
What RNA World? Why a Peptide/RNA Partnership Merits Renewed Experimental Attention

Abstract
:1. Introduction
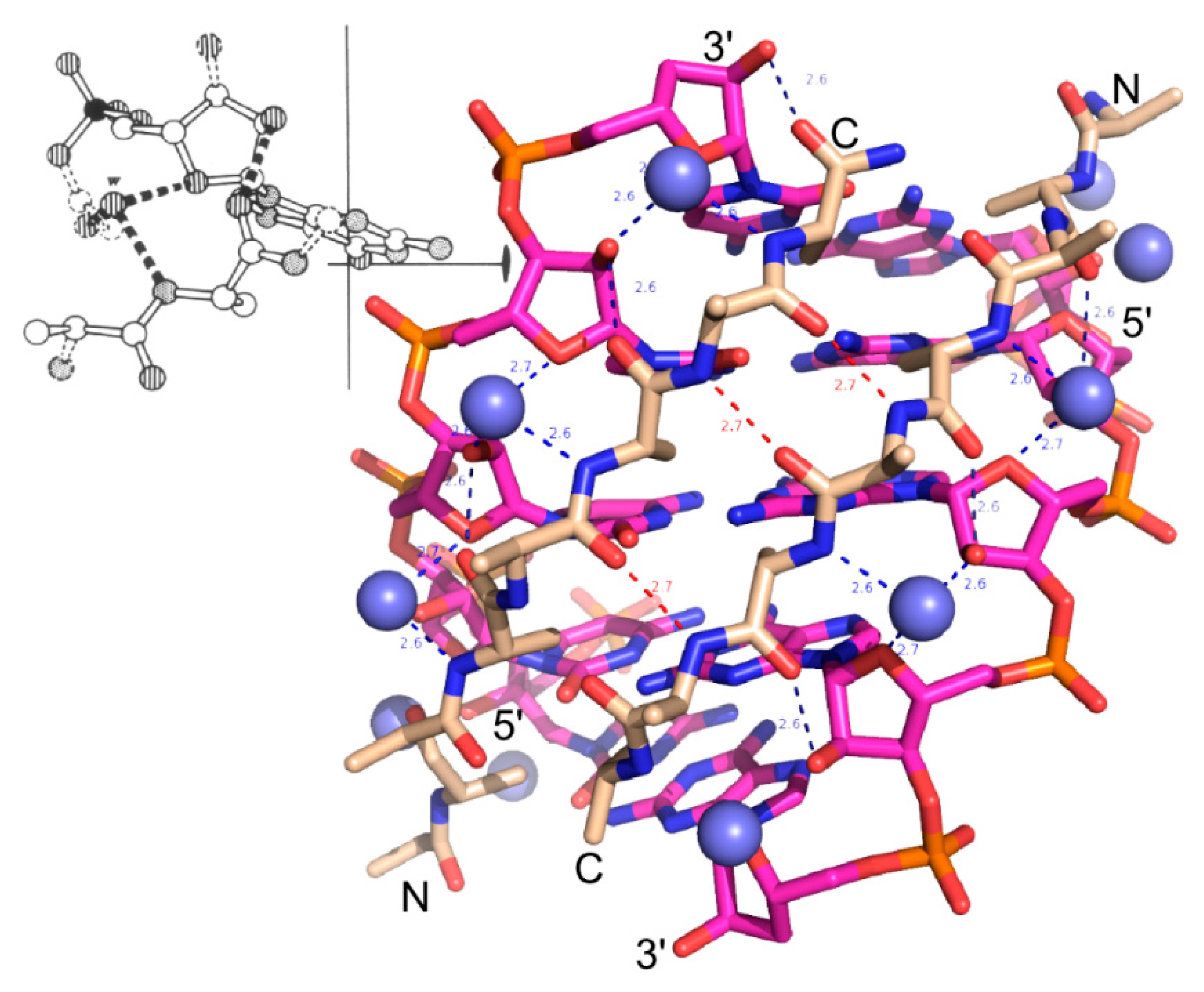

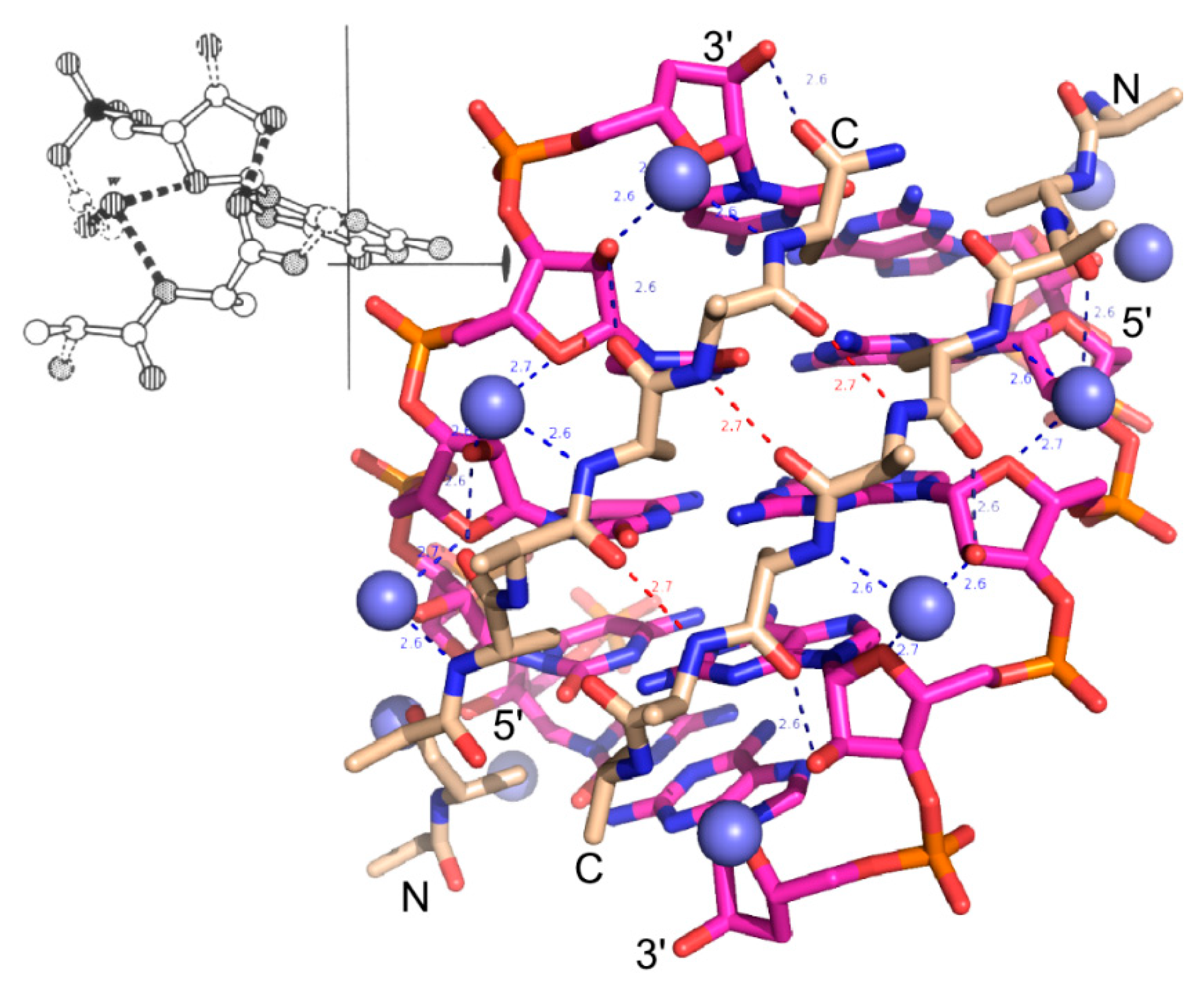
2. Experimental Section
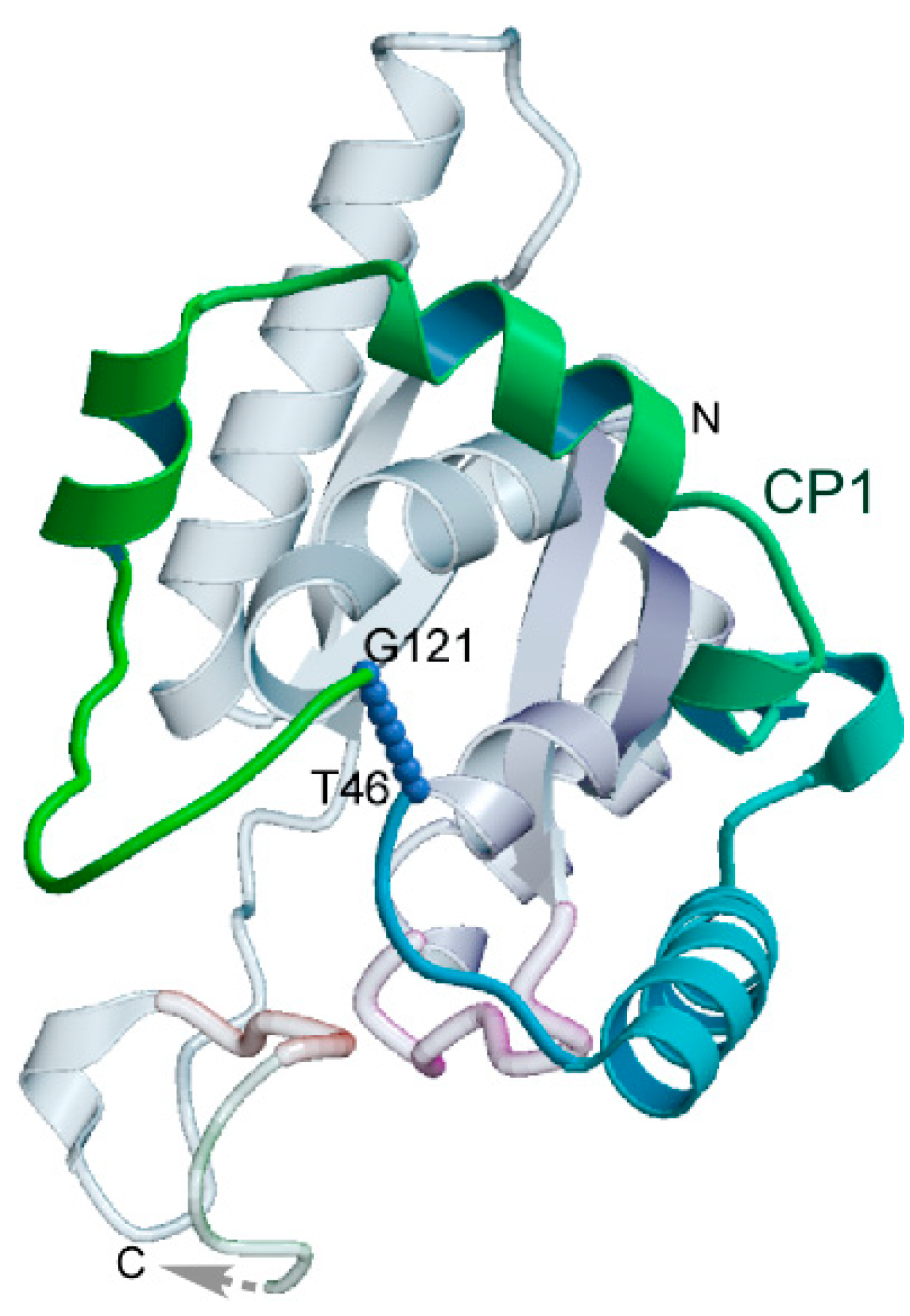

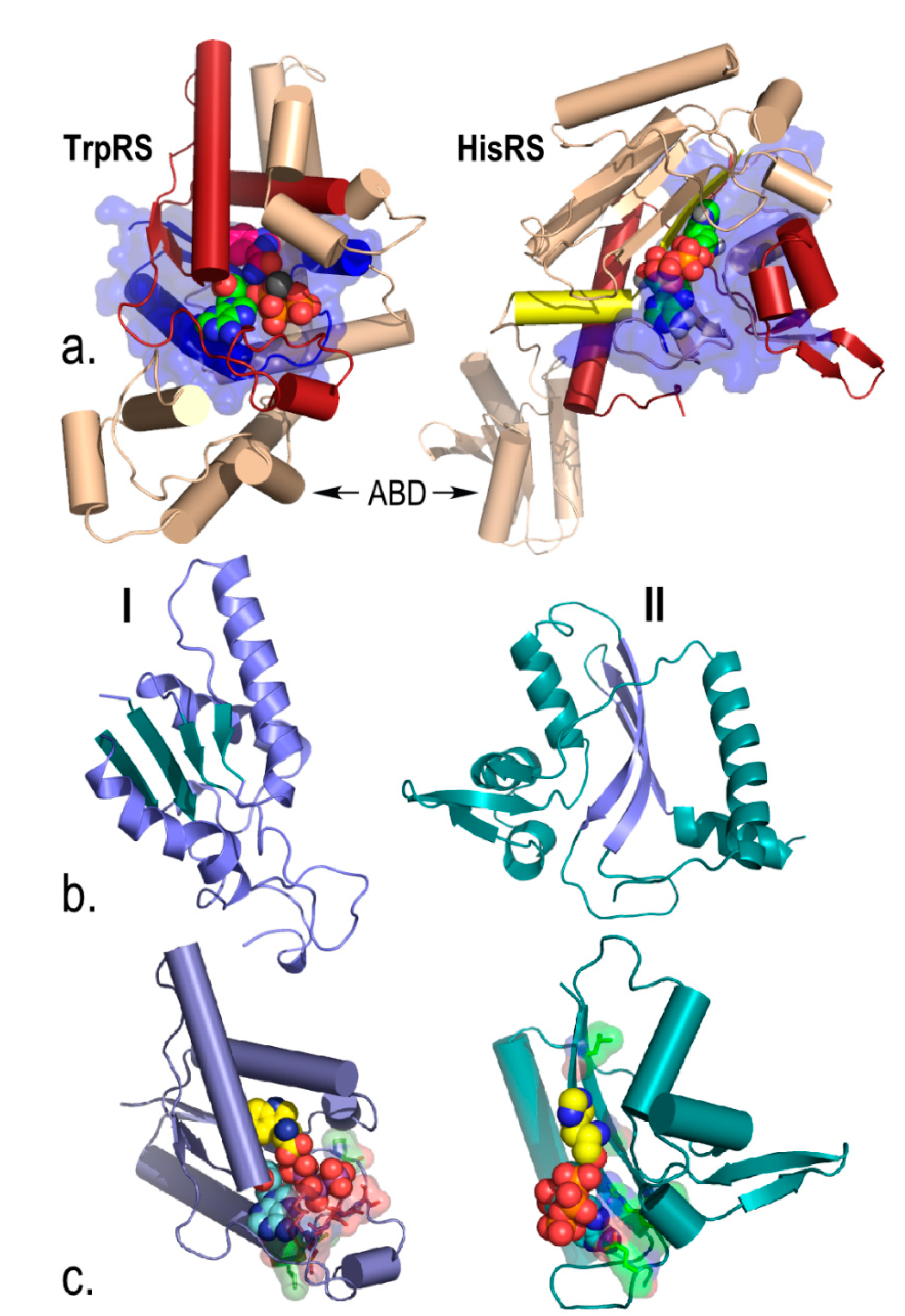
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | kcat/KM, /s/M |
|---|---|
| Uncatalyzed | 8.30 × 10−9 |
| Class I SAS 46mer | 2.70× 10−7 |
| Class II SAS 46mer | 2.90× 10−7 |
| HisRS 46mer | 3.1× 10−7 |
| TrpRS 46mer | 1.9× 10−6 |
| HisRS-1 Urzyme | 7.26× 10−1 |
| HisRS-2 Urzyme | 1.13 |
| TrpRS Urzyme | 1.36 |
| HisRS-3 Urzyme | 2.02 |
| HisRS-4 Urzyme | 2.76 |
| TrpRS Urzyme D146A | 2.65 × 10 |
| LeuRS Urzyme | 7.84 × 10 |
| HisRS_Cat_domain | 4.13 × 103 |
| Full-length TrpRS | 6.49 × 106 |
| Full-length HisRS | 9.96 × 106 |

3. Results and Discussion
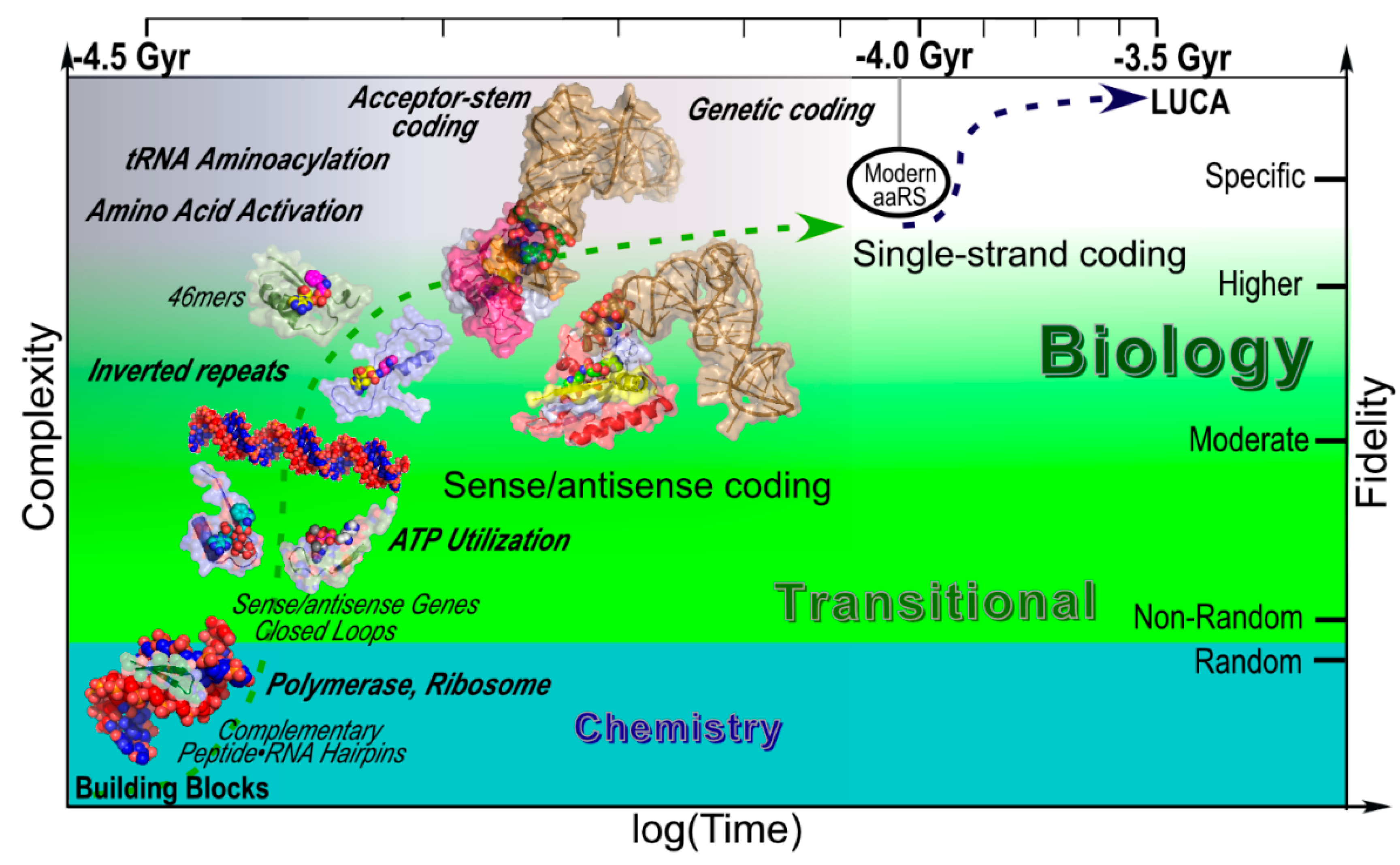
3.1. Urzymes are a Logarithmic Mean between the Earliest Catalysts and Contemporary aaRS
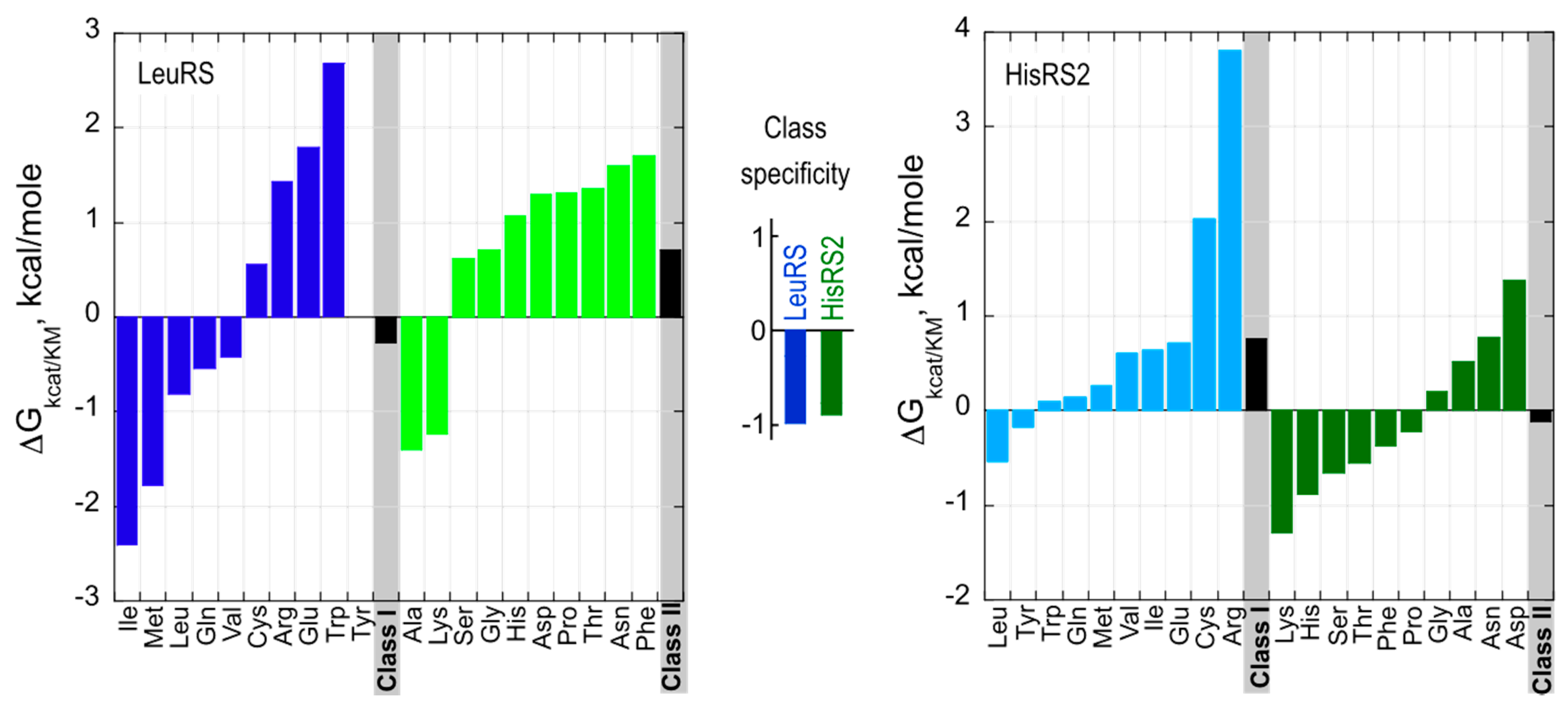
3.2. Urzyme Specificities are Consistent with Implementing Statistical Peptide Ensembles
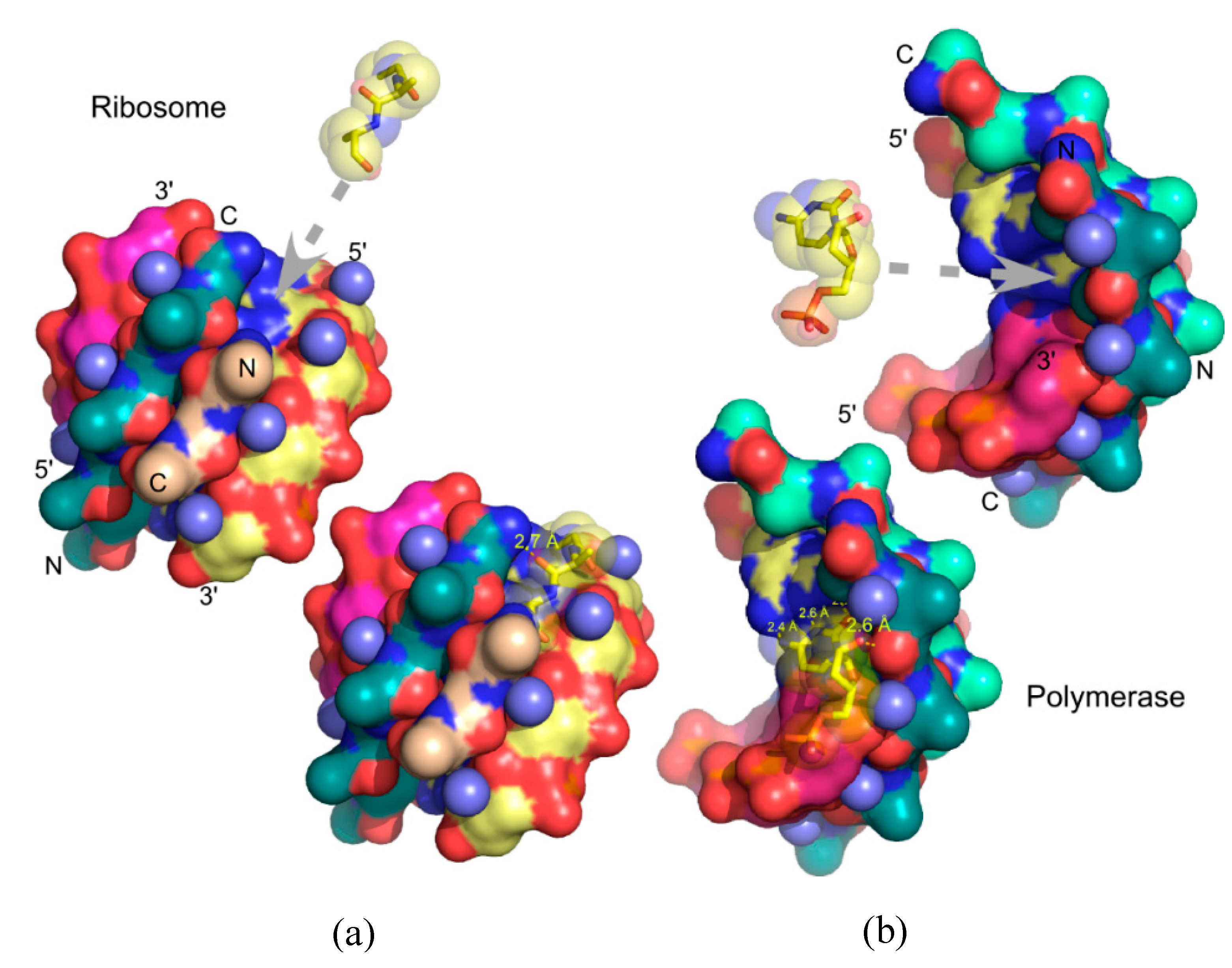
3.3. Urzymology in the Context of Similar Analyses of Ribosome Evolution
3.4. Sense/Antisense Ancestry Furnishes Key Links Backward to Simpler Genetics
3.5. Links Connecting the Sense/Antisense 46mer Gene to the Carter and Kraut Model
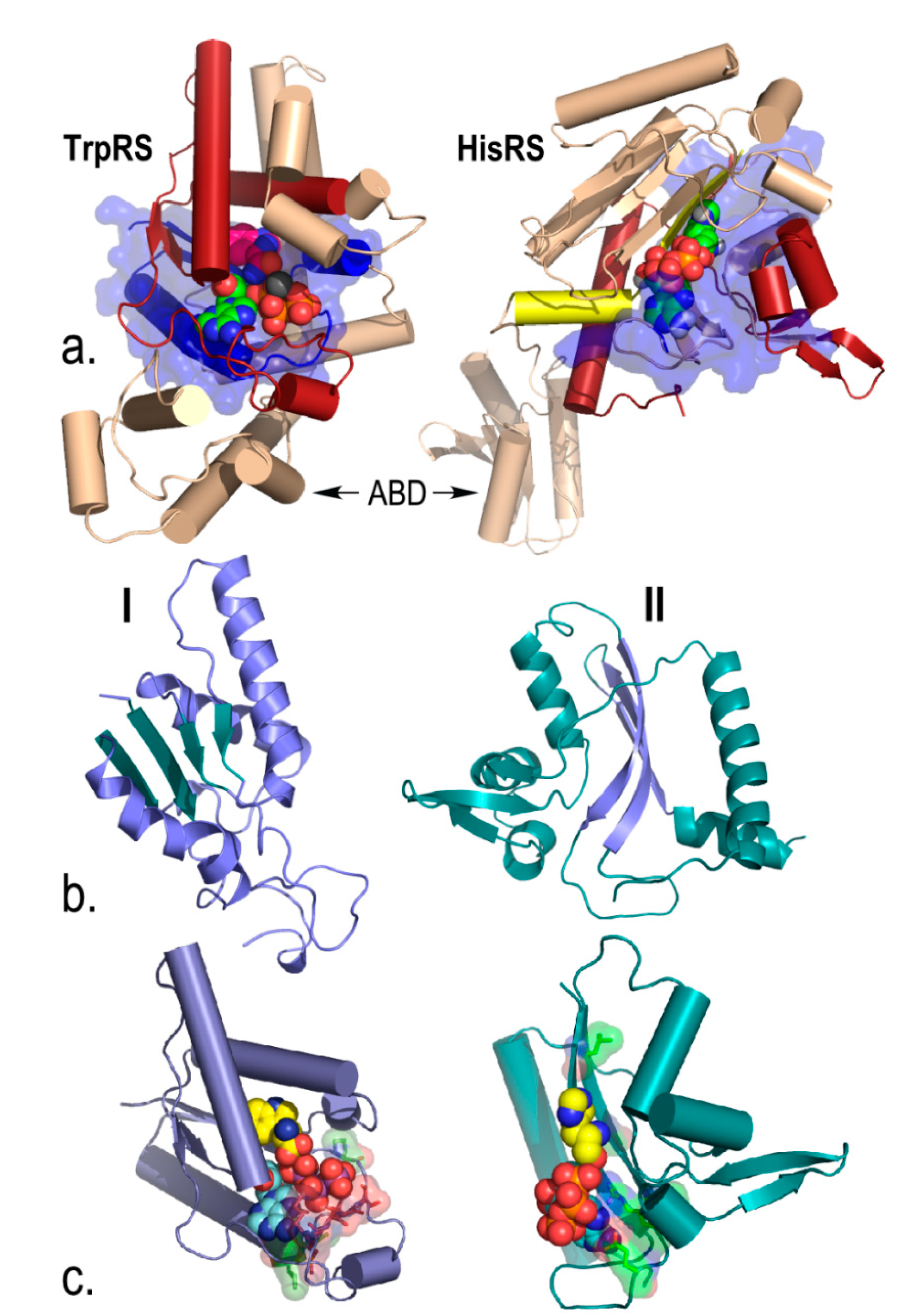
3.5.1. Amino Acid Activation Is Accelerated by 46-Residue ATP Binding Sites from Both aaRS Classes

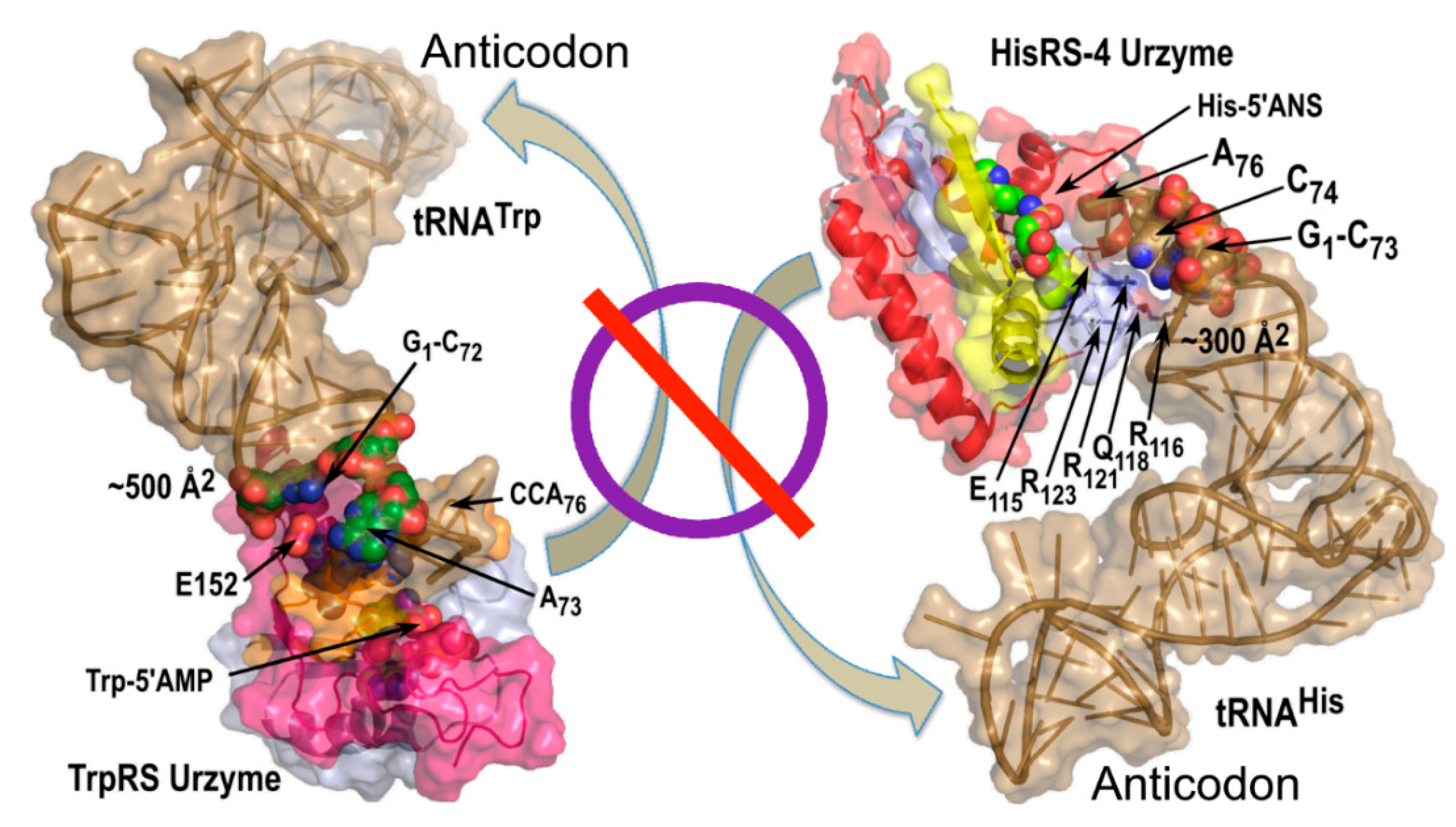
3.5.2. tRNA Anticodon and Acceptor Stem Bases Form Complementary, Non-Overlapping Codes for the 20 Amino Acids
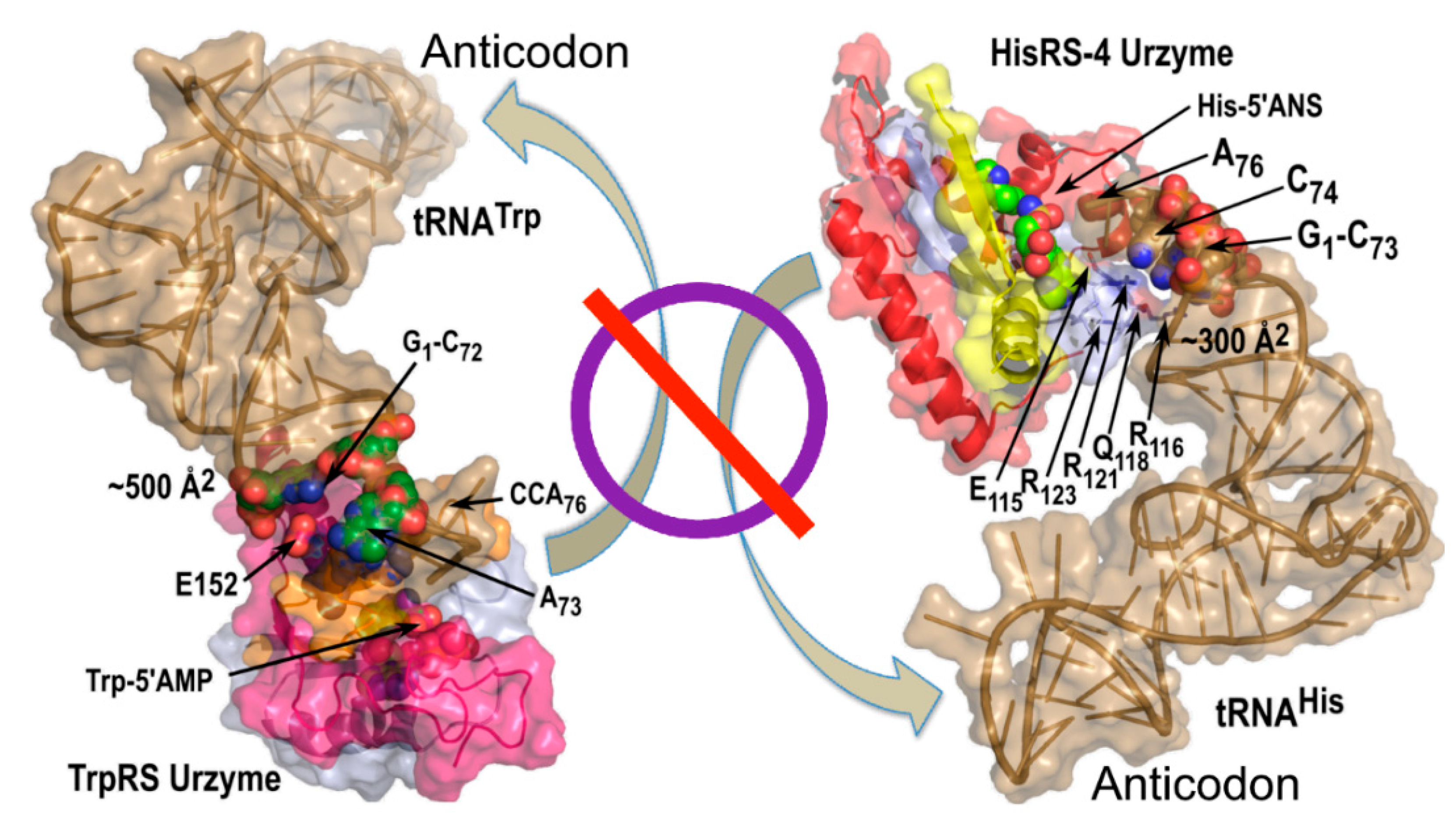
