
Archaeal Clusters of Orthologous Genes (arCOGs): An Update and Application for Analysis of Shared Features between Thermococcales, Methanococcales, and Methanobacteriales




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Sequences and Basic Sequence Analysis
2.2. Genome Weighting
2.3. Clade Representation in arCOGs
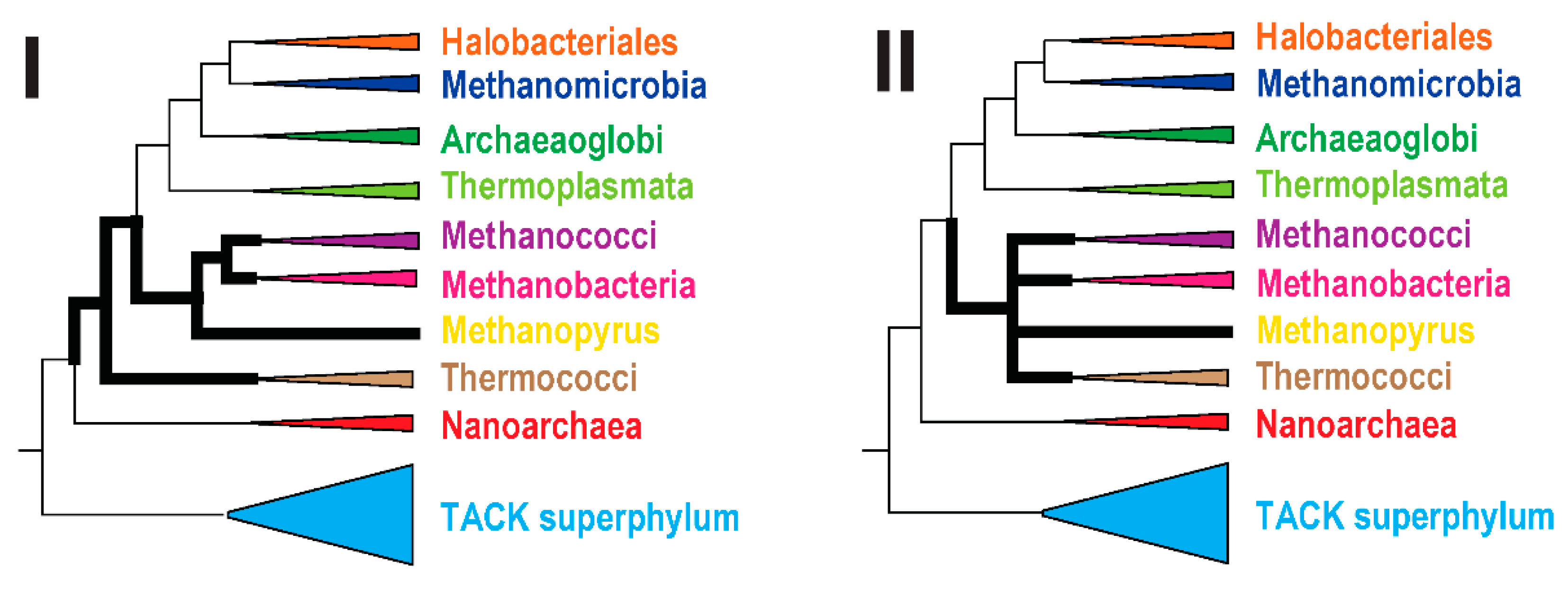
2.4. Reconstruction of Archaeal Phylogeny Using arCOGs
- weights of leaves descending from a node include at least 0.75 of the total weight of the given clade;
- among the leaves descending from a node, at least one other clade is represented by at least 0.75 of the total weight of this clade;
- among the internal nodes descending from a node, there is no other node satisfying the two criteria described above.
3. Results and Discussion
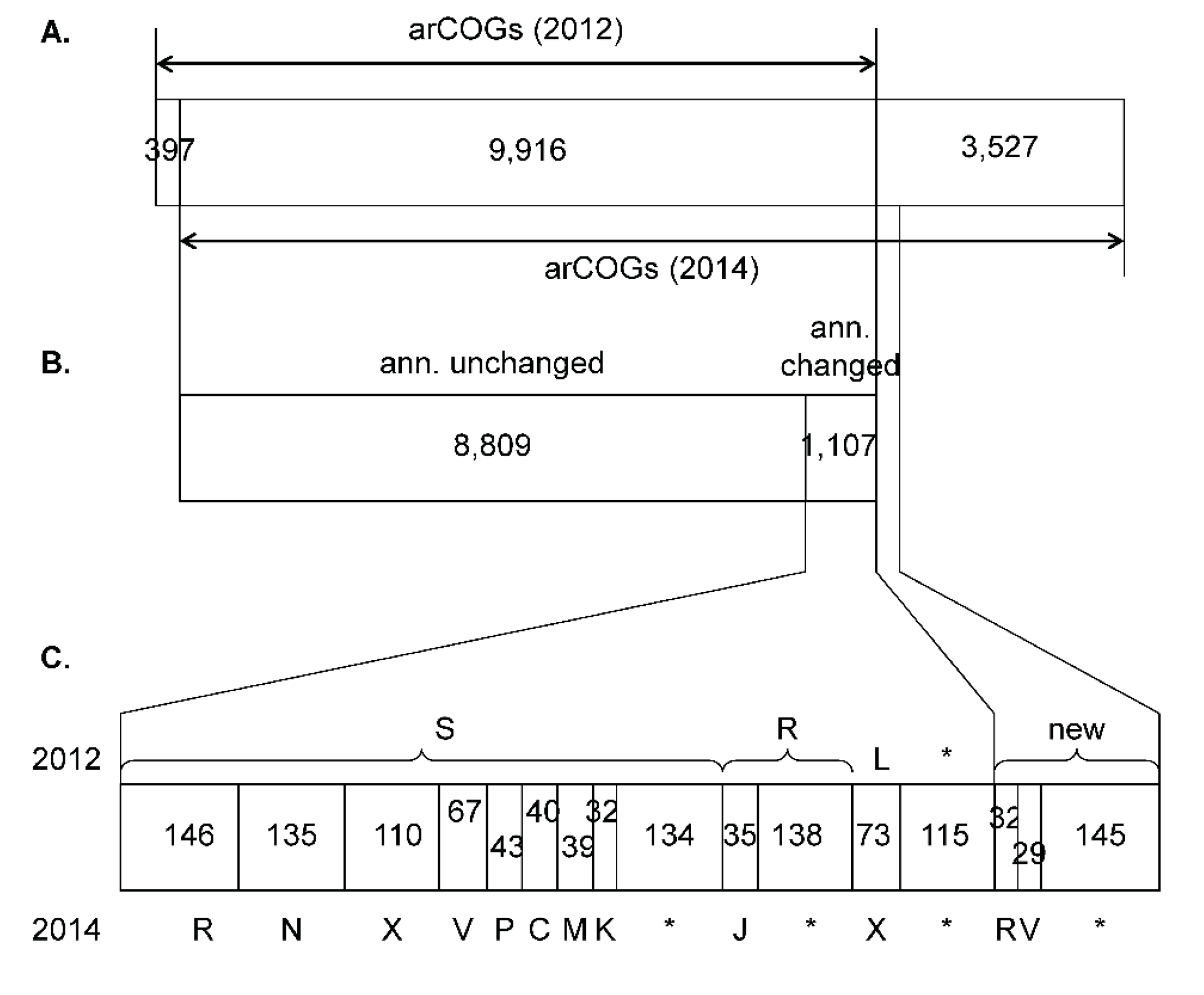
3.1. Update of the arCOGs
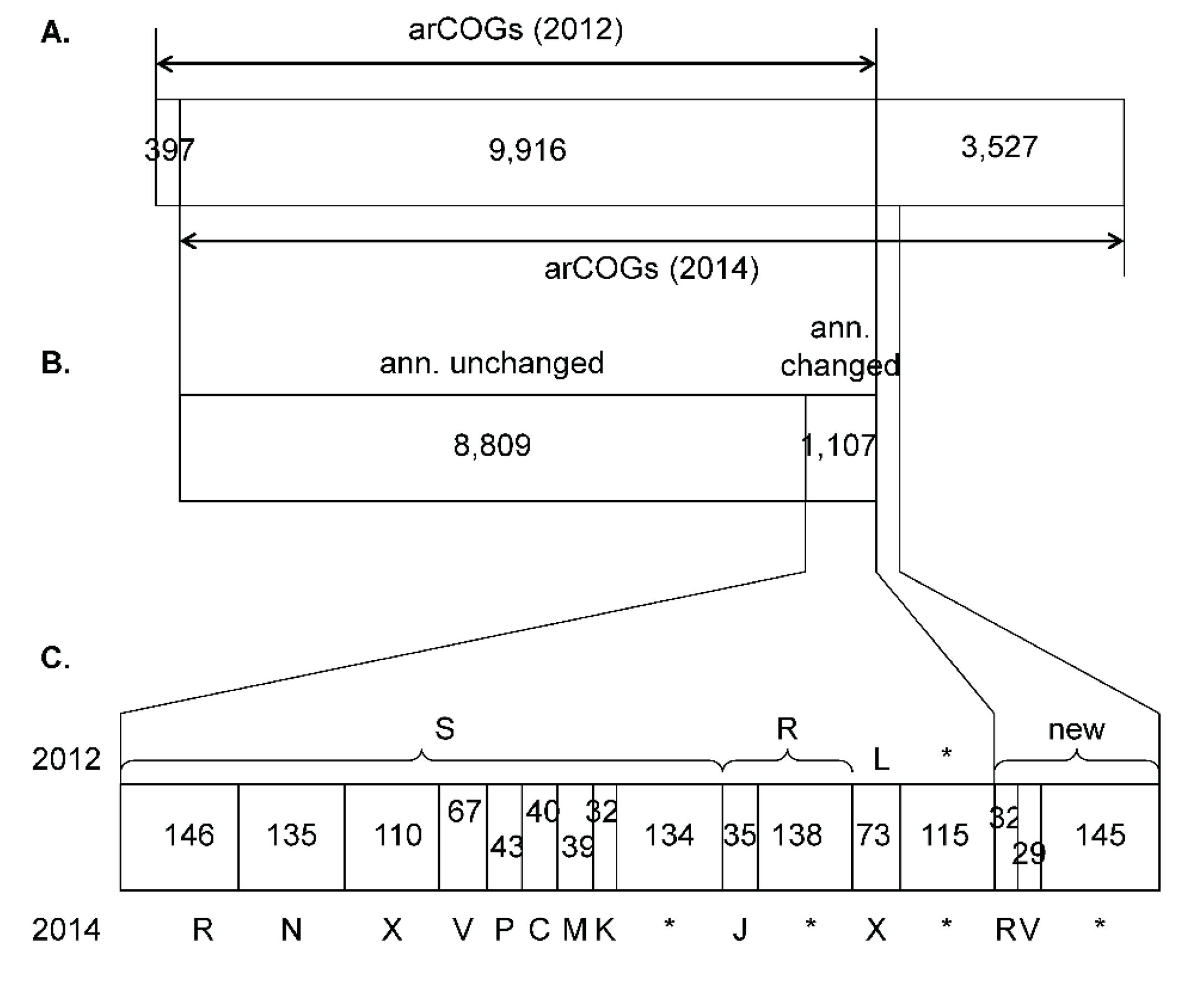
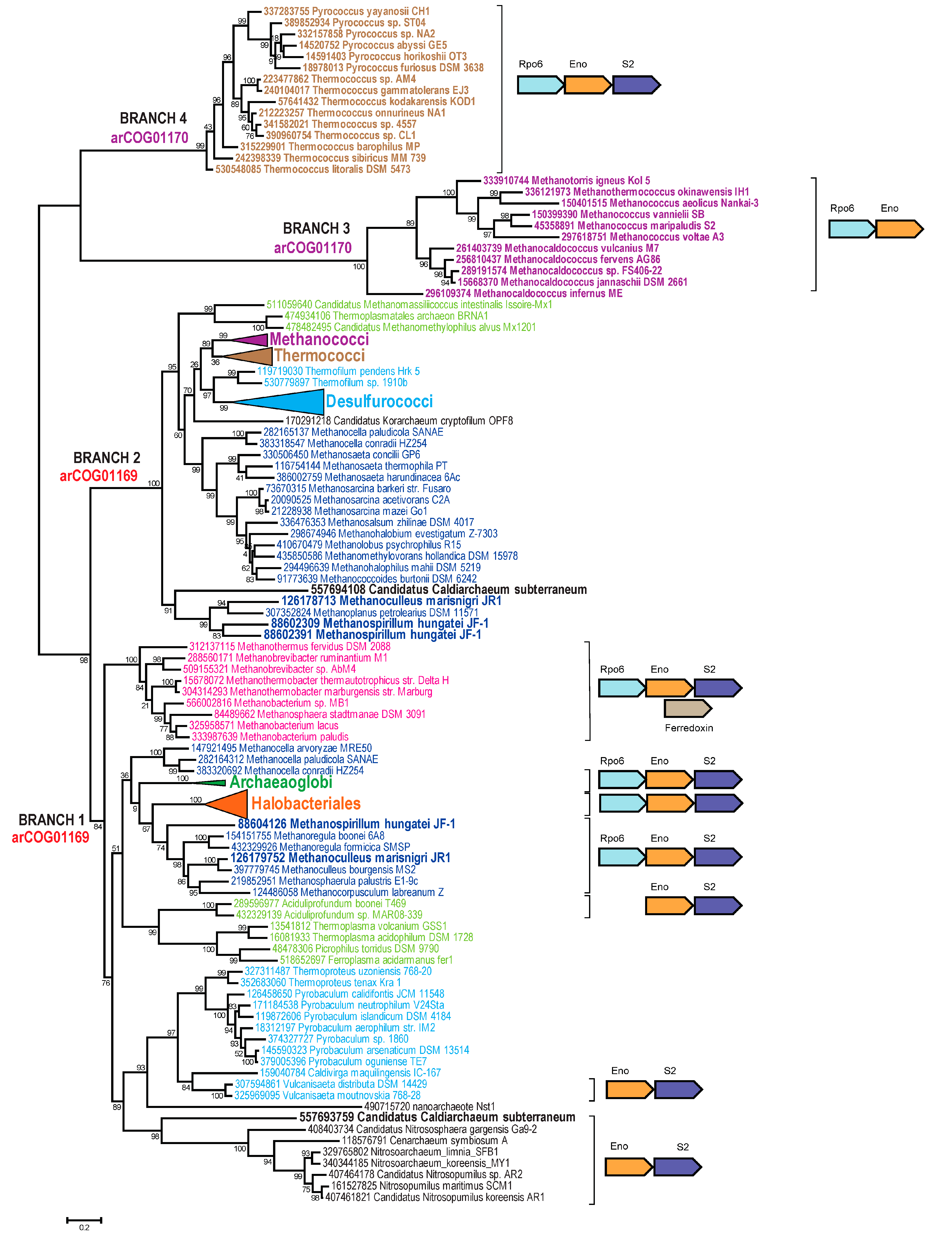
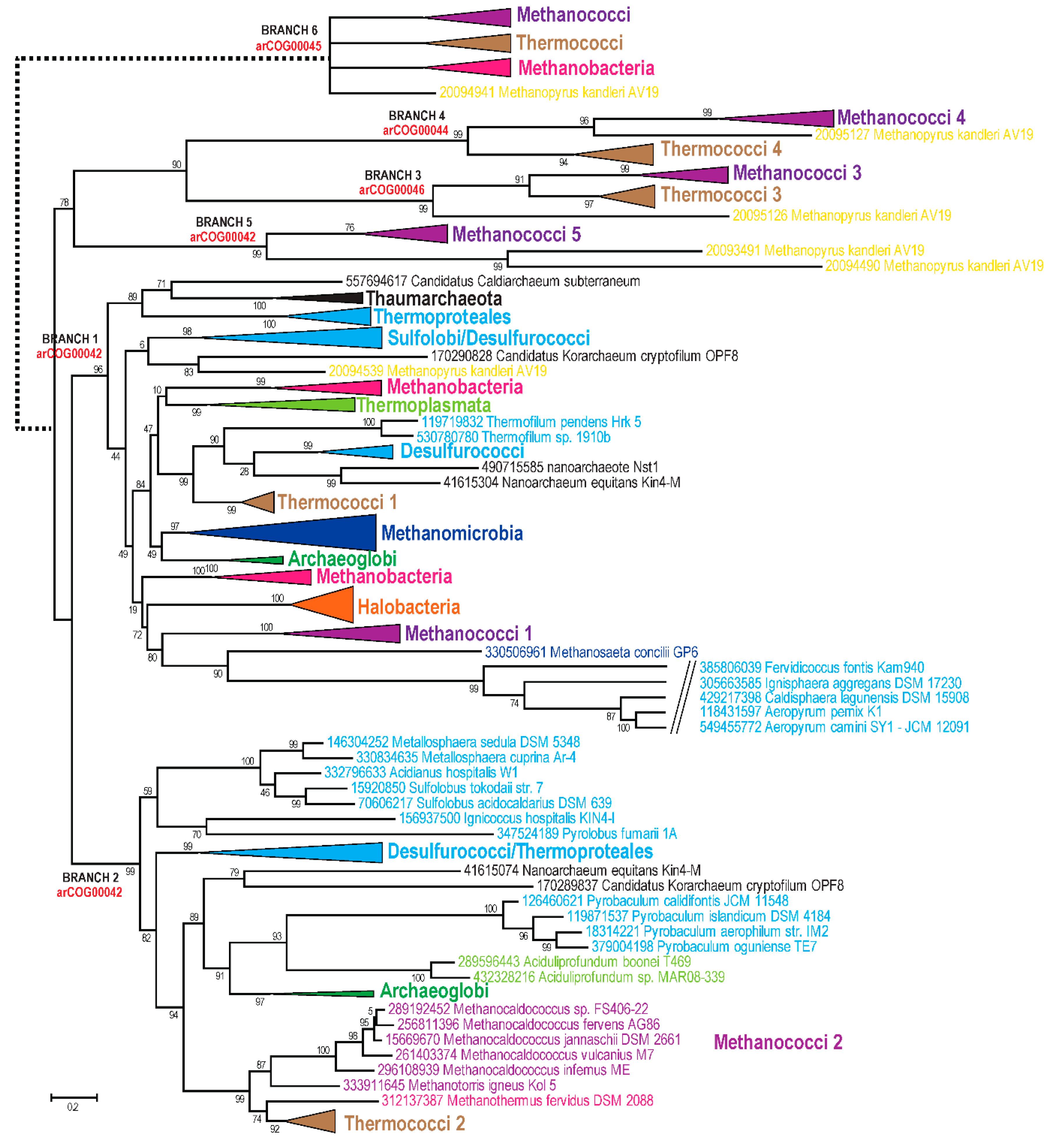
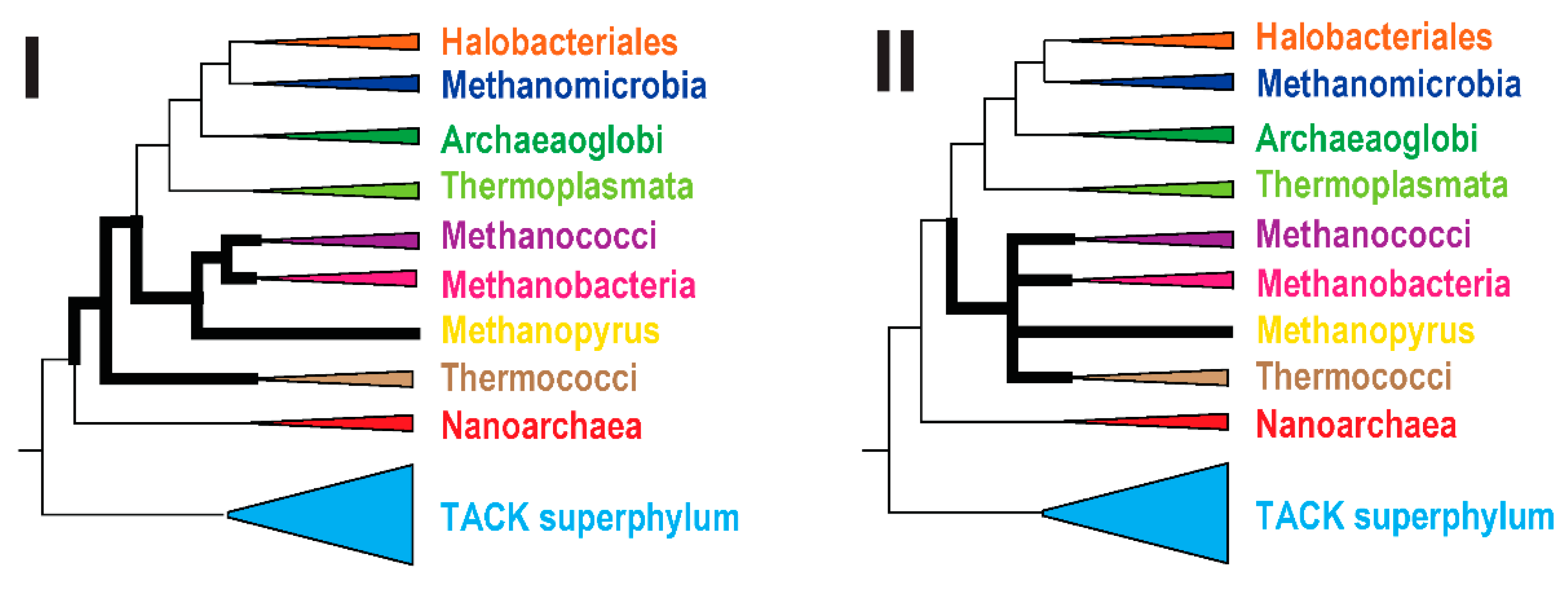
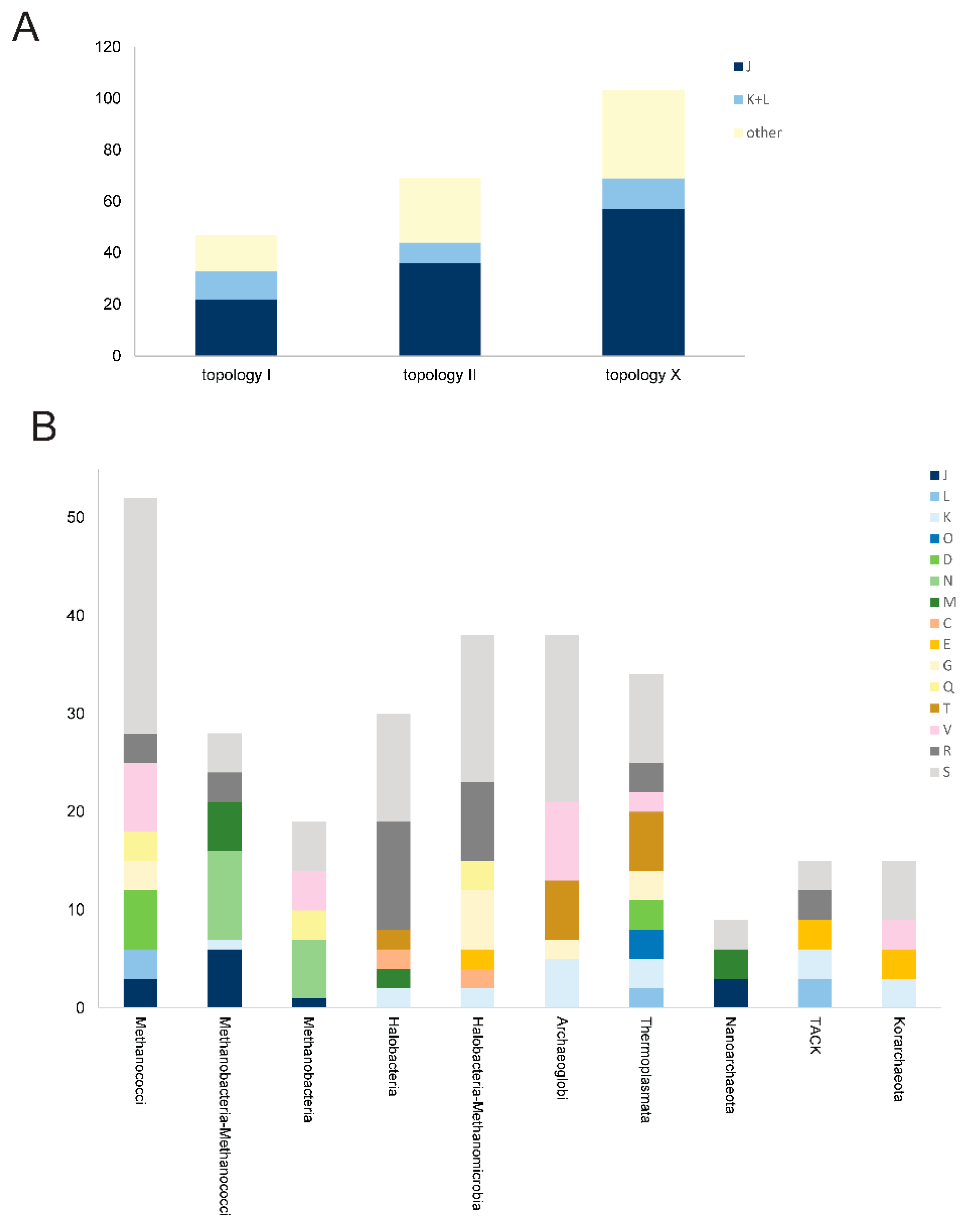
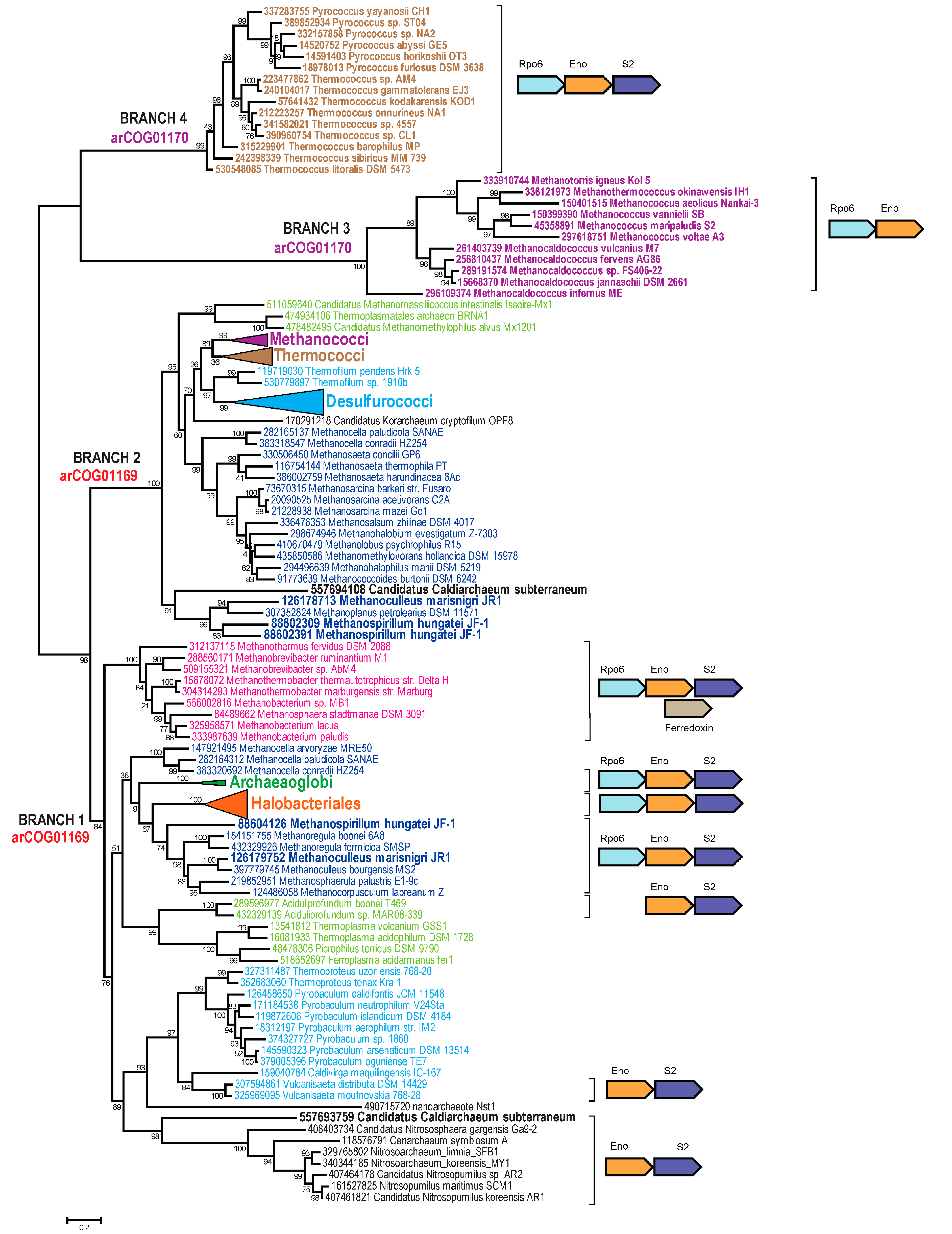
3.2. Analysis of the Shared Features of the Thermococci with Methanococci, Methanobacteria, and Methonopyrus
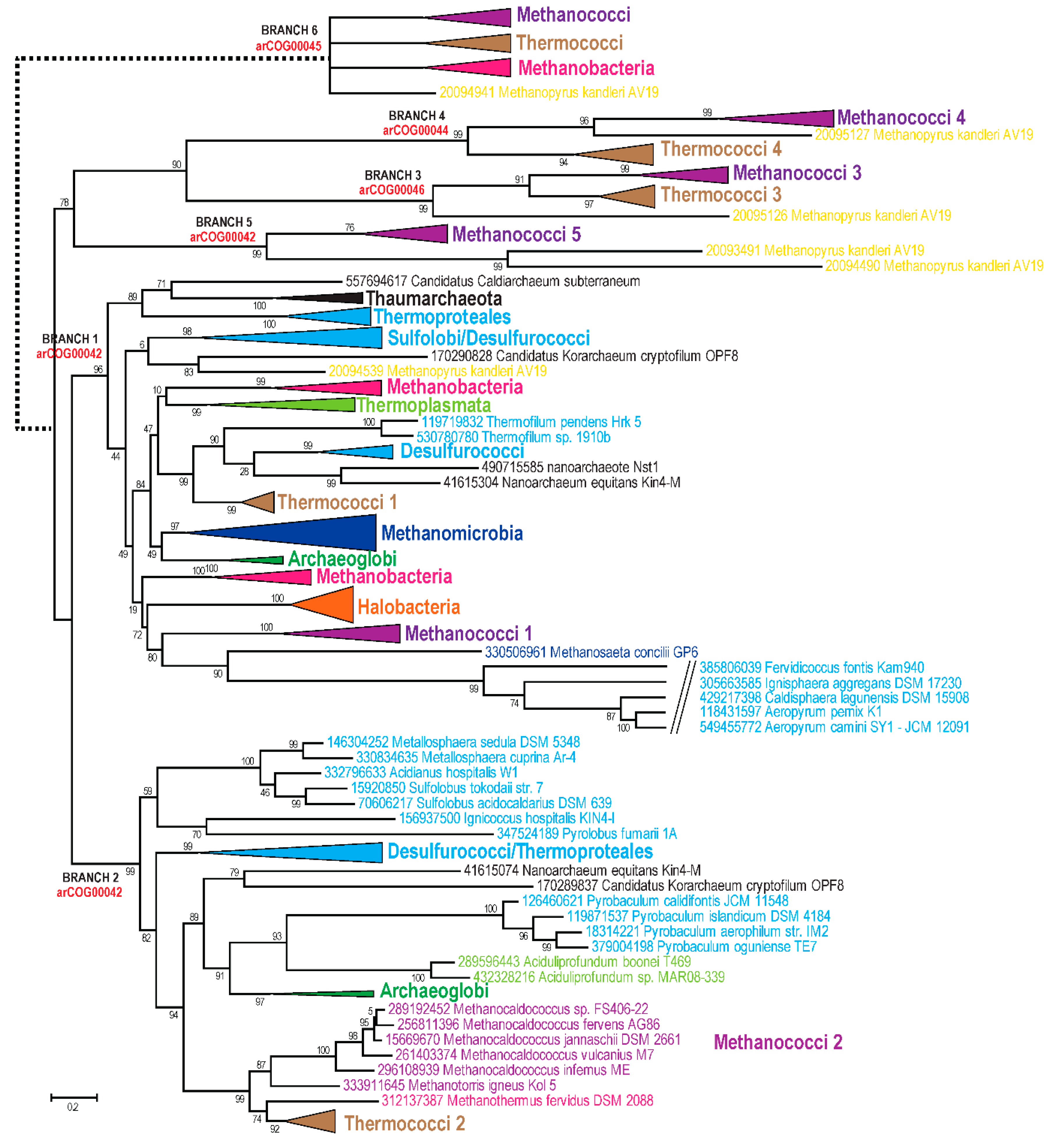
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Halachev, M.R.; Loman, N.J.; Pallen, M.J. Calculating orthologs in bacteria and Archaea: A divide and conquer approach. PLoS One 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, D.M.; Wolf, Y.I.; Mushegian, A.R.; Koonin, E.V. Computational methods for Gene Orthology inference. Brief. Bioinform. 2011, 12, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Altenhoff, A.M.; Dessimoz, C. Inferring orthology and paralogy. Methods Mol. Biol. 2012, 855, 259–279. [Google Scholar] [PubMed]
