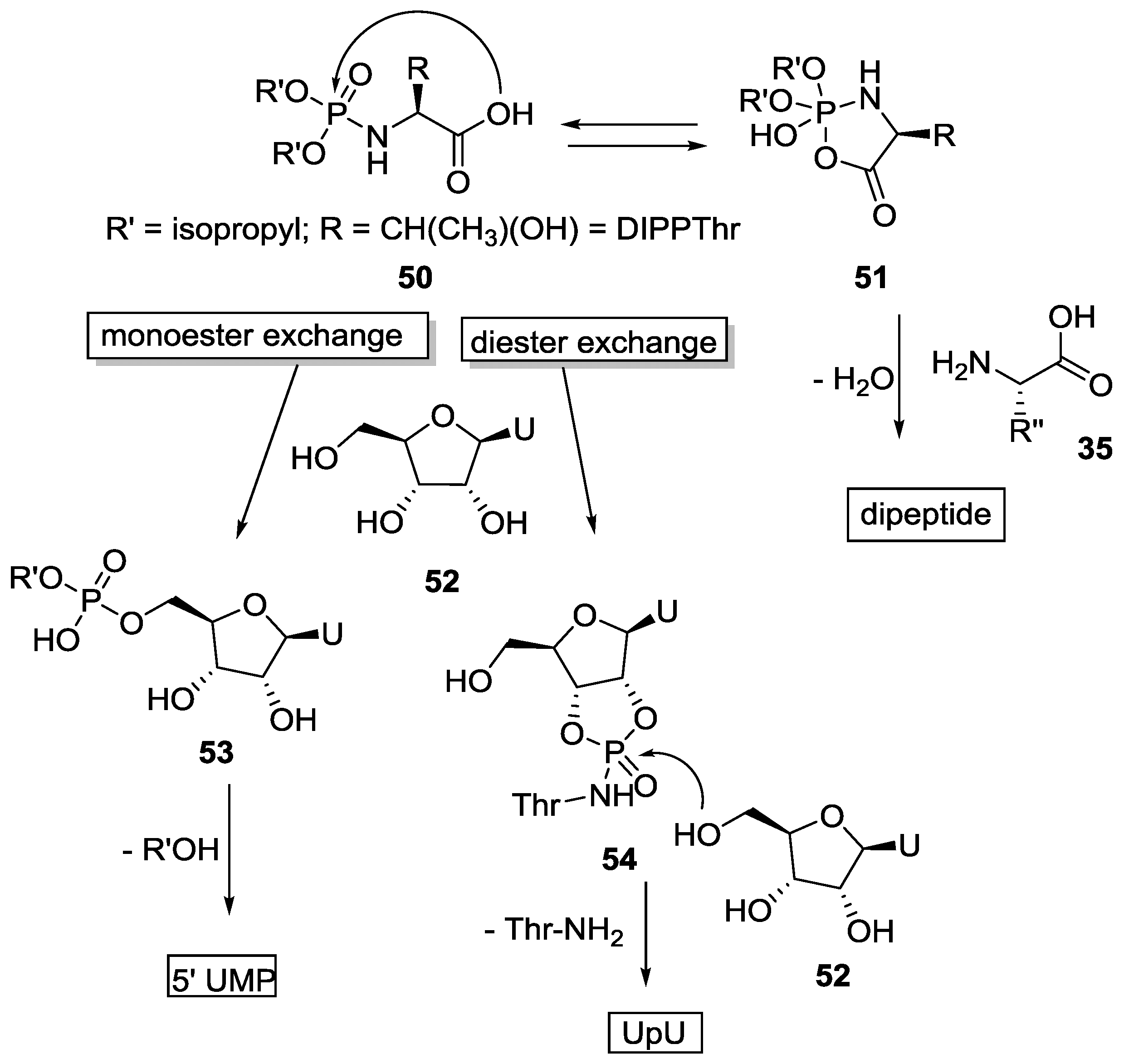
4. Formation of N-Phosphoryl Amino Acids and Their Reactivity: Condensation of Amino Acid and (Poly, Cyclic)Phosphates
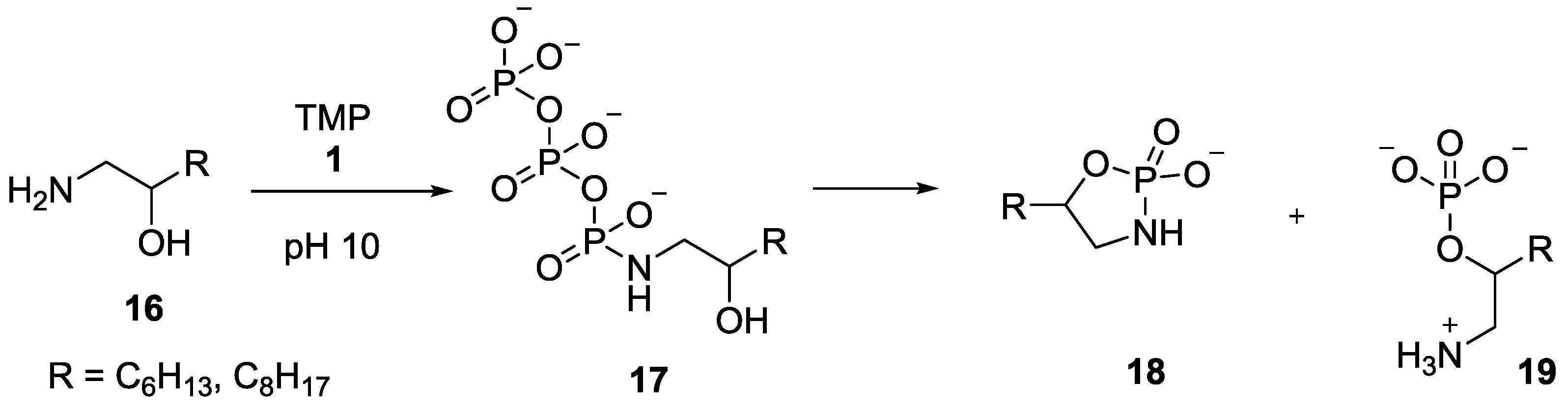
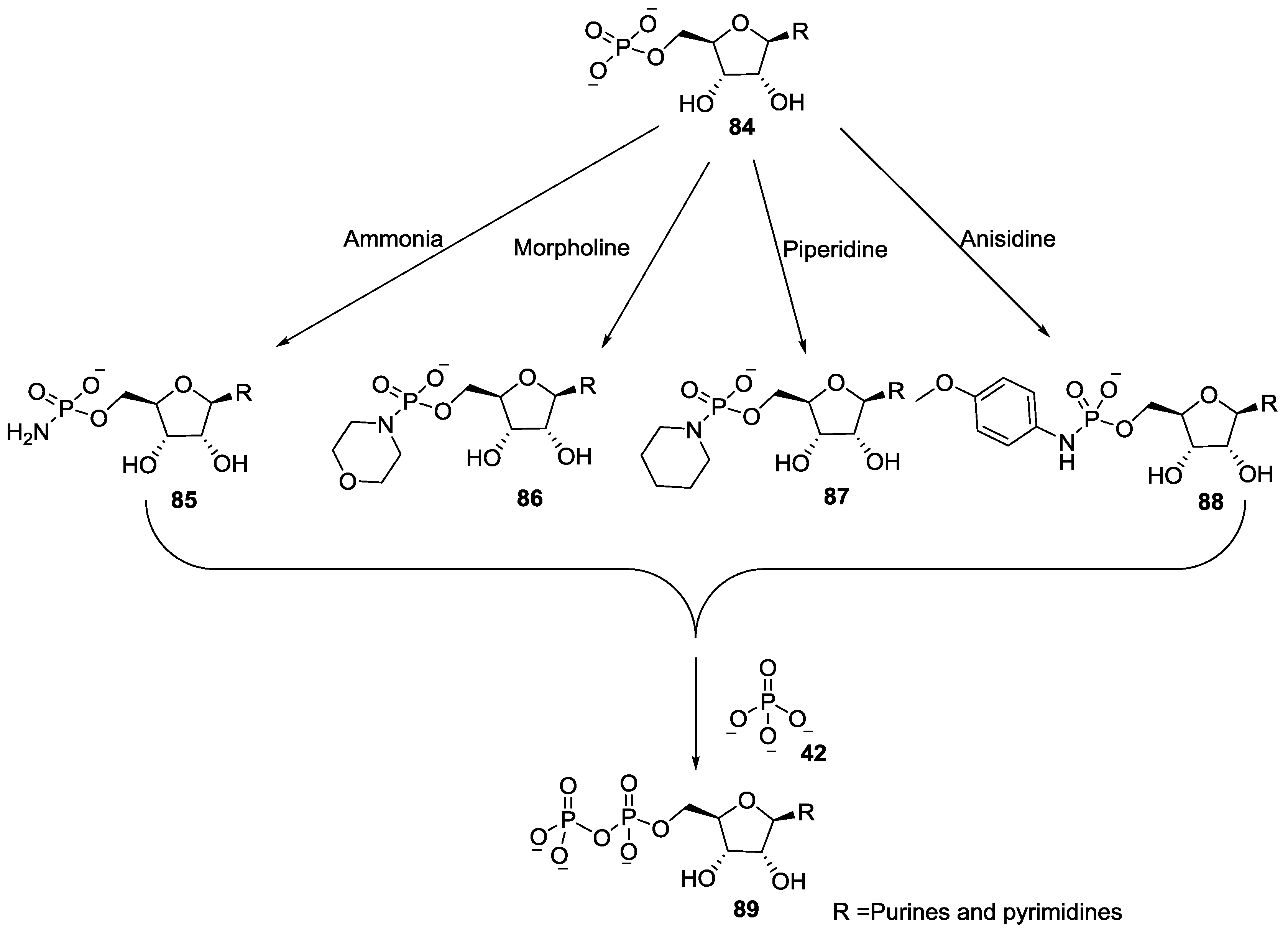
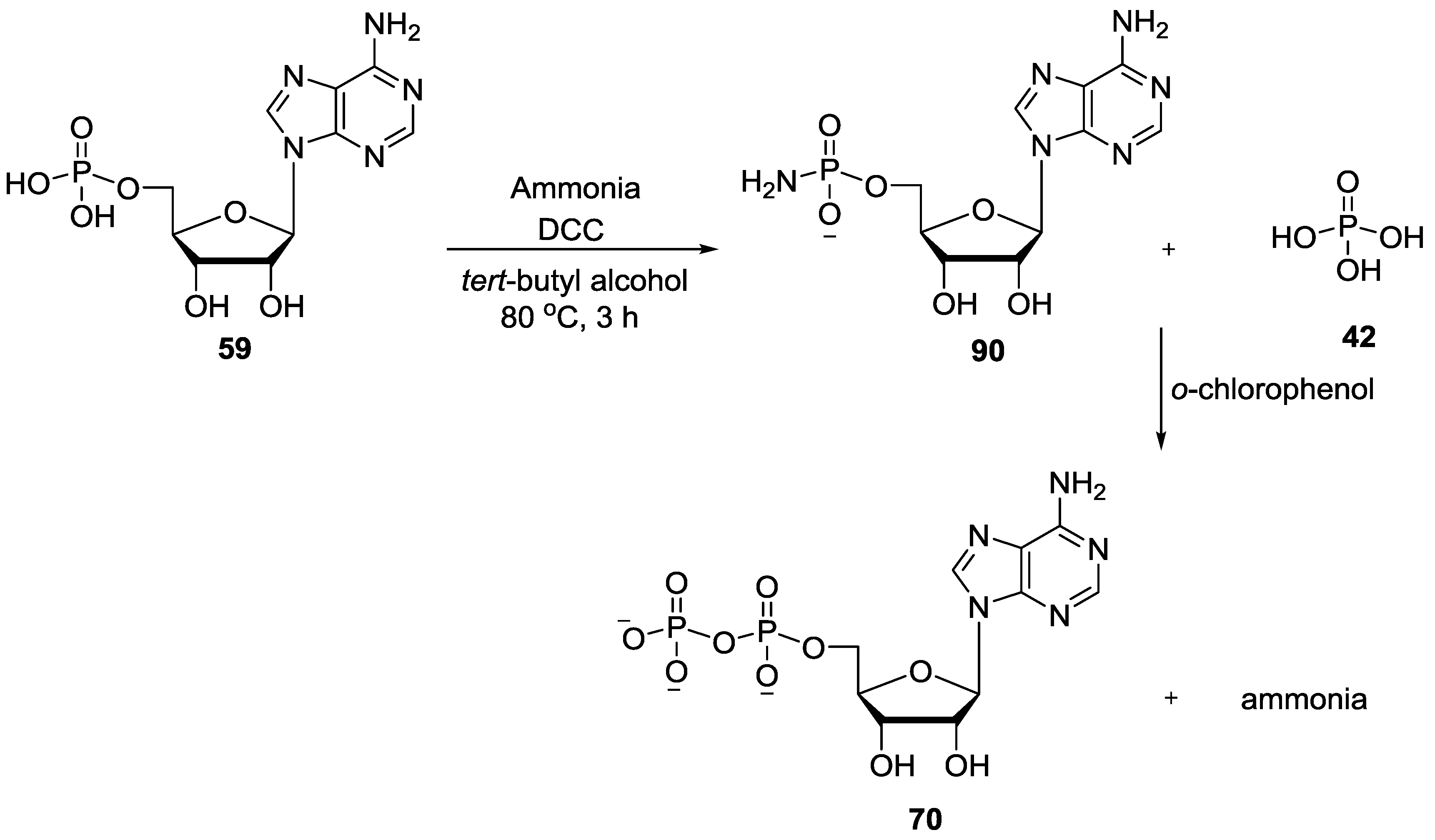
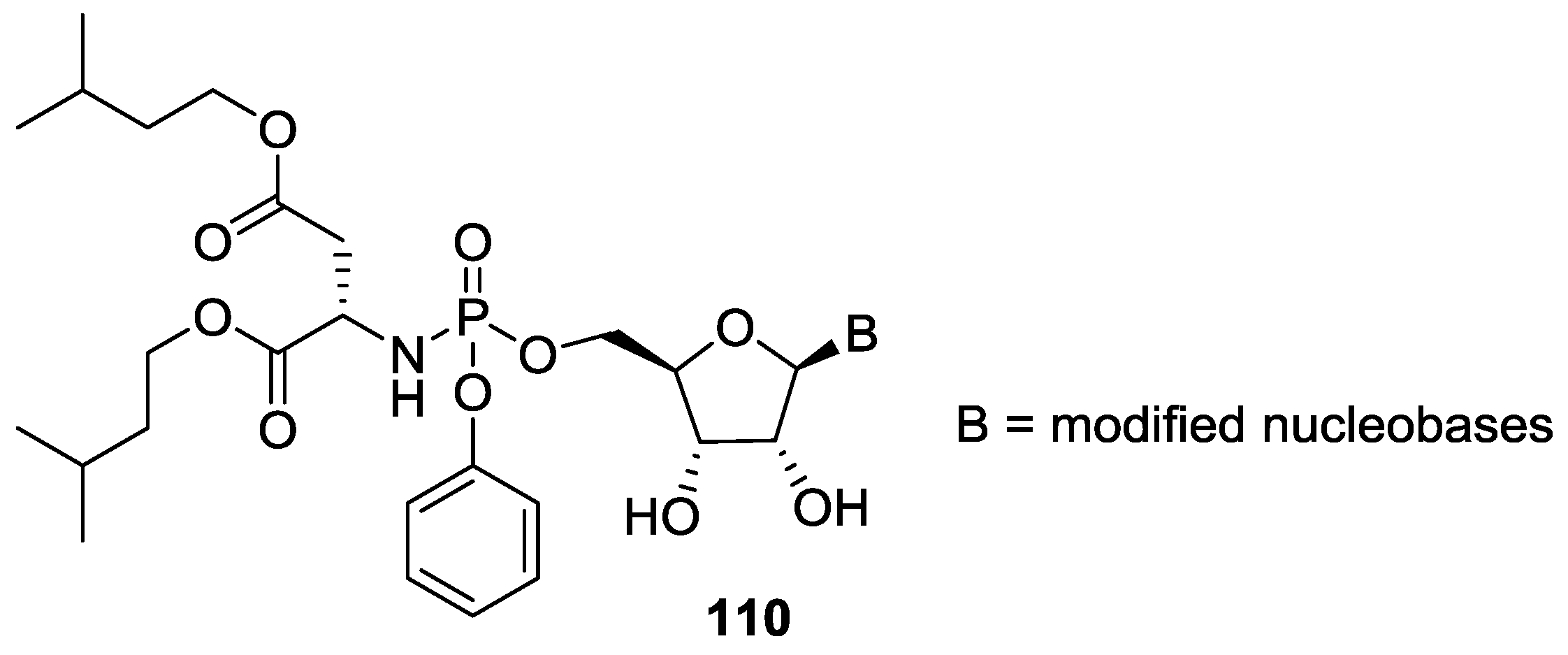
5. Implementation of P–N Nucleoside Phosphoramidate Derivatives
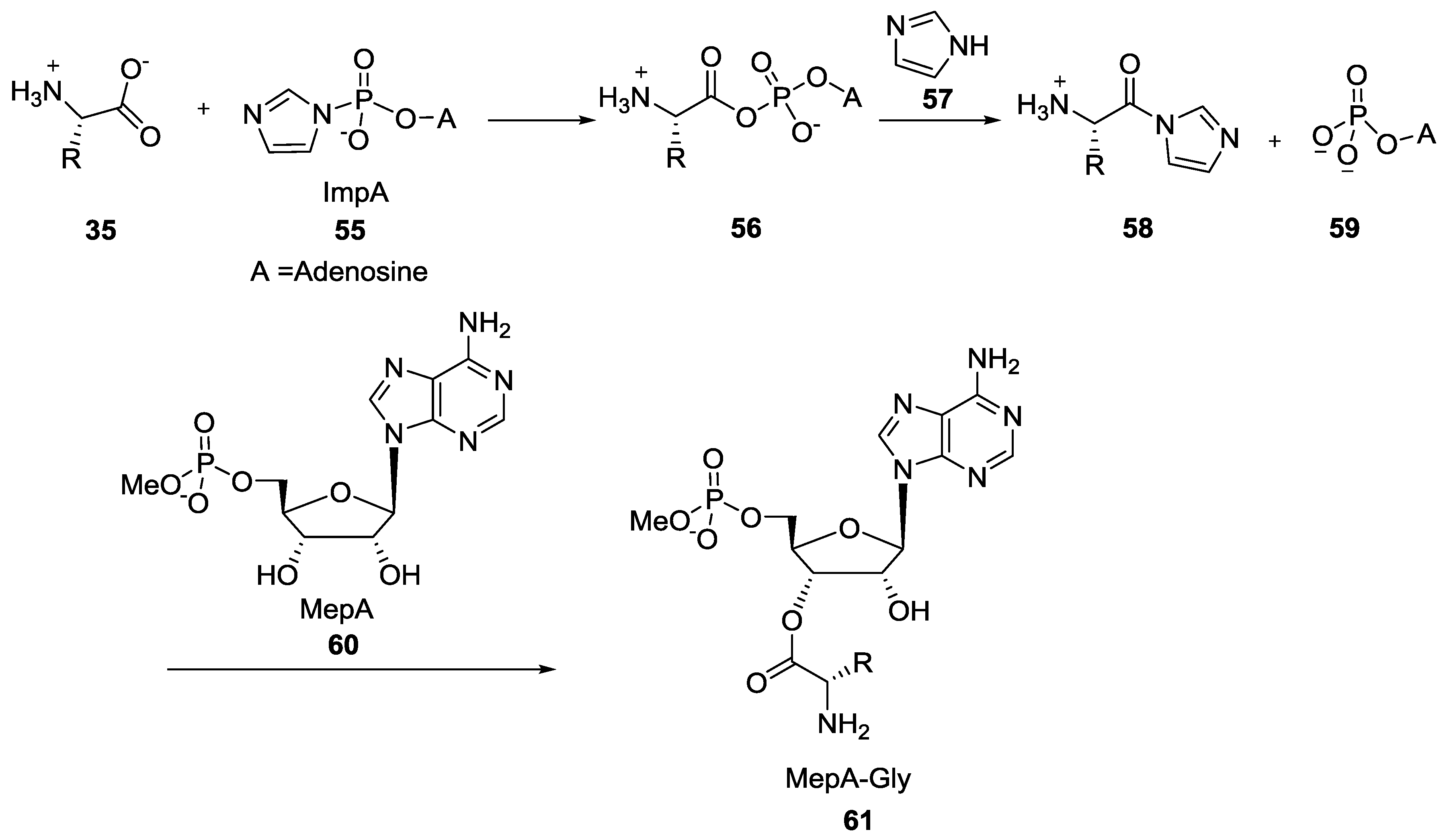
6. Use of Phosphoroimidazolides for the Polymerization of Nucleotides in the Origin of Life Context
7. Role of P–N Species in Oligonucleotide Chemistry
8. Miscellaneous Application of Compounds Containing P–N Bonds
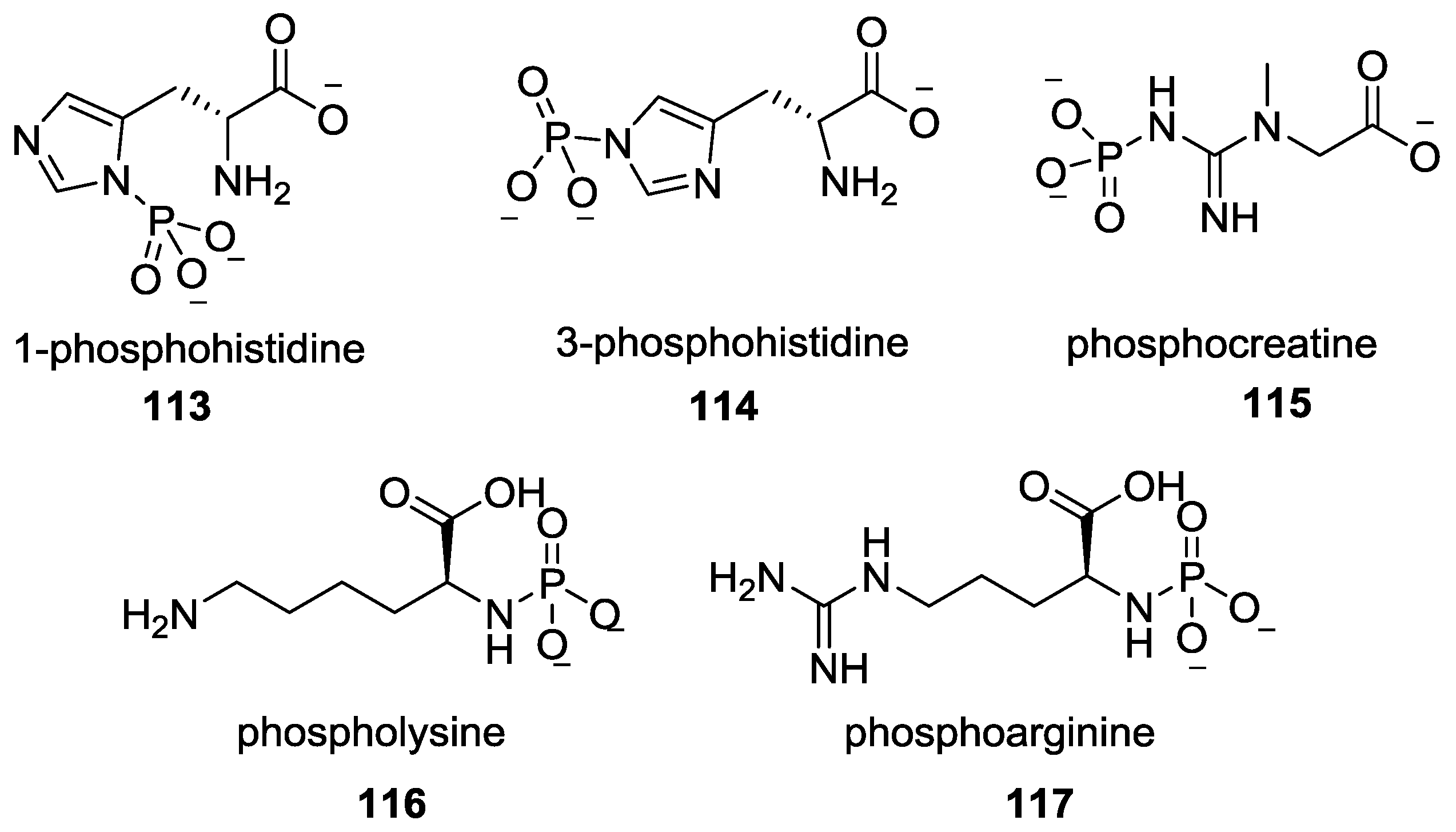
9. P–N Phosphorylation in Biology
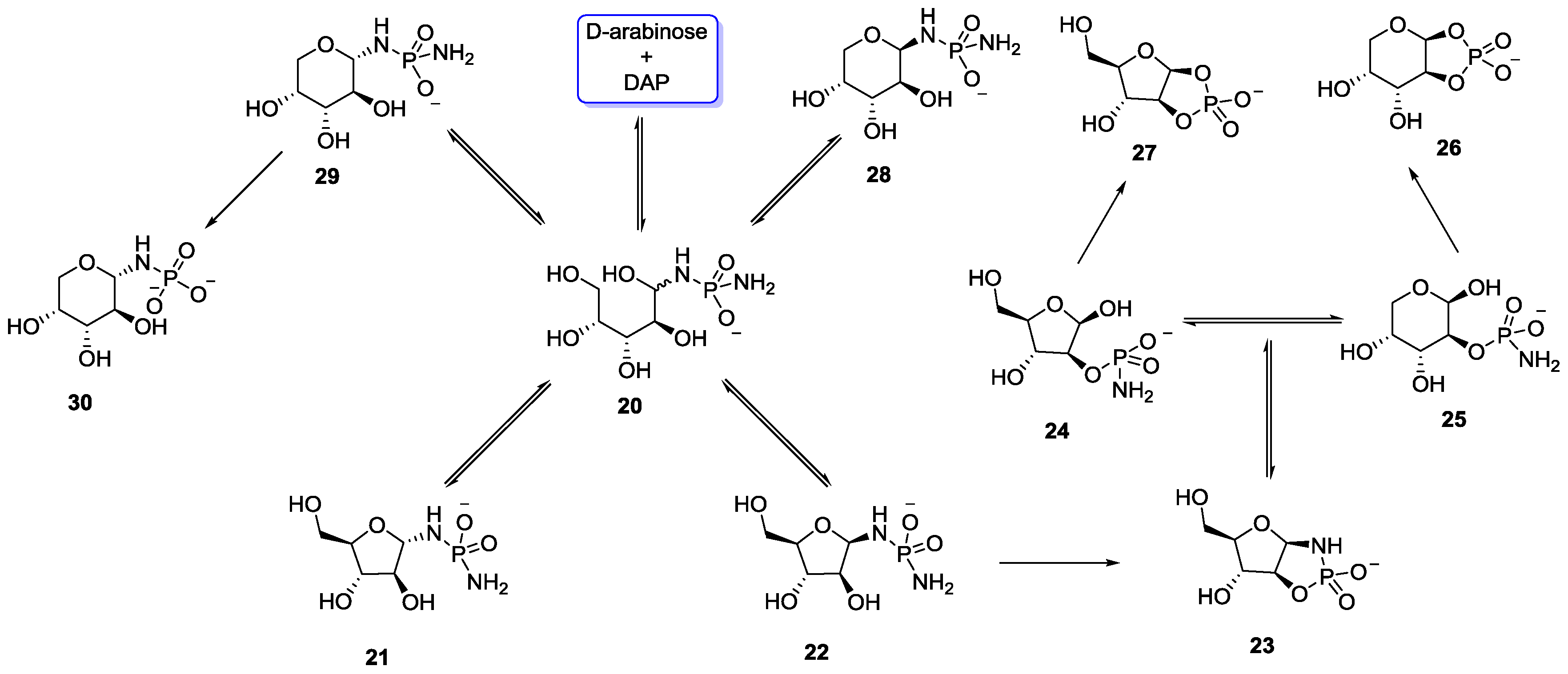
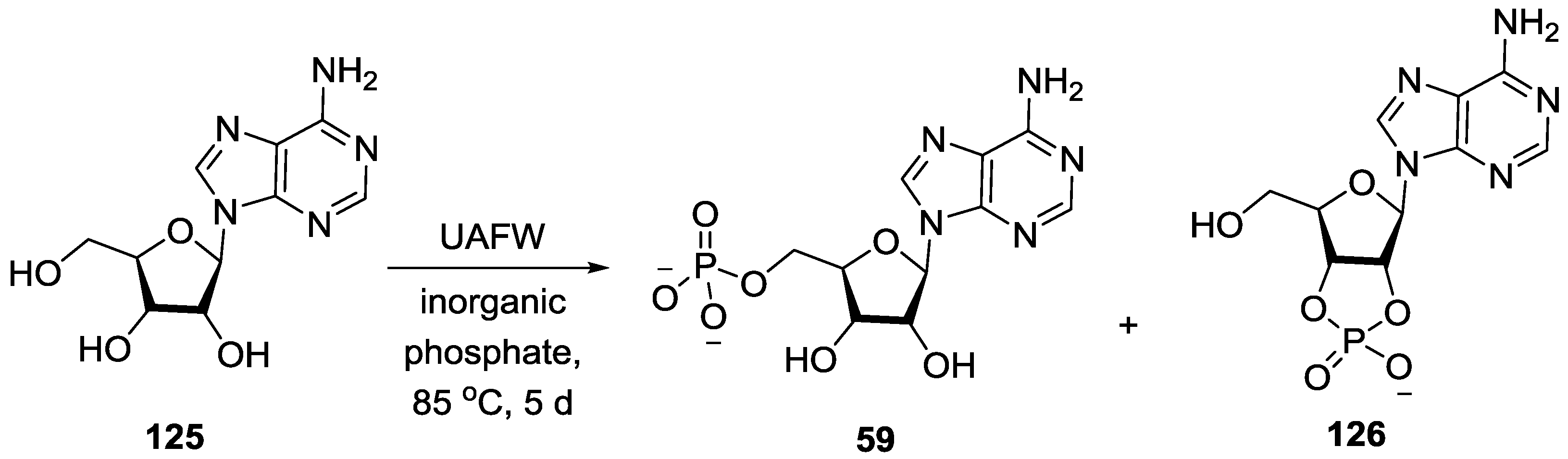
10. Plausible P–N Sources in a Prebiotic Context?
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gulick, A. Phosphorus as a factor in the origin of life. Am. Sci. 1955, 43, 479–489. [Google Scholar]
- Schwartz, A.W. Phosphorus in prebiotic chemistry. Phil. Trans. R. Soc. B 2006, 361, 1743–1749. [Google Scholar] [CrossRef] [PubMed]
- Holm, N.G. Glasses as sources of condensed phosphates on the early earth. Geochem. Trans. 2014, 15, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Gull, M. Prebiotic phosphorylation reactions on the early Earth. Challenges 2014, 5, 193–212. [Google Scholar] [CrossRef]
- De Graaf, R.M.; Visscher, J.; Schwartz, A.W. A plausibly prebiotic synthesis of phosphonic acids. Nature 1995, 378, 474. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, R.M.; Visscher, J.; Schwartz, A.W. Reactive phosphonic acids as prebiotic carriers of phosphorus. J. Mol. Evol. 1997, 44, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Pasek, M.A. Rethinking early Earth phosphorus geochemistry. Proc. Natl. Acad. Sci. USA 2008, 105, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Pasek, M.A.; Harnmeijer, J.P.; Buick, R.; Gull, M.; Atlas, Z. Evidence for reactive reduced phosphorus species in the early archean ocean. Proc. Natl. Acad. Sci. USA 2013, 110, 10089–10094. [Google Scholar] [CrossRef] [PubMed]
- Pasek, M.A.; Dworkin, J.P.; Lauretta, D.S. A radical pathway for organic phosphorylation during schreibersite corrosion with implications for the origin of life. Geochim. Cosmochim. Acta 2007, 71, 1721–1736. [Google Scholar] [CrossRef]
- Pasek, M.A.; Lauretta, D.S. Aqueous corrosion of phosphide minerals from iron meteorites: A highly reactive source of prebiotic phosphorus on the surface of the early Earth. Astrobiology 2005, 5, 515–535. [Google Scholar] [CrossRef] [PubMed]
- Kee, T.P.; Bryant, D.E.; Herschy, B.; Marriott, K.E.; Cosgrove, N.E.; Pasek, M.A.; Atlas, Z.D.; Cousins, C.R. Phosphate activation via reduced oxidation state phosphorus (P). Mild routes to condensed-P energy currency molecules. Life 2013, 3, 386–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohrmann, R.; Orgel, L.E. Prebiotic activation processes. Nature 1973, 244, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Osterberg, R.; Orgel, L.E. Polyphosphate and trimetaphosphate formation under potentially prebiotic conditions. J. Mol. Evol. 1972, 1, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Etaix, E.; Orgel, L.E. Phosphorylation of Nucleosides in Aqueous-Solution Using Trimetaphosphate-Formation of Nucleoside Triphosphates. J. Carbohydr. Nucleos. Nucleot. 1978, 5, 91–110. [Google Scholar]
- Saffhill, R. Selective phosphorylation of the cis-2', 3'-diol of unprotected ribonucleosides with trimetaphosphate in aqueous solution. J. Org. Chem. 1970, 35, 2881–2883. [Google Scholar] [CrossRef] [PubMed]