3.6. tRNA Acceptor-Stem Coding Preserves Peptide RNA Interactions of the Carter and Kraut Model
3.7. A Coherent Scenario Links the Carter & Kraut Model to Contemporary aaRS
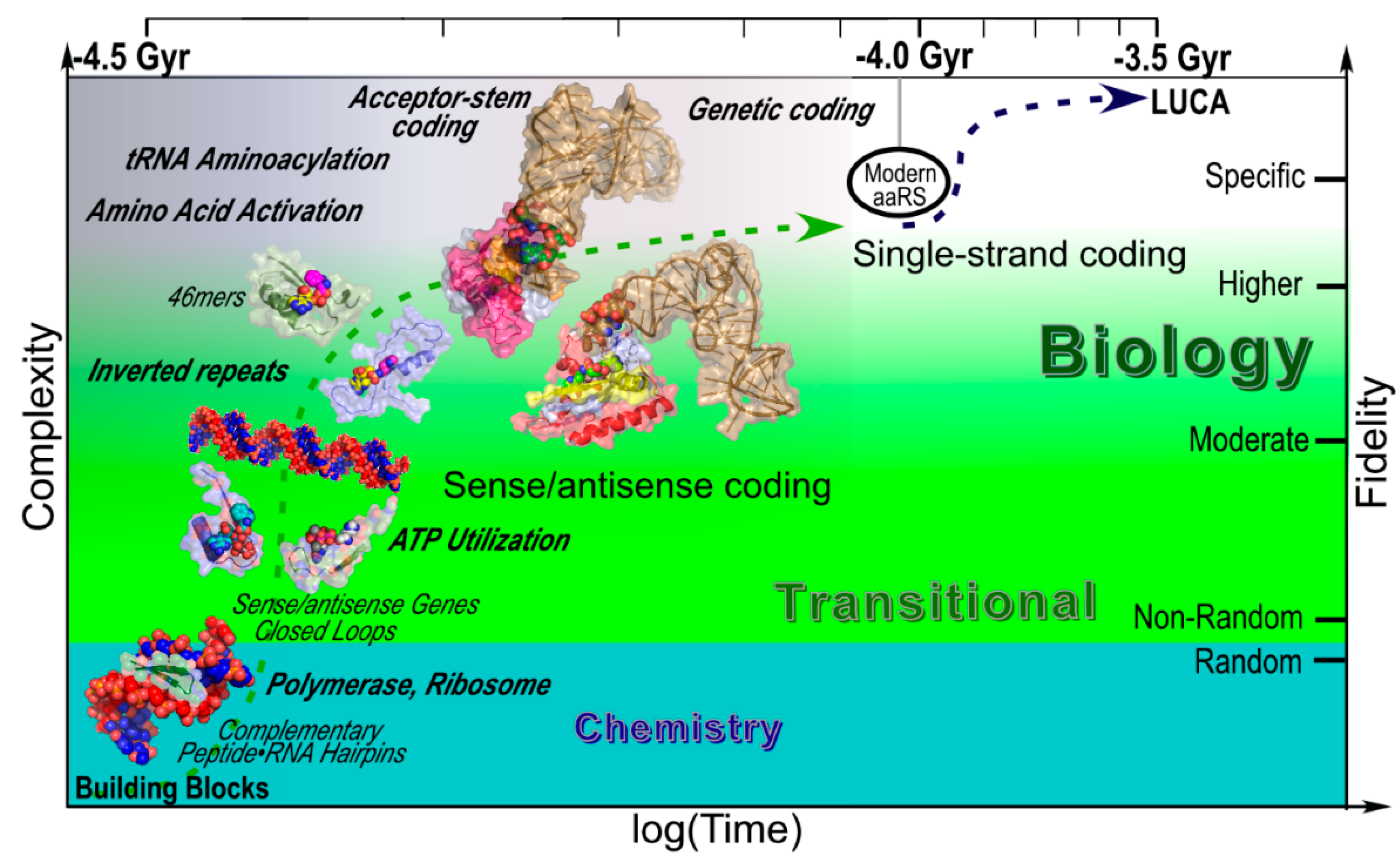
3.8. The Carter and Kraut Model Makes More Powerful, Successful Predictions than the RNA World
3.8.1. Predictions Arising from the RNA World Hypothesis Are Closely-Related and Self-Fulfilling
3.8.2. The Carter and Kraut Hypothesis Correctly Predicts Novel, Unexpected Aspects of Biology
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Carter, C.W., Jr.; Kraut, J. A Proposed Model for Interaction of Polypeptides with RNA. Proc. Natl. Acad. Sci. USA 1974, 71, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W., Jr. Cradles for Molecular Evolution. New Scientist 1975, 784–787. [Google Scholar]
- Joyce, G.; Orgel, L.E. Progress Toward Understanding the Origin of the RNA World. In The RNA World, 3rd ed.; Gesteland, R.F., Cech, T.R., Atkins, J., Eds.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2006. [Google Scholar]
- Akst, J. RNA World 2.0. The Scientist 2014, 34–40. [Google Scholar]
- Watson, J.D.; Crick, F.H.C. A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W. The RNA World. Nature 1986, 319, 618. [Google Scholar] [CrossRef]
- Koonin, E.V. The Logic of Chance: The Nature and Origin of Biological Evolution; Pearson Education; FT Press Science: Upper Saddle River, NJ, USA, 2011. [Google Scholar]
- Rodin, S.N.; Ohno, S. Two Types of Aminoacyl-tRNA Synthetases Could be Originally Encoded by Complementary Strands of the Same Nucleic Acid. Orig. Life Evol. Biosph. 1995, 25, 565–589. [Google Scholar] [CrossRef] [PubMed]