- Trachana, K.; Larsson, T.A.; Powell, S.; Chen, W.H.; Doerks, T.; Muller, J.; Bork, P. Orthology prediction methods: A quality assessment using curated protein families. Bioessays 2011, 33, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Poux, S.; Magrane, M.; Arighi, C.N.; Bridge, A.; O’Donovan, C.; Laiho, K. Expert curation in UniProtKB: A case study on dealing with conflicting and erroneous data. Database 2014. [Google Scholar] [CrossRef]
- Matsuya, A.; Sakate, R.; Kawahara, Y.; Koyanagi, K.O.; Sato, Y.; Fujii, Y.; Yamasaki, C.; Habara, T.; Nakaoka, H.; Todokoro, F.; et al. Evola: Ortholog database of all human genes in H-InvDB with manual curation of phylogenetic trees. Nucleic Acids Res. 2008, 36, D787–D792. [Google Scholar] [CrossRef] [PubMed]
- Mazandu, G.K.; Mulder, N.J. The use of semantic similarity measures for optimally integrating heterogeneous Gene Ontology data from large scale annotation pipelines. Front. Genet. 2014, 5. [Google Scholar] [CrossRef]
- Trachana, K.; Forslund, K.; Larsson, T.; Powell, S.; Doerks, T.; von Mering, C.; Bork, P. A phylogeny-based benchmarking test for orthology inference reveals the limitations of function-based validation. PLoS One 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Bocs, S.; Danchin, A.; Medigue, C. Re-annotation of genome microbial coding-sequences: Finding new genes and inaccurately annotated genes. BMC Bioinform. 