- Lohrmann, R. Formation of nucleoside 5′-polyphosphates from nucleotides and trimetaphosphate. J. Mol. Evol. 1975, 6, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.; Flores, J.; Krebsbach, R.; Rogers, G. Peptide formation in the presence of linear or cyclic polyphosphates. Nature 1969, 224, 795–796. [Google Scholar] [CrossRef] [PubMed]
- Chung, N.M.; Lohrmann, R.; Orgel, L.E.; Rabinowitz, J. The mechanism of the trimetaphosphate-induced peptide synthesis. Tetrahedron 1971, 27, 1205–1210. [Google Scholar] [CrossRef]
- Feldmann, W. Ringaufspaltung des Trimetaphosphations, P3O93-, durch Aminosäuren in wäßriger Lösung; Über die Knüpfung von Peptidbindungen mit P3O93-. Z. Chem. 1969, 9, 154–155. [Google Scholar] [CrossRef]
- Inoue, H.; Furukawa, T.; Maeda, Y.; Tsuhako, M. Formation of dipeptide in the reaction of amino acids with cyclo-triphosphate. Chem. Pharm. Bull. 1993, 41, 1895–1899. [Google Scholar] [CrossRef]
- Yamagata, Y.; Inoue, H.; Inomata, K. Specific effect of magnesium ion on 2′, 3′-cyclic AMP synthesis from adenosine and trimetaphosphate in aqueous solution. Orig. Life Evol. Biosph. 1995, 25, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Kolb, V.; Zhang, S.; Xu, Y.; Arrhenius, G. Mineral induced phosphorylation of glycolate ion—a metaphor in chemical evolution. Orig. Life Evol. Biosph. 1997, 27, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Miller, S.L. Are polyphosphates or phosphate esters prebiotic reagents? Orig. Life Evol. Biosph. 1995, 41, 693–702. [Google Scholar] [CrossRef]
- Yamagata, Y.; Watanabe, H. Volcanic production of polyphosphates and its relevance to prebiotic evolution. Nature 1991, 352, 516. [Google Scholar] [CrossRef] [PubMed]
- Griffith, E.J.; Ponnamperuma, C.; Gabel, N.W. Phosphorus, a key to life on the primitive earth. Orig. Life Evol. Biosph. 1977, 8, 71–85. [Google Scholar] [CrossRef]
- Baltscheffsky, H.; von Stedingk, L.V.; Heldt, H.W.; Klingenberg, M. Inorganic pyrophosphate: Formation in bacterial photophosphorylation. Science 1966, 153, 1120–1122. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, A.; Kornberg, S.R.; Simms, E.S. Metaphosphate synthesis by an enzyme from Escherichia coli. Biochim. Biophys. Acta. 1956, 20, 215–227. [Google Scholar] [CrossRef]
- Kumble, K.D.; Kornberg, A. Inorganic polyphosphate in mammalian cells and tissues. J. Biol. Chem. 1995, 270, 5818–5822. [Google Scholar] [CrossRef] [PubMed]
- Kumble, K.D.; Kornberg, A. Endopolyphosphatases for long chain inorganic polyphosphate in yeast and mammals. J. Biol. Chem. 1996, 271, 27146–27151. [Google Scholar] [CrossRef]
- Kornberg, S.R. Adenosine triphosphate synthesis from polyphosphate by an enzyme from Escherichia coli. Biochim. Biophys. Acta 1957, 26, 294–300. [Google Scholar] [CrossRef]
- Kornberg, A.; Rao, N.N.; Ault-Riche, D. Inorganic polyphosphate: A molecule of many functions. Ann. Rev. Biochem. 1999, 68, 89–125. [Google Scholar] [CrossRef] [PubMed]
- Beier, M.; Reck, F.; Wagner, T.; Krishnamurthy, R.; Eschenmoser, A. Chemical etiology of nucleic acid structure: Comparing pentopyranosyl-(2'→ 4') oligonucleotides with RNA. Science 1999, 283, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Pitsch, S.; Wendeborn, S.; Krishnamurthy, R.; Holzner, A.; Minton, M.; Bolli, M.; Miculka, C.; Windhab, N.; Micura, R.; Stanek, M.; et al. The β-d-ribopyranosyl-(4→2)-oligonucleotide system (“pyranosyl-RNA”): Synthesis and resume of base-pairing properties. Helv. Chim. Acta 2003, 86, 4270–4363. [Google Scholar] [CrossRef]
- Krishnamurthy, R.; Arrhenius, G.; Eschenmoser, A. Formation of glycolaldehyde phosphate from glycolaldehyde in aqueous solution. Orig. Life Evol. Biosph. 1999, 29, 333–354. [Google Scholar] [CrossRef] [PubMed]