- Eriani, G.; Delarue, M.; Poch, O.; Gangloff, J.; Moras, D. Partition of tRNA Synthetases into Two Classes Based on Mutually Exclusive Sets of Sequence Motifs. Nature 1990, 347, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Cusack, S.; Berthet-Colominas, C.; Härtlein, M.; Nassar, N.; Leberman, R. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 Å. Nature 1990, 347, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Ruff, M.; Krishnaswamy, S.; Boeglin, M.; Poterszman, A.; Mitschler, A.; Podjarny, A.; Rees, B.; Thierry, J.C.; Moras, D. Class II Aminoacyl Transfer RNA Synthetases: Crystal Structure of Yeast Aspartyl-tRNA Synthetase Complexed with Trna (Asp). Science 1991, 252, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W., Jr. Urzymology: Experimental Access to a Key Transition in the Appearance of Enzymes. J. Biol. Chem. 2014, 289, 30213–30220. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Francklyn, C.; Carter, C.W., Jr. Aminoacylating Urzymes Challenge the RNA World Hypothesis. J. Biol. Chem. 2013, 288, 26856–26863. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Weinreb, V.; Francklyn, C.; Carter, C.W., Jr. Histidyl-tRNA Synthetase Urzymes: Class I and II Aminoacyl-tRNA Synthetase Urzymes have Comparable Catalytic Activities for Cognate Amino Acid Activation. J. Biol. Chem. 2011, 286, 10387–10395. [Google Scholar] [CrossRef] [PubMed]
- Pham, Y.; Li, L.; Kim, A.; Erdogan, O.; Weinreb, V.; Butterfoss, G.; Kuhlman, B.; Carter, C.W., Jr. A Minimal TrpRS Catalytic Domain Supports Sense/Antisense Ancestry of Class I and II Aminoacyl-tRNA Synthetases. Mol. Cell 2007, 25, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W., Jr.; Duax, W.L. Did tRNA Synthetase Classes Arise on Opposite Strands of the Same Gene? Mol. Cell 2002, 10, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.N.; Yardimci, G.; Erdogan, O.; Roach, J.M.; Carter, C.W., Jr. Statistical Evaluation of the Rodin-Ohno Hypothesis: Sense/Antisense Coding of Ancestral Class I and II Aminoacyl-tRNA Synthetases. Mol. Biol. Evol. 2013, 30, 1588–1604. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.; Jimenez-Rodriguez, M.; Gonzalez-Rivera, K.; Williams, T.; Li, L.; Weinreb, V.; Niranj Chandrasekaran, S.; Collier, M.; Ambroggio, X.; Kuhlman, B.; Erdogan, O.; Carter, C.W., Jr. Functional Class I and II Amino Acid Activating Enzymes Can Be Coded by Opposite Strands of the Same Gene. J. Biol. Chem. 2015. Submitted for publication. [Google Scholar]
- Jimenez, M.; Williams, T.; González-Rivera, A.K.; Li, L.; Erdogan, O.; Carter, C.W., Jr. Did Class 1 and Class 2 Aminoacyl-tRNA Synthetases Descend from Genetically Complementary, Catalytically Active ATP-Binding Motifs? Biophys. J. 2014, 106, 675a. [Google Scholar]
- Carter, C.W., Jr.; Li, L.; Weinreb, V.; Collier, M.; Gonzales-Rivera, K.; Jimenez-Rodriguez, M.; Erdogan, O.; Chandrasekharan, S.N. The Rodin-Ohno Hypothesis That Two Enzyme Superfamilies Descended from One Ancestral Gene: An Unlikely Scenario for the Origins of Translation That Will Not Be Dismissed. Biol. Direct 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Pham, Y.; Kuhlman, B.; Butterfoss, G.L.; Hu, H.; Weinreb, V.; Carter, C.W., Jr. Tryptophanyl-tRNA synthetase Urzyme: A model to recapitulate molecular evolution and investigate intramolecular complementation. J. Biol. Chem. 2010, 285, 38590–38601. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, G.K.; Wolfenden, R. The Rate Enhancement Produced by the Ribosome: An Improved Model. Biochemisty 2007, 46, 4037–4044. [Google Scholar] [CrossRef]
- Sievers, A.; Beringer, M.; Rodnina, M.V.; Wolfenden, R. The ribosome as an entropy trap. Proc. Natl. Acad. Sci. USA 2004, 101, 7897–7901. [Google Scholar] [CrossRef] [PubMed]