2002, 3. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Sorokin, A.V.; Novichkov, P.S.; Wolf, Y.I.; Koonin, E.V. Clusters of orthologous genes for 41 archaeal genomes and implications for evolutionary genomics of archaea. Biol. Direct 2007, 2. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Makarova, K.S.; Yutin, N.; Koonin, E.V. Updated clusters of orthologous genes for Archaea: A complex ancestor of the Archaea and the byways of horizontal gene transfer. Biol. Direct 2012, 7. [Google Scholar] [CrossRef]
- Yutin, N.; Puigbo, P.; Koonin, E.V.; Wolf, Y.I. Phylogenomics of prokaryotic ribosomal proteins. PLoS One 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Marinsek, N.; Barry, E.R.; Makarova, K.S.; Dionne, I.; Koonin, E.V.; Bell, S.D. GINS, a central nexus in the archaeal DNA replication fork. EMBO Rep. 2006, 7, 539–545. [Google Scholar] [PubMed]
- Makarova, K.; Kelman, Z.; Koonin, E.V. The archaeal CMG (CDC45/RecJ, MCM, GINS) complex is a conserved component of the DNA replication system in all archaea and eukaryotes. Biol. Direct 2012, 13. [Google Scholar] [CrossRef]
- Makarova, K.S.; Anantharaman, V.; Grishin, N.V.; Koonin, E.V.; Aravind, L. CARF and WYL domains: Ligand-binding regulators of prokaryotic defense systems. Front. Genet. 2014, 5. [Google Scholar] [CrossRef]
- Makarova, K.S.; Aravind, L.; Wolf, Y.I.; Koonin, E.V. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Direct 2011, 6. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E.V. Two new families of the FtsZ-tubulin protein superfamily implicated in membrane remodeling in diverse bacteria and archaea. Biol. Direct 2010, 5. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E.V. Archaeology of eukaryotic DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Forterre, P.; Prangishvili, D.; Krupovic, M.; Koonin, E.V. Dark matter in archaeal genomes: A rich source of novel mobile elements, defense systems and secretory complexes. Extremophiles 2014, 18, 877–893. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Snir, S.; Koonin, E.V. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 2011, 193, 6039–6056. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Yutin, N.; Bell, S.D.; Koonin, E.V. Evolution of diverse cell division and vesicle formation systems in Archaea. Nat. Rev. Microbiol. 2010, 8, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Krupovic, M.; Koonin, E.V. Evolution of replicative DNA polymerases in archaea and their contributions to the eukaryotic replication machinery. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Esser, D.; Pham, T.K.; Reimann, J.; Albers, S.V.; Siebers, B.; Wright, P.C. Change of carbon source causes dramatic effects in the phospho-proteome of the archaeon Sulfolobus solfataricus. J. Proteome Res. 2012, 11, 4823–4833. [Google Scholar] [CrossRef] [PubMed]
- Podar, M.; Makarova, K.S.; Graham, D.E.; Wolf, Y.I.; Koonin, E.V.; Reysenbach, A.L. Insights into archaeal evolution and symbiosis from the genomes of a nanoarchaeon and its inferred crenarchaeal host from Obsidian Pool, Yellowstone National Park. Biol. Direct 2013, 8. [Google Scholar] [CrossRef]