- Quimby, O.T.; Flautt, T.J. Ammonolyse des Trimetaphosphats. Z. Anorg. Allg. Chem. 1958, 296, 220–228. [Google Scholar] [CrossRef]
- Feldmann, W.; Thilo, E. Zur Chemie der kondensierten Phosphate und Arsenate. XXXVIII. Amidotriphosphat. Z. Anorg. Chem. 1964, 328, 113–126. [Google Scholar] [CrossRef]
- Krishnamurthy, R.; Guntha, S.; Eschenmoser, A. Regioselective α-phosphorylation of aldoses in aqueous solution. Angew. Chem. Int. Ed. 2000, 39, 2281–2285. [Google Scholar] [CrossRef]
- Mullen, L.B.; Sutherland, J.D. Formation of Potentially Prebiotic Amphiphiles by Reaction of β-Hydroxy-n-alkylamines with Cyclotriphosphate. Angew. Chem. Int. Ed. 2007, 46, 4166–4168. [Google Scholar] [CrossRef] [PubMed]
- Anastasi, C.; Buchet, F.F.; Crowe, M.A.; Helliwell, M.; Raftery, J.; Sutherland, J.D. The Search for a Potentially Prebiotic Synthesis of Nucleotides via Arabinose-3-phosphate and Its Cyanamide Derivative. Chem. Eur. J. 2008, 14, 2375–2388. [Google Scholar] [CrossRef] [PubMed]
- Coggins, A.J.; Powner, M. W. Prebiotic synthesis of phosphoenol pyruvate by α-phosphorylation-controlled triose glycolysis. Nat. Chem. 2016, 9, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, R. Recherches sur la formation et la transformation des esters LXXXIII [l] Réactions de condensation et/ou de phosphorylation, en solution aqueuse, de divers composés organiques a fonctions -OH, COOH, NH, ou autres, à l’aide de polyphosphates linéaires ou cycliques. Helv. Chim. Acta 1969, 52, 2663–2671. [Google Scholar]
- Rabinowitz, R.; Flores, J.; Krebsbacg, R.; Rogers, G. Peptide Formation in the Presence of Linear or Cyclic polyphosphates. Nature 1969, 224, 795–796. [Google Scholar] [CrossRef] [PubMed]
- Thilo, E. Zur Strukturchemie der kondensierten anorganischen Phosphate. Angew. Chem. 1965, 77, 1056–1066. [Google Scholar] [CrossRef]
- Chung, N.M.; Lohrmann, R.; Orgel, L.E. The mechanism of the trimetaphosphate-induced peptide synthesis. Tetrahedron 1971, 27, 1205–1210. [Google Scholar] [CrossRef]
- Li, Y.M.; Yin, Y.W.; Zhao, Y.F. Phosphoryl group participation leads to peptide formation from N-phosphorylamino acids. Int. J. Pept. Prot. Res. 1992, 39, 375–381. [Google Scholar] [CrossRef]
- Ji, G.J.; Xue, C.B.; Zeng, J.N.; Li, L.P.; Chai, W.G.; Zhao, Y.F. Synthesis of N-(Diisopropyloxyphosphoryl)amino acids and peptides. Synthesis 1988, 6, 444–448. [Google Scholar] [CrossRef]
- Ni, F.; Gao, X.; Zhao, Z.X.; Huang, C.; Zhao, Y.F. On the Electrophilicity of Cyclic Acylphosphoramidates (CAPAs) Postulated as Intermediates. Eur. J. Org. Chem. 2009, 3026–3035. [Google Scholar] [CrossRef]
- Lia, H.; Fu, A.; Jianga, Y.; Zhao, Y.; Liuaa, J. New and Efficient Approach to Aryl Phosphoramidate Derivatives of AZT/d4T as Anti-HIV Prodrugs. Synlett 2004, 14, 2600–2602. [Google Scholar]
- Ju, Y.; Zhao, Y.F.; Sha, Y.; Tan, B. Phosphoryl Promotion and Differentiation Effect on Amino Acids and Prebiotic Synthesis of Protein. Phosphorus Sulfur Silicon Relat. Elem. 1995, 101, 117–123. [Google Scholar] [CrossRef]
- Ni, F.; Fu, C.; Gao, X.; Liu, Y.; Xu, P.; Liu, L.; Lv, Y.; Fu, S.; Sun, Y.; Han, D.; et al. N-phosphoryl amino acid models for P–N bonds in prebiotic chemical evolution. Sci. China Chem. 2015, 58, 374–382. [Google Scholar] [CrossRef]
- Zhou, W.; Ju, Y.; Zhao, Y.F.; Wang, Q.; Luo, G. Simultaneous Formation of Peptides and Nucleotides. Orig. Life Evol. Biosph. 1996, 26, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.L.; Orgel, L.E. Amino Acid Activation with Adenosine -Phosphorimidazolide. J. Mol. Evol. 1978, 11, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.L.; Orgel, L.E. The formation of peptides from the 2’(3’)-Glycyl Ester of a Nucleotide. J. Mol. Evol. 1978, 11, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Jauker, M.; Griesser, H.; Richert, C. Copying of RNA Sequences without Pre-Activation. Angew. Chem. Int. Ed. 2015, 54, 14559–14563. [Google Scholar] [CrossRef] [PubMed]