- Rodin, A.; Rodin, S.N.; Carter, C.W., Jr. On Primordial Sense-Antisense Coding. J. Mol. Evol. 2009, 69, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, V.; Li, L.; Chandrasekaran, S.N.; Koehl, P.; Delarue, M.; Carter, C.W., Jr. Enhanced Amino Acid Selection in Fully-Evolved Tryptophanyl-tRNA Synthetase, Relative to its Urzyme, Requires Domain Movement Sensed by the D1 Switch, a Remote, Dynamic Packing Motif. J. Biol. Chem. 2014, 289, 4367–4376. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Carter, C.W., Jr. Full Implementation of the Genetic Code by Tryptophanyl-tRNA Synthetase Requires Intermodular Coupling. J. Biol. Chem. 2013, 288, 34736–34745. [Google Scholar] [CrossRef] [PubMed]
- Burbaum, J.; Schimmel, P. Structural Relationships and the Classification of Aminoacyl-tRNA Synthetases. J. Biol. Chem. 1991, 266, 16965–16968. [Google Scholar] [PubMed]
- Burbaum, J.J.; Schimmel, P. Assembly of a Class I tRNA Synthetase from Products of an Artificially Split Gene. Biochemtry 1991, 30, 319–324. [Google Scholar] [CrossRef]
- Burbaum, J.J.; Starzyk, R.M.; Schimmel, P. Understanding Structural Relationships in Proteins of Unsolved Three-Dimensional Structure. Protein. Struct. Funct. Genet. 1990, 7, 99–111. [Google Scholar] [CrossRef]
- Liu, Y.; Kuhlman, B. RosettaDesign server for protein design. Nucleic Acids Res. 2006, 34, 235–238. [Google Scholar] [CrossRef]
- Wolfenden, R.; Snider, M.J. The Depth of Chemical Time and the Power of Enzymes as Catalysts. Acc. Chem. Res. 2001, 34, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.J.; Younas, M. The Reactivity of Phosphate Esters. Reactions of Diesters with Nucleophiles. J. Chem. Soc. B Phys. Org. 1970. [Google Scholar] [CrossRef]