- Siebers, B.; Zaparty, M.; Raddatz, G.; Tjaden, B.; Albers, S.V.; Bell, S.D.; Blombach, F.; Kletzin, A.; Kyrpides, N.; Lanz, C.; et al. The complete genome sequence of Thermoproteus tenax: A physiologically versatile member of the Crenarchaeota. PLoS One 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podar, M.; Anderson, I.; Makarova, K.S.; Elkins, J.G.; Ivanova, N.; Wall, M.A.; Lykidis, A.; Mavromatis, K.; Sun, H.; Hudson, M.E.; et al. A genomic analysis of the archaeal system Ignicoccus hospitalis-Nanoarchaeum equitans. Genome Biol. 2008, 9. [Google Scholar] [CrossRef]
- Elkins, J.G.; Podar, M.; Graham, D.E.; Makarova, K.S.; Wolf, Y.; Randau, L.; Hedlund, B.P.; Brochier-Armanet, C.; Kunin, V.; Anderson, I.; et al. A korarchaeal genome reveals insights into the evolution of the Archaea. Proc. Natl. Acad. Sci. USA 2008, 105, 8102–8107. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Parisot, N.; Harris, H.M.; Peyretaillade, E.; Gaci, N.; Tottey, W.; Bardot, O.; Raymann, K.; Gribaldo, S.; Peyret, P.; et al. Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genomics 2014, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimann, J.; Esser, D.; Orell, A.; Amman, F.; Pham, T.K.; Noirel, J.; Lindas, A.C.; Bernander, R.; Wright, P.C.; Siebers, B.; et al. Archaeal signal transduction: Impact of protein phosphatase deletions on cell size, motility, and energy metabolism in Sulfolobus acidocaldarius. Mol. Cell. Proteomics 2013, 12, 3908–3923. [Google Scholar] [CrossRef] [PubMed]
- Goncearenco, A.; Berezovsky, I.N. Exploring the evolution of protein function in Archaea. BMC Evol. Biol. 2012, 12. [Google Scholar] [CrossRef]