- Griesser, H.; Bechthold, M.; Tremmel, P.; Kervio, E.; Richert, C. Amino Acid-Specific, Ribonucleotide-Promoted Peptide Formation in the Absence of Enzymes. Angew. Chem. Int. Ed. 2017, 56, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Griesser, H.; Tremmel, P.; Kervio, E.; Pfeffer, C.; Steiner, H.E.; Richert, C. Ribonucleotides and RNA Promote Peptide Chain Growth. Angew. Chem. Int. Ed. 2017, 56, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, J.M.; Schatz, M.N. The Nonenzymatic Activation of Acetate by Adenosine Triphosphate-Bivalent Metal Chelates. J. Biol. Chem. 1961, 236, 305–307. [Google Scholar] [PubMed]
- Lohrmann, R.; Orgel, L.E. Prebiotic Activation Process. Nature 1973, 244, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L.E.; Lohrmann, R. Prebiotic Chemistry and Nucleic Acid Replication. Acc. Chem. Res. 1974, 7, 368–377. [Google Scholar] [CrossRef]
- Sleeper, H.L.; Orgel, L.E. The Catalysis of Nucleotides Polymerization by Compounds of Divalent Lead. J. Mol. Evol. 1979, 12, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H. Catalysis of Internucleotide Bond Formation by Divalent Metal Ions. J. Am. Chem. Soc. 1976, 98, 7837–7839. [Google Scholar] [CrossRef]
- Lohrmann, R.; Orgel, L.E. Preferential formation of (2'–5’)-linked internucleotide bonds in non-enzymatic reactions. Tetrahedron 1978, 34, 853–855. [Google Scholar] [CrossRef]
- Burton, F.G.; Lohrmann, R.; Orgel, L.E. On the possible role of crystals in the origins of life. J. Mol. Evol. 1974, 3, 141–150. [Google Scholar] [PubMed]
- Fahrenbach, A.C. Template-directed nonenzymatic oligonucleotide synthesis: Lessons from synthetic chemistry. Pure Appl. Chem. 2015, 87, 205–218. [Google Scholar] [CrossRef]
- Li, L.; Lelyveld, V.S.; Prywes, N.; Szostak, J.W. Experimental and Computational Evidence for a Loose Transition State in Phosphoroimidazolide Hydrolysis. J. Am. Chem. Soc. 2016, 138, 3986–3989. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H.; Orgel, L.E. Oligonucleotide synthesis catalyzed by the zinc(2+) ion. J. Am. Chem. Soc. 1975, 97, 3532–3533. [Google Scholar] [CrossRef] [PubMed]
- Sleeper, H.L.; Lohrmann, R.; Orgel, L.E. Template-directed Synthesis of Oligoadenylates Catalyzed by Pb2+ ions. J. Mol. Evol. 1979, 13, 203–314. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Orgel, L.E. Substituent Control of the Poly©-Directed Oligomerization of Guanosine 5’-Phosphoroimidazolide. J. Am. Chem. Soc. 1981, 103, 7666–7667. [Google Scholar] [CrossRef]
- Kunio, K.; Ferris, J.P. Clay Catalysis of Oligonucleotide Formation: Kinetics of the Reaction of the 5'-Phosphorimidazolides of Nucleotides with the Non-Basic Heterocycles Uracil and Hypoxanthine. Orig. Life Evol. Biosph. 1999, 29, 563–591. [Google Scholar]
- Joshi, P.C.; Aldersley, M.F.; Zagorevskii, D.V.; Ferris, J.P. A Nucleotide Dimer Synthesis Without Protecting Groups Using Montmorillonite as Catalyst. Nucleosides Nucleotides Nucleic Acids 2012, 31, 536–566. [Google Scholar] [CrossRef] [PubMed]
- Prabahar, K.J.; Cole, T.D.; Ferris, J.P. Effect of Phosphate Activating Group on Oligonucleotide Formation on Montmorillonite: The regioselective Formation of 3’,5’-Linked Oligoadenylates. J. Am. Chem. Soc. 1994, 116, 10914–10920. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Prywes, N.; Tam, C. P.; O’Flaherty, D.K.; Lelyveld, V.S.; Izgu, E.C.; Pal, A.; Szostak, J.W. Enhanced Nonenzymatic RNA Copying with 2-Aminoimidazole Activated Nucleotides. J. Am. Chem. Soc. 2017, 139, 1810–1813. [Google Scholar] [CrossRef] [PubMed]