- Stockbridge, R.B.; Wolfenden, R. The Intrinsic Reactivity of ATP and the Catalytic Proficiencies of Kinases Acting on Glucose, N-Acetylgalactosamine, and Homeserine: A Thermodynamic Analysis. J. Biol. Chem. 2009, 284, 22747–22757. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.K.; Yarus, M. RNA-catalyzed amino acid activation. Biochemtry 2001, 40, 6998–7004. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Posada, D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Meth. 2012, 9. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: A program package for phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Traut, T.W. Allosteric Regulatory Enzymes; Springer: New York, NY, USA, 2007. [Google Scholar]
- Traut, T.W. Are proteins made of modules. Mol. Cell. Biochem. 1986, 7, 3–10. [Google Scholar]
- Peters, J.W.; Williams, L.D. The Origin of Life: Look Up and Look Down. Astrobiology 2012, 12, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Vestigian, K.; Woese, C.R.; Goldenfeld, N. Collective Evolution and the Genetic Code. Proc. Natl. Acad. Sci. USA 2006, 103, 10696–10701. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R. On the Origin of the Genetic Code. Proc. Natl. Acad. Sci. USA 1965, 54, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.S.; Bernier, C.R.; Hsiao, C.; Norris, A.M.; Kovacs, N.A.; Waterbury, C.C.; Stepanov, V.G.; Harvey, S.C.; Fox, G.E.; Wartell, R.M.; et al. Evolution of the Ribosome at Atomic Resolution. Proc. Natl. Acad. Sci. USA 2014, 111, 10251–10256. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.; Lenz, T.K.; Peters, J.K.; Fang, P.-Y.; Schneider, D.M.; Anderson, E.J.; Preeprem, T.; Bowman, J.C.; O’Neill, E.B.; Lie, L.; et al. Molecular paleontology: A biochemical model of the ancestral ribosome. Nucleic Acids Res. 2013, 41, 3373–3385. [Google Scholar] [CrossRef] [PubMed]