- Leahy, S.C.; Kelly, W.J.; Altermann, E.; Ronimus, R.S.; Yeoman, C.J.; Pacheco, D.M.; Li, D.; Kong, Z.; McTavish, S.; Sang, C.; et al. The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS One 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Csuros, M.; Miklos, I. Streamlining and large ancestral genomes in Archaea inferred with a phylogenetic birth-and-death model. Mol. Biol. Evol. 2009, 26, 2087–2095. [Google Scholar] [CrossRef] [PubMed]
- Hooper, S.D.; Anderson, I.J.; Pati, A.; Dalevi, D.; Mavromatis, K.; Kyrpides, N.C. Integration of phenotypic metadata and protein similarity in Archaea using a spectral bipartitioning approach. Nucleic Acids Res. 2009, 37, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Lindas, A.C.; Karlsson, E.A.; Lindgren, M.T.; Ettema, T.J.; Bernander, R. A unique cell division machinery in the Archaea. Proc. Natl. Acad. Sci. USA 2008, 105, 18942–18946. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Holmes, A.D.; Smith, A.M.; Tran, D.; Lowe, T.M. The UCSC Archaeal Genome Browser: 2012 update. Nucleic Acids Res. 2012, 40, D646–D652. [Google Scholar] [CrossRef] [PubMed]
- Anderson, I.; Ulrich, L.E.; Lupa, B.; Susanti, D.; Porat, I.; Hooper, S.D.; Lykidis, A.; Sieprawska-Lupa, M.; Dharmarajan, L.; Goltsman, E.; et al. Genomic characterization of methanomicrobiales reveals three classes of methanogens. PLoS One 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- NCBI FTP site. Available online: ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/ (accessed on 6 March 2015).