- Burcar, T.B.; Jawed, M.; Shah, H.; McGown, L.B. In situ Imidazole Activation of Ribonucleotides for Abiotic RNA Oligomerization Reactions. Orig. Life Evol. Biosph. 2015, 45, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Duvernay, F.; Chiavassa, T.; Borget, B.; Aycard, J.P. Experimental Study of Water−Ice Catalyzed Thermal Isomerization of Cyanamide into Carbodiimide: Implication for Prebiotic Chemistry. J. Am. Chem. Soc. 2004, 126, 7772–7773. [Google Scholar] [CrossRef] [PubMed]
- Aylward, N. A plausible Route to a Prebiotic Synthesis of L-Histidine. WSEAS Trans. Biol. Biomed. 2012, 9, 14–23. [Google Scholar]
- Le Corre, S.S.; Berchel, M.; Couthon-Gourvès, H.; Haelters, J.P.; Jaffrès, P.A. Atherton-Todd reaction: Mechanism, scope and applications. Beilstein J. Org. Chem. 2014, 10, 1166–1196. [Google Scholar] [CrossRef] [PubMed]
- Atherton, F.R.; Openshaw, H.T.; Todd, A.R. Studies on phosphorylation. Part II. The reaction of dialkyl phosphites with polyhalogen compounds in presence of bases. A new method for the phosphorylation of amines. J. Chem. Soc. 1945, 660–663. [Google Scholar] [CrossRef]
- Moffatt, J.G.; Khorana, H.G. Nucleoside Polyphosphates. X. The Synthesis and Some Reactions of Nucleoside-5'Phosphoromorpholidates and Related Compounds. Improved Methods for the Preparation of Nucleoside-5'Polyphosphates. J. Am. Chem. Soc. 1961, 83, 649–658. [Google Scholar] [CrossRef]
- Chambers, R.W.; Moffatt, J.G.; Khorana, H.G. The preparation of nucleoside 5'-phosphoramidates and the specific synthesis of nucleotide coenzymes. J. Am. Chem. Soc. 1957, 79, 4240–4241. [Google Scholar] [CrossRef]
- Clark, V.M.; Kirby, G.W.; Todd, A. Studies on phosphorylation. Part XV. The use of phosphoramidic esters in acylation. A new preparation of adenosine-5′ pyrophosphate and adenosine-5′ triphosphate. J. Chem. Soc. 1957, 1497–1501. [Google Scholar] [CrossRef]
- Letsinger, R.L.; Ogilvie, K.K. Nucleotide chemistry. XIII. Synthesis of oligothymidylates via phosphotriester intermediates. J. Am. Chem. Soc. 1961, 91, 3350–3355. [Google Scholar] [CrossRef]
- McBride, L.J.; Caruthers, M.H. An investigation of several deoxynucleoside phosphoramidites useful for synthesizing deoxyoligonucleotides. Tetrahedron Lett. 1983, 24, 245–248. [Google Scholar] [CrossRef]
- Mohamady, M.; Taylor, S.D. Synthesis of Nucleoside 5′-Tetraphosphates Containing Terminal Fluorescent Labels via Activated Cyclic Trimetaphosphate. J. Org. Chem. 2014, 79, 2308–2313. [Google Scholar] [CrossRef] [PubMed]
- Mohamady, M.; Taylor, S.D. Synthesis of Nucleoside Triphosphates from 2′-3′-Protected Nucleosides Using Trimetaphosphate. Org. Lett. 2016, 18, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Schramm, G.; Wissmann, H. Peptidsynthesen mit Hilfe von Polyphosphorsäureestern. Chem. Ber. 1958, 91, 1073–1082. [Google Scholar] [CrossRef]
- Fahmy, M.A.; Fukuto, T.R.; Myers, R.O.; March, R.B. Selective toxicity of new N-phosphorothioylarbamate esters. J. Agric. Food Chem. 1970, 18, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.S. Phosphazenes and Phosphazanes—The Nature of the PN Bond. Phosphorus Sulfur Silicon Relat. Elem. 1994, 87, 101–111. [Google Scholar] [CrossRef]
- Rupper, P.; Gaan, S.; Salimova, V.; Heuberger, M. Characterization of chars obtained from cellulose treated with phosphoramidate flame retardants. J. Anal. Appl. Pyrolysis 2010, 87, 93–98. [Google Scholar] [CrossRef]
- Hamon, N.; Quintiliani, M.; Balzarini, J.; McGuigan, C. Synthesis and biological evaluation of prodrugs of 2-fluoro-2-deoxyribose-1-phosphate and 2,2-difluoro-2-deoxyribose-1-phosphate. Bioorg. Med. Chem. Lett. 2013, 23, 2555–2559. [Google Scholar] [CrossRef] [PubMed]