- Noller, H.F.; Hoffarth, V.; Zimniak, L. Unusual Resistance of Peptidyl Transferase to Protein Extraction Procedures. Science 1992, 256, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Shakhnovich, B.E.; Dokholyan, N.V.; DeLisi, C.; Shacknovich, E. Functional Fingerprints of Folds: Evidence for Correlated Structure-Function Evolution. J. Mol. Biol. 2003, 326, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dokholyan, N.V.; Shakhnovich, B.; Shacknovich, E.I. Expanding protein universe and its origin from the biological big bang. Proc. Natl. Acad. Sci. USA 2002, 99, 14132–14136. [Google Scholar] [CrossRef] [PubMed]
- Dokholyan, N.V.; Shakhnovich, E.I. Understanding hierarchical protein evolution from first principles. J. Mol. Biol. 2001, 312, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Mullen, G.P.; Vaughn, J.B., Jr.; Mildvan, A.S. Sequential Proton NMR Resonance Assignments, Circular Dichroism, and Structural Properties of a 50-Residue Substrate-Binding Peptide from DNA Polymerase I. Arch. Biochem. Biophys. 1993, 301, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Chuang, W.-J.; Abeygunawardana, C.; Pedersen, P.L.; Mildvan, A.S. Two-Dimensional NMR, Circular Dichroism, and Fluorescence Studies of PP-50, a Synthetic ATP-Binding Peptide from the β-Subunit of Mitochondrial ATP Synthase. Biochem. 1992, 31, 7915–7921. [Google Scholar] [CrossRef]
- Chuang, W.-J.; Abeygunawardana, C.; Gittis, A.G.; Pedersen, P.L.; Mildvan, A.S. Solution Structure and Function in Trifluoroethanol of PP-50, an ATP-Binding Peptide from F1ATPase. Arch. Biochem. Biophys. 1992, 319, 110–122. [Google Scholar] [CrossRef]