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2014, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; et al. CDD: Specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009, 37, D205–D210. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Haft, D.H.; Loftus, B.J.; Richardson, D.L.; Yang, F.; Eisen, J.A.; Paulsen, I.T.; White, O. TIGRFAMs: A protein family resource for the functional identification of proteins. Nucleic Acids Res. 2001, 29, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Makarova, K.S.; Mekhedov, S.L.; Wolf, Y.I.; Koonin, E.V. The deep archaeal roots of eukaryotes. Mol. Biol. Evol. 2008, 25, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS One 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, C.; Deschamps, P.; Lopez-Garcia, P.; Moreira, D. Rooting the domain archaea by phylogenomic analysis supports the foundation of the new kingdom proteoarchaeota. Genome Biol. Evol. 2014, 7, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Aravind, L.; Grishin, N.V.; Koonin, E.V. Evolution of aminoacyl-tRNA synthetases—Analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999, 9, 689–710. [Google Scholar] [PubMed]
- NCBI arCOG database. Available online: ftp://ftp.ncbi.nih.gov/pub/wolf/COGs/arCOG/ (accessed on 6 March 2015).
- Koonin, E.V.; Wolf, Y.I. Genomics of bacteria and archaea: The emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008, 36, 6688–6719. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Galperin, M.Y.; Koonin, E.V. Comparative genomic analysis of evolutionarily conserved but functionally uncharacterized membrane proteins in archaea: Prediction of novel components of secretion, membrane remodeling and glycosylation systems. Biochimie 2015. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S. CRISPR-Cas: Evolution of an RNA-based adaptive immunity system in prokaryotes. RNA Biol. 2013, 10, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. The basic building blocks and evolution of CRISPR-Cas systems. Biochem. Soc. Trans. 2013, 41, 1392–1400. [Google Scholar] [PubMed]
- Marquez, V.; Frohlich, T.; Armache, J.P.; Sohmen, D.; Donhofer, A.; Mikolajka, A.; Berninghausen, O.; Thomm, M.; Beckmann, R.; Arnold, G.J.; et al. Proteomic characterization of archaeal ribosomes reveals the presence of novel archaeal-specific ribosomal proteins. J. Mol. Biol. 2011, 405, 1215–1232. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Brockenbrough, J.S.; Paddy, M.R.; Aris, J.P. NCL1, a novel gene for a non-essential nuclear protein in Saccharomyces cerevisiae. Gene 1998, 220, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Mekhedov, S.L.; Mirkin, B.G.; Koonin, E.V. Ancestral paralogs and pseudoparalogs and their role in the emergence of the eukaryotic cell. Nucleic Acids Res. 2005, 33, 4626–4638. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Carl Woese’s vision of cellular evolution and the domains of life. RNA Biol. 2014, 11, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Puigbo, P.; Wolf, Y.I.; Koonin, E.V. Seeing the Tree of Life behind the phylogenetic forest. BMC Biol. 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Beauregard-Racine, J.; Bicep, C.; Schliep, K.; Lopez, P.; Lapointe, F.J.; Bapteste, E. Of woods and webs: Possible alternatives to the tree of life for studying genomic fluidity in E. coli. Biol. Direct 2011, 6. [Google Scholar] [CrossRef]
- Bapteste, E.; O’Malley, M.A.; Beiko, R.G.; Ereshefsky, M.; Gogarten, J.P.; Franklin-Hall, L.; Lapointe, F.J.; Dupre, J.; Dagan, T.; Boucher, Y.; et al. Prokaryotic evolution and the tree of life are two different things. Biol. Direct 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Wolf, Y.I. The fundamental units, processes and patterns of evolution, and the tree of life conundrum. Biol. Direct 2009, 4. [Google Scholar] [CrossRef]
- Bapteste, E.; Susko, E.; Leigh, J.; Ruiz-Trillo, I.; Bucknam, J.; Doolittle, W.F. Alternative methods for concatenation of core genes indicate a lack of resolution in deep nodes of the prokaryotic phylogeny. Mol. Biol. Evol. 2008, 25, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, W.F.; Bapteste, E. Pattern pluralism and the Tree of Life hypothesis. Proc. Natl. Acad. Sci. USA 2007, 104, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Dagan, T.; Martin, W. The tree of one percent. Genome Biol. 2006, 7. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.A.; Embley, T.M. Archaeal “dark matter” and the origin of eukaryotes. Genome Biol. Evol. 2014, 6, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Nelson-Sathi, S.; Sousa, F.L.; Roettger, M.; Lozada-Chavez, N.; Thiergart, T.; Janssen, A.; Bryant, D.; Landan, G.; Schonheit, P.; Siebers, B.; et al. Origins of major archaeal clades correspond to gene acquisitions from bacteria. Nature 2014, 517, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Matte-Tailliez, O.; Brochier, C.; Forterre, P.; Philippe, H. Archaeal phylogeny based on ribosomal proteins. Mol. Biol. Evol. 2002, 19, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Raymann, K.; Forterre, P.; Brochier-Armanet, C.; Gribaldo, S. Global phylogenomic analysis disentangles the complex evolutionary history of DNA replication in archaea. Genome Biol. Evol. 2014, 6, 192–212. [Google Scholar] [CrossRef] [PubMed]
- Brochier, C.; Forterre, P.; Gribaldo, S. An emerging phylogenetic core of Archaea: Phylogenies of transcription and translation machineries converge following addition of new genome sequences. BMC Evol. Biol. 2005, 5. [Google Scholar] [CrossRef] [Green Version]
- Huber, H.; Hohn, M.J.; Rachel, R.; Fuchs, T.; Wimmer, V.C.; Stetter, K.O. A new phylum of Archaea represented by a nanosized hyperthermophilic symbiont. Nature 2002, 417, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.; Hohn, M.J.; Ahel, I.; Graham, D.E.; Adams, M.D.; Barnstead, M.; Beeson, K.Y.; Bibbs, L.; Bolanos, R.; Keller, M.; et al. The genome of Nanoarchaeum equitans: Insights into early archaeal evolution and derived parasitism. Proc. Natl. Acad. Sci. USA 2003, 100, 12984–12988. [Google Scholar] [CrossRef] [PubMed]