- Pradere, U.; Garnier-Amblard, E.C.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis of Nucleoside Phosphate and Phosphonate Prodrugs. Chem. Rev. 2014, 114, 9154–9218. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.-J.; Jonghe, S.D.; Herdewijn, P. Synthesis of a Nucleobase-Modified ProTide Library. Org. Lett. 2016, 18, 5816–5819. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Groaz, E.; Margamuljana, L.; Herdewijn, P. Syntheses of 5’-Nucleoside Monophosphate Derivatives with Unique Aminal, Hemiaminal, and Hemithioaminal Functionalities: A New Class of 5’-Peptidyl Nucleotides. Chem. Eur. J. 2016, 22, 8167–8180. [Google Scholar] [CrossRef] [PubMed]
- Derouiche, A.; Cousin, C.; Mijakovic, I. Protein phosphorylation from the perspective of systems biology. Curr. Opin. Biotechnol. 2012, 23, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. Why nature chose phosphate to modify proteins. Phil. Trans. R. Soc. B 2012, 367, 2513–2516. [Google Scholar] [CrossRef] [PubMed]
- Ciesla, J.; Fraczyk, T.; Rode, W. Phosphorylation of basic amino acid residues in proteins: Important but easily missed. Acta Biochim. Polon. 2011, 58, 137–148. [Google Scholar] [PubMed]
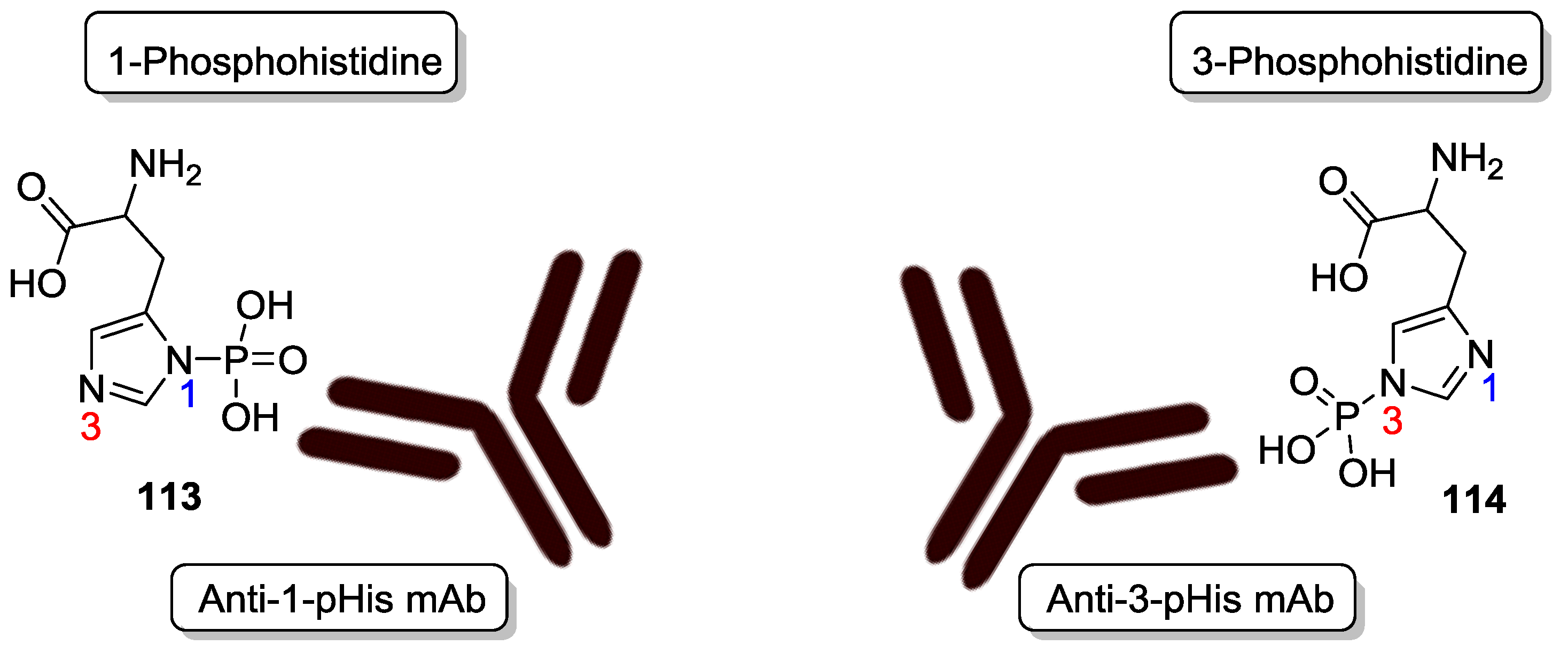
- Attwood, P.V.; Piggott, M.J.; Zu, X.L.; Besant, P.G. Focus on phosphohistidine. Amino Acids 2007, 32, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Crovello, C.S.; Furie, B.C.; Furie, B. Histidine phosphorylation of P-selectin upon stimulation of human platelets: A novel pathway for activation-dependent signal transduction. Cell 1995, 82, 279–286. [Google Scholar] [CrossRef]
- Fuhs, S.R.; Meisenhelder, J.; Aslanian, A.; Ma, L.; Zagorska, A.; Stankova, M.; Binnie, A.; Al-Obeidi, F.; Mauger, J.; Lemke, G.; et al. Monoclonal 1-and 3-phosphohistidine antibodies: New tools to study histidine phosphorylation. Cell 2015, 162, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Fiske, C.H.; Subbarow, Y. The nature of the "inorganic phosphate" in voluntary muscle. Science 1927, 65, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Eggleton, P.; Eggleton, G.P. The Inorganic Phosphate and a Labile Form of Organic Phosphate in the Gastrocnemius of the Frog. Biochem. J. 1927, 21, 190–195. [Google Scholar] [CrossRef] [PubMed]
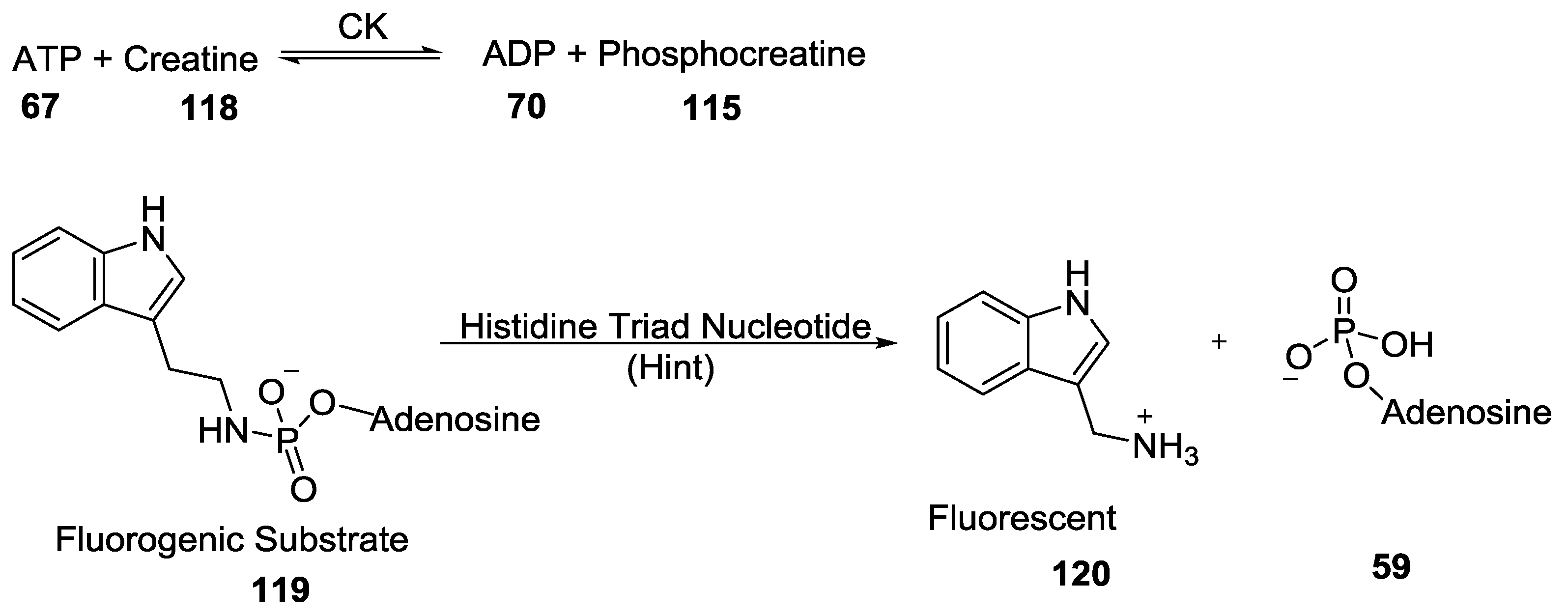
- Chou, T.F.; Baraniak, J.; Kaczmarek, R.; Zhou, X.; Cheng, J.; Ghosh, B.; Wagner, C.R. Phosphoramidate pronucleotides: A comparison of the phosphoramidase substrate specificity of human and Escherichia coli histidine triad nucleotide binding proteins. Mol. Pharm. 2007, 4, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Marcia, E. The role of phosphorus in chemical evolution. Chem. Soc. Rev. 2005, 34, 691–701. [Google Scholar]
- Fegley, B.; Lewis, J.S. Volatile element chemistry in the solar nebula: Na, K, F, Cl, Br, and P. Icarus 1980, 41, 439–455. [Google Scholar] [CrossRef]
- Sutton, E.C.; Blake, G.A.; Masson, C.R.; Phillips, T.G. Molecular line survey of Orion A from 215 to 247 GHz. Ap. J. Suppl. 1985, 58, 341. [Google Scholar] [CrossRef]
- Ziurys, L.M. Detection of interstellar P–N: The first phosphorus-bearing species observed in molecular clouds. Astrophys. J. 1987, 321, L81–L85. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.E.; Bally, J. Detection of interstellar P-N—The first identified phosphorus compound in the interstellar medium. Astrophys. J. 1987, 321, L75–L79. [Google Scholar] [CrossRef]
- Rivilla, V.M.; Fontani, F.; Beltrán, M.T.; Vasyunin, A.; Caselli, P.; Martín-Pintado, J.; Cesaroni, R. The first detections of the key prebiotic molecule PO in star-forming regions. Astrophys. J. 2016, 826, 161. [Google Scholar] [CrossRef]
- Ferris, J.P.; Khwaja, H. Laboratory simulations of PH3 photolysis in the atmospheres of Jupiter and Saturn. Icarus 1985, 62, 415–424. [Google Scholar] [CrossRef]
- Kim, H.-J.; Furukawa, Y.; Kakegawa, T.; Bita, A.; Scorei, R.; Benner, S.A. Evaporite Borate-Containing Mineral Ensembles Make Phosphate Available and Regiospecifically Phosphorylate Ribonucleosides: Borate as a Multifaceted Problem Solver in Prebiotic Chemistry. Angew. Chem. Int. Ed. 2016, 55, 15816–15820. [Google Scholar] [CrossRef] [PubMed]
- Burcar, B.; Pasek, M.; Gull, M.; Cafferty, B.J.; Velasco, F.; Hud, N.V.; Menor-Salvan, C. Darwin’s Warm Little Pond: A One-Pot Reaction for Prebiotic Phosphorylation and the Mobilization of Phosphate from Minerals in a Urea-Based Solvent. Angew. Chem. Int. Ed. 2016, 55, 13249–13253. [Google Scholar] [CrossRef] [PubMed]
- Pasek, M.A.; Kee, T.P.; Bryant, D.E.; Pavlov, A.A.; Lunine, J.I. Production of potentially prebiotic condensed phosphates by phosphorus redox chemistry. Angew. Chem. Int. Ed. 2008, 47, 7918–7920. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.E.; Kee, T.P. Direct evidence for the availability of reactive, water soluble phosphorus on the early earth. H-phosphinic acid from the Nantan meteorite. Chem. Commun. 2006, 22, 2344–2346. [Google Scholar] [CrossRef] [PubMed]
- Gorrell, I.B.; Wang, L.; Marks, A.J.; Bryant, D.E.; Bouillot, F.; Goddard, A.; Heard, D.E.; Kee, T.P. On the origin of Murchison meteorite phosphonates. Implications for prebiotic chemistry. Chem. Commun. 2006, 21, 1643–1645. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.E.; Greenfield, D.; Walshaw, R.D.; Evans, S. M.; Nimmo, A.E.; Smith, C.; Wang, L.; Pasek, M.A.; Kee, T.P. Electrochemical studies of iron meteorites. Phosphorus redox chemistry on the early earth. Int. J. Astrobiol. 2009, 8, 27–36. [Google Scholar] [CrossRef]
- Bryant, D.E.; Marriott, K.E.R.; Macgregor, S.A.; Kilner, C.; Pasek, M.A.; Kee, T.P. On the prebiotic potential of reduced oxidation state phosphorus: The H-phosphinate-pyruvate system. Chem. Commun. 2010, 46, 3726–3728. [Google Scholar] [CrossRef] [PubMed]
- Gull, M.; Zhou, M.; Fernández, F.M.; Pasek, M.A. Prebiotic phosphate ester syntheses in a deep eutectic solvent. J. Mol. Evol. 2014, 78, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Powner, M.W.; Sutherland, J.D. Prebiotic chemistry: A new modus operandi. Phil. Trans. R. Soc. B 2011, 366, 2870–2877. [Google Scholar] [CrossRef] [PubMed]
- Kluger, R.; Davis, P.P.; Adawadkar, P.D. Mechanism of urea participation in phosphonate ester hydrolysis. Mechanistic and stereochemical criteria for enzymic formation and reaction of phosphorylated biotin. J. Am. Chem. Soc. 1979, 101, 5995–6000. [Google Scholar] [CrossRef]
- Gull, M.; Mojica, M.A.; Fernández, F.M.; Gaul, D.A.; Orlando, T.M.; Liotta, C.L.; Pasek, M.A. Nucleoside phosphorylation by the mineral schreibersite. Sci. Rep. 2015, 5, 17198. [Google Scholar] [CrossRef] [PubMed]
- Hazen, R.M. Paleomineralogy of the Hadean Eon: A preliminary species list. Am. J. Sci. 2013, 313, 807–843. [Google Scholar] [CrossRef]
- Gull, M.; Pasek, M.A. Is struvite a prebiotic mineral? Life 2013, 3, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Handschuh, G.J.; Orgel, L.E. Struvite and prebiotic phosphorylation. Science 1973, 179, 483–484. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P.; Nicodem, D.E. Ammonia photolysis and the Role of ammonia in chemical revolution. Nature 1972, 238, 268–269. [Google Scholar] [CrossRef]