- Brochier, C.; Gribaldo, S.; Zivanovic, Y.; Confalonieri, F.; Forterre, P. Nanoarchaea: Representatives of a novel archaeal phylum or a fast-evolving euryarchaeal lineage related to Thermococcales? Genome Biol. 2005, 6. [Google Scholar] [CrossRef] [Green Version]
- Brochier, C.; Forterre, P.; Gribaldo, S. Archaeal phylogeny based on proteins of the transcription and translation machineries: Tackling the Methanopyrus kandleri paradox. Genome Biol. 2004, 5, R17. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Sun, Z.; Arndt, W.; Shi, J.; Friedman, R.; Tang, J. Gene order phylogeny and the evolution of methanogens. PLoS One 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Slesarev, A.I.; Mezhevaya, K.V.; Makarova, K.S.; Polushin, N.N.; Shcherbinina, O.V.; Shakhova, V.V.; Belova, G.I.; Aravind, L.; Natale, D.A.; Rogozin, I.B.; et al. The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens. Proc. Natl. Acad. Sci. USA 2002, 99, 4644–4649. [Google Scholar] [CrossRef] [PubMed]
- Guy, L.; Ettema, T.J. The archaeal “TACK” superphylum and the origin of eukaryotes. Trends Microbiol. 2011, 19, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.A.; Foster, P.G.; Nye, T.M.; Cox, C.J.; Embley, T.M. A congruent phylogenomic signal places eukaryotes within the Archaea. Proc. Biol. Sci. 2012, 279, 4870–4879. [Google Scholar] [CrossRef] [PubMed]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.J.; Comolli, L.R.; Dick, G.J.; Hauser, L.J.; Hyatt, D.; Dill, B.D.; Land, M.L.; Verberkmoes, N.C.; Hettich, R.L.; Banfield, J.F.; et al. Enigmatic, ultrasmall, uncultivated Archaea. Proc. Natl. Acad. Sci. USA 2010, 107, 8806–8811. [Google Scholar] [CrossRef] [PubMed]
- Gatesy, J.; Springer, M.S. Phylogenetic analysis at deep timescales: Unreliable gene trees, bypassed hidden support, and the coalescence/concatalescence conundrum. Mol. Phylogenet. Evol. 2014, 80, 231–266. [Google Scholar] [CrossRef] [PubMed]
- Lanier, H.C.; Huang, H.; Knowles, L.L. How low can you go? The effects of mutation rate on the accuracy of species-tree estimation. Mol. Phylogenet. Evol. 2014, 70, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Roettger, M.; Lockhart, P.J. A reality check for alignments and trees. Trends Genet. 2007, 23, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Brochier-Armanet, C.; Forterre, P.; Gribaldo, S. Phylogeny and evolution of the Archaea: One hundred genomes later. Curr. Opin. Microbiol. 2011, 14, 274–281. [Google Scholar] [CrossRef] [PubMed]
- NCBI FTP site. Available online: ftp://ftp.ncbi.nih.gov/pub/wolf/_suppl/arCOG2014/ (accessed on 6 March 2015).
- Ciccarelli, F.D.; Doerks, T.; von Mering, C.; Creevey, C.J.; Snel, B.; Bork, P. Toward automatic reconstruction of a highly resolved tree of life. Science 2006, 311, 1283–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnik-Habrink, M.; Lewis, R.J.; Mader, U.; Stulke, J. RNA degradation in Bacillus subtilis: An interplay of essential endo- and exoribonucleases. Mol. Microbiol. 2012, 84, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Kaberdin, V.R.; Lin-Chao, S. Unraveling new roles for minor components of the E. coli RNA degradosome. RNA Biol. 2009, 6, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Makarova, K.S.; Murvai, J.; Czabarka, E.; Wolf, Y.I.; Tatusov, R.L.; Szekely, L.A.; Koonin, E.V. Connected gene neighborhoods in prokaryotic genomes. Nucleic Acids Res. 2002, 30, 2212–2223. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Force, A. The probability of duplicate gene preservation by subfunctionalization. Genetics 2000, 154, 459–473. [Google Scholar] [PubMed]
- Bork, P.; Koonin, E.V. A P-loop-like motif in a widespread ATP pyrophosphatase domain: Implications for the evolution of sequence motifs and enzyme activity. Proteins 1994, 20, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, Y.; Soma, A.; Ote, T.; Kato, J.; Sekine, Y.; Suzuki, T. molecular mechanism of lysidine synthesis that determines tRNA identity and codon recognition. Mol. Cell 2005, 19, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Valverde, R.; Edwards, L.; Regan, L. Structure and function of KH domains. FEBS J. 2008, 275, 2712–2726. [Google Scholar] [CrossRef] [PubMed]
- Planet, P.J.; Kachlany, S.C.; DeSalle, R.; Figurski, D.H. Phylogeny of genes for secretion NTPases: Identification of the widespread tadA subfamily and development of a diagnostic key for gene classification. Proc. Natl. Acad. Sci. USA 2001, 98, 2503–2508. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Z.; Stahl, A.O.; Albers, S.V.; Kissinger, J.C.; Driessen, A.J.; Pohlschroder, M. Identification of diverse archaeal proteins with class III signal peptides cleaved by distinct archaeal prepilin peptidases. J. Bacteriol. 2007, 189, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Puigbo, P.; Wolf, Y.I.; Koonin, E.V. Search for a “Tree of Life” in the thicket of the phylogenetic forest. J. Biol. 2009, 8. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Archaeal Clusters of Orthologous Genes (arCOGs): An Update and Application for Analysis of Shared Features between Thermococcales, Methanococcales, and Methanobacteriales. Life 2015, 5, 818-840. https://doi.org/10.3390/life5010818
Makarova KS, Wolf YI, Koonin EV. Archaeal Clusters of Orthologous Genes (arCOGs): An Update and Application for Analysis of Shared Features between Thermococcales, Methanococcales, and Methanobacteriales. Life. 2015; 5(1):818-840. https://doi.org/10.3390/life5010818
Chicago/Turabian StyleMakarova, Kira S., Yuri I. Wolf, and Eugene V. Koonin. 2015. "Archaeal Clusters of Orthologous Genes (arCOGs): An Update and Application for Analysis of Shared Features between Thermococcales, Methanococcales, and Methanobacteriales" Life 5, no. 1: 818-840. https://doi.org/10.3390/life5010818
APA StyleMakarova, K. S., Wolf, Y. I., & Koonin, E. V. (2015). Archaeal Clusters of Orthologous Genes (arCOGs): An Update and Application for Analysis of Shared Features between Thermococcales, Methanococcales, and Methanobacteriales. Life, 5(1), 818-840. https://doi.org/10.3390/life5010818