- Smirnov, A.; Hausner, D.; Laffers, R.; Strongin, D.R.; Schoonen, M.A.A. Abiotic ammonium formation in the presence of Ni-Fe metals and alloys and its implications for the Hadean nitrogen cycle. Geochem. Trans. 2008, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Cruz, N.L.L.; Qasim, D.; Abbott-Lyon, H.; Pirim, C.; McKee, A.D.; Orlando, T.; Gull, M.; Lindsay, D.; Pasek, M.A. The evolution of the surface of the mineral schreibersite in prebiotic chemistry. Phys. Chem. Chem. Phys. 2016, 18, 20160–20167. [Google Scholar]
- Summers, D.P. Sources and sinks for ammonia and nitrite on the early Earth and the reaction of nitrite with ammonia. Orig. Life Evol. Biosph. 1999, 29, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Henderson-Sellers, A.; Schwartz, A.W. Chemical evolution and ammonia in the early Earth's atmosphere. Nature 1980, 287, 526–528. [Google Scholar] [CrossRef]
- Summer, D.P.; Chang, S. Prebiotic ammonia from reduction of nitrite by iron (II) on the early Earth. Nature 1993, 365, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Cleaves, H.J.; Chalmers, J.H.; Lazcano, A.; Miller, S.L.; Bada, J.L. A reassessment of prebiotic organic synthesis in neutral planetary atmospheres. Orig. Life Evol. Biosph. 2008, 38, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Pizzarello, S.; Williams, L.B.; Lehman, J.; Holland, G.P.; Yarger, J.L. Abundant ammonia in primitive asteroids and the case for a possible exobiology. Proc. Natl. Acad. Sci. USA 2011, 108, 4303–4306. [Google Scholar] [CrossRef] [PubMed]
- Singireddy, S.; Gordon, A.D.; Smirnov, A.; Vance, M.A.; Schoonen, M.A.; Szilagyi, R.K.; Strongin, D.R. Reduction of nitrite and nitrate to ammonium on pyrite. Orig. Life Evol. Biosph. 2012, 42, 275–294. [Google Scholar] [CrossRef] [PubMed]
- Brandes, J.A.; Boctor, N.Z.; Cody, G.D.; Cooper, B.A.; Hazen, R.M.; Yoder, H.S., Jr. Abiotic nitrogen reduction on the early Earth. Nature 1998, 395, 365. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.H. Earth's Early Atmosphere and Oceans, and The Origin of Life; Springer: Cham (ZG), Switzerland, 2016. [Google Scholar]
- Buchwald, V.F. Jerslev, the first iron meteorite from Denmark. Meteorit. Planet. Sci. 1986, 21, 47–58. [Google Scholar] [CrossRef]
- Rabinowitz, J.; Woeller, F.; Flores, J.; Krebsbach, R. Electric discharge reactions in mixtures of phosphine, methane, ammonia and water. Nature 1969, 224, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Glindemann, D.; Edwards, M.; Schrems, O. Phosphine and methylphosphine production by simulated lightning—A study for the volatile phosphorus cycle and cloud formation in the earth atmosphere. Atmos. Environ. 2004, 38, 6867–6874. [Google Scholar] [CrossRef]
- Glindemann, D.; Edwards, M.; Morgenstern, P. Phosphine from rocks: Mechanically driven phosphate reduction? Environ. Sci. Technol. 2005, 39, 8295–8299. [Google Scholar] [CrossRef] [PubMed]

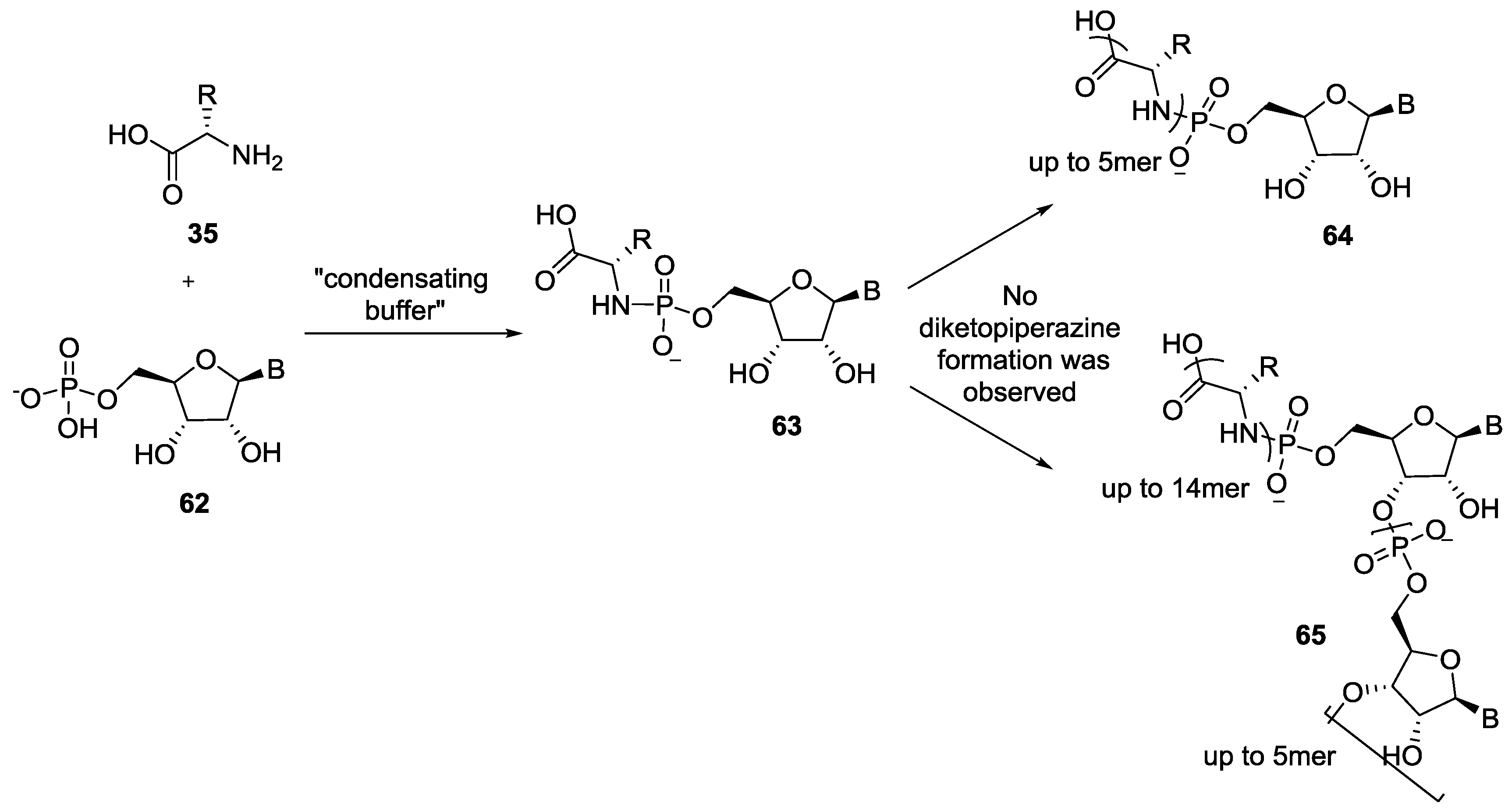
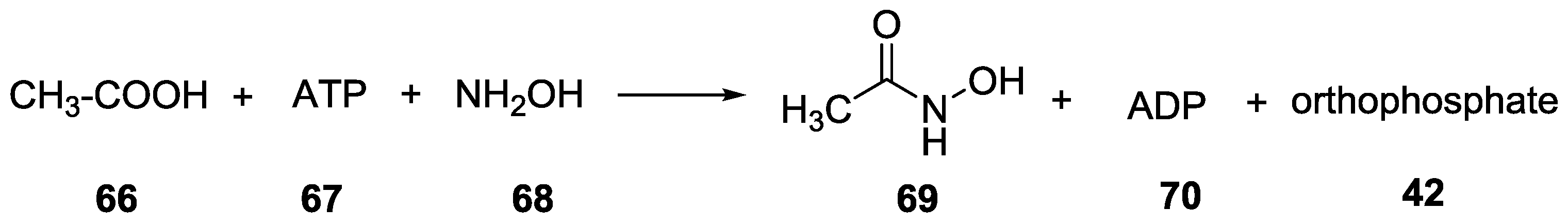
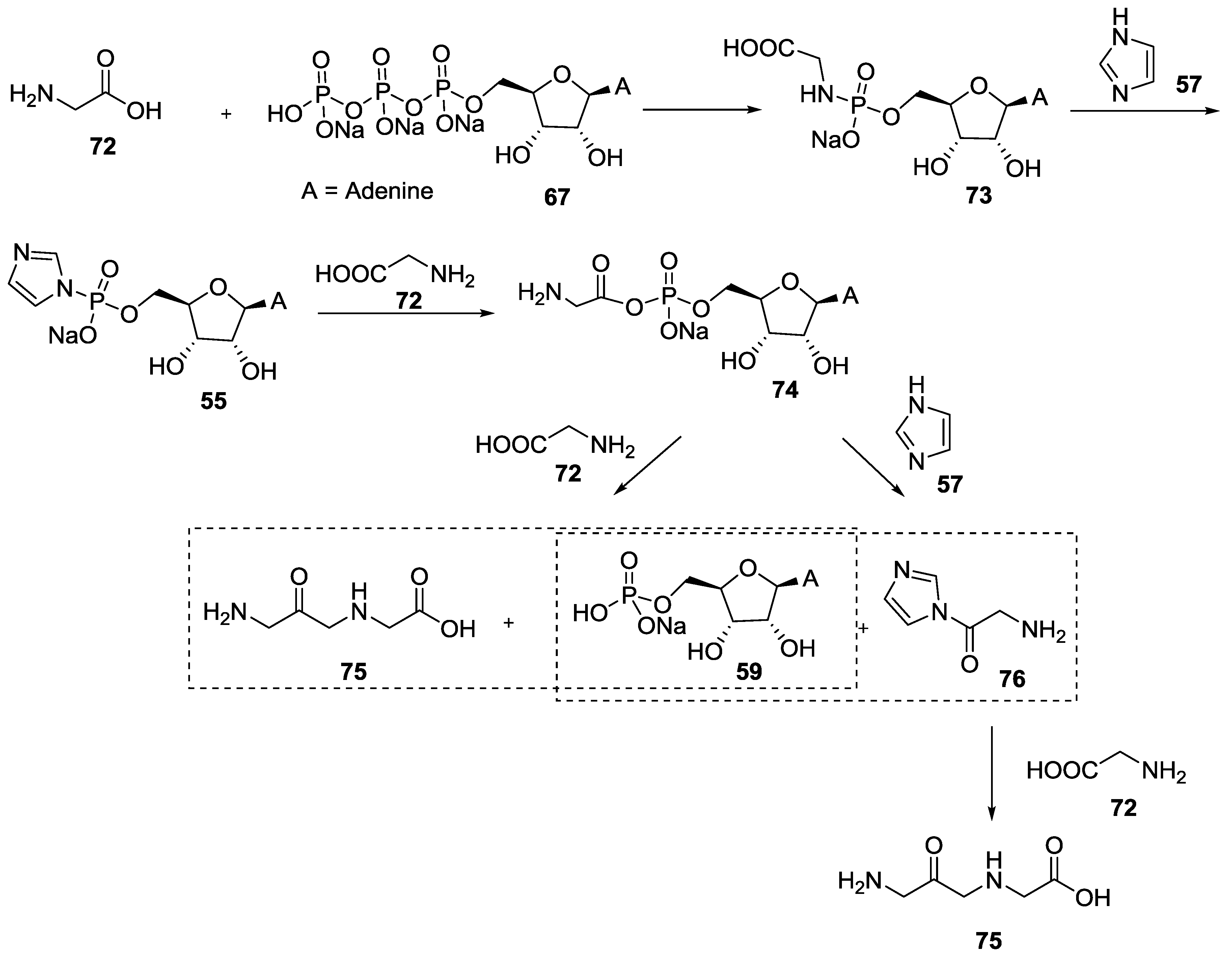
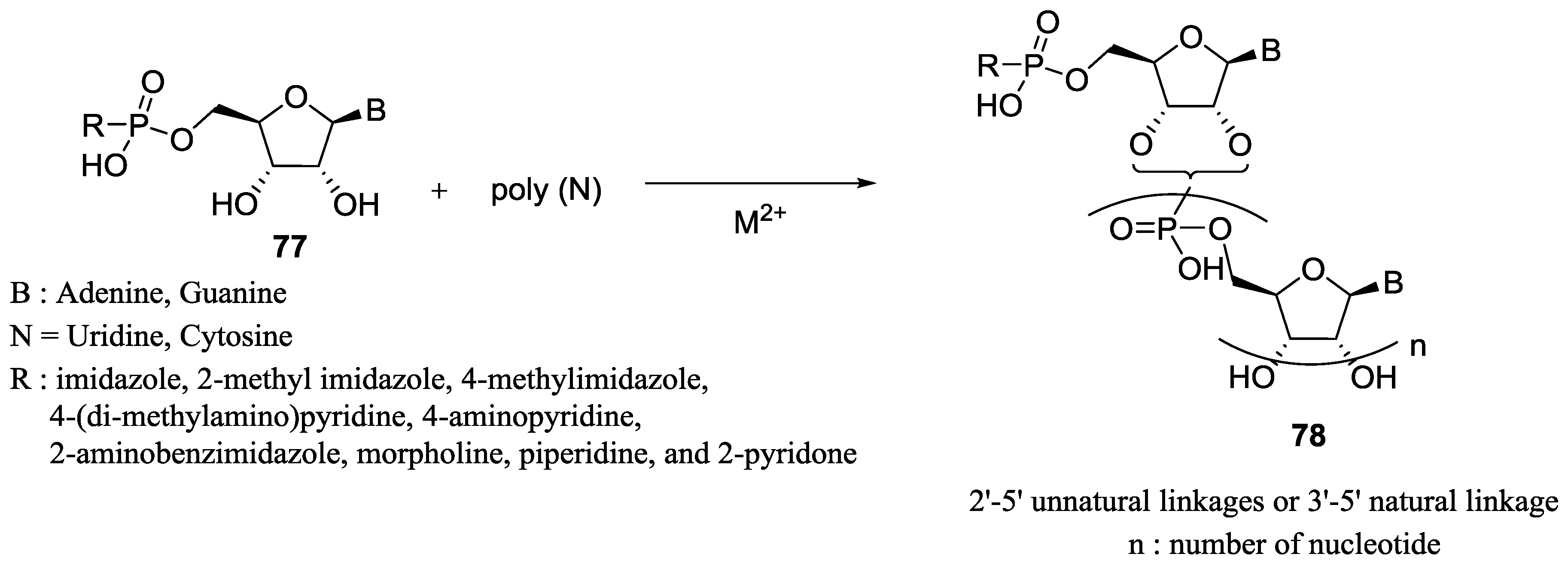


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karki, M.; Gibard, C.; Bhowmik, S.; Krishnamurthy, R. Nitrogenous Derivatives of Phosphorus and the Origins of Life: Plausible Prebiotic Phosphorylating Agents in Water. Life 2017, 7, 32. https://doi.org/10.3390/life7030032
Karki M, Gibard C, Bhowmik S, Krishnamurthy R. Nitrogenous Derivatives of Phosphorus and the Origins of Life: Plausible Prebiotic Phosphorylating Agents in Water. Life. 2017; 7(3):32. https://doi.org/10.3390/life7030032
Chicago/Turabian StyleKarki, Megha, Clémentine Gibard, Subhendu Bhowmik, and Ramanarayanan Krishnamurthy. 2017. "Nitrogenous Derivatives of Phosphorus and the Origins of Life: Plausible Prebiotic Phosphorylating Agents in Water" Life 7, no. 3: 32. https://doi.org/10.3390/life7030032