- Fry, D.C.; Byler, D.M.; Sisu, H.; Brown, E.M.; Kuby, S.A.; Mildvan, A.S. Solution Structure of the 45-Residue MgATP-Binding Peptide of Adenylate Kinase As Examined by 2-d NMR, FTIR, and CD Spectroscopy. Biochem. 1988, 27, 3588–3598. [Google Scholar] [CrossRef]
- Fry, D.C.; Kuby, S.A.; Mildvan, A.S. NMR Studies of the MgATP Binding Site of Adenylate Kinase and of a 45-Residue Peptide Fragment of the Enzyme. Biochemtry 1985, 24, 4680–4694. [Google Scholar] [CrossRef]
- Schimmel, P.; Giegé, R.; Moras, D.; Yokoyama, S. An operational RNA code for amino acids and possible relationship to genetic code. Proc. Natl. Acad. Sci. USA 1993, 90, 8763–8768. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W., Jr.; Wolfenden, R. tRNA Acceptor-Stem and Anticodon Bases Form Independent Codes Related to Protein Folding. Proc. Natl. Acad. Sci. USA 2015. Submitted for publication. [Google Scholar]
- Muñoz, V.; Serrano, L. Intrinsic Secondary Structure Propensities of the Amino Acids, Using Statistical Φ-Ψ matrices: Comparison with Experimental Scales. Protein. Struct. Funct. Gen. 1994, 20, 301–311. [Google Scholar] [CrossRef]
- Kramer, R.M.; Shende, V.R.; Motl, N.; Pace, C.N.; Scholtz, J.M. Toward a Molecular Understanding of Protein Solubility: Increased Negative Surface Charge Correlates with Increased Solubility. Biophys. J. 2014, 102, 1907–1915. [Google Scholar] [CrossRef]
- Franzen, K.L.; Kinsella, J.E. Functional Properties of Succinylated and Acetylated Soy Protein. J. Agric. Food Chem. 1976, 24, 788–795. [Google Scholar] [CrossRef]
- Szostak, J. The eightfold path to non-enzymatic RNA replication. J. Syst. Chem. 2012, 3. [Google Scholar] [CrossRef]
- Glusker, J.P.; Katz, A.K.; Bock, C.W. METAL IONS IN BIOLOGICAL SYSTEMS. Rigaku J. 1999, 16, 8–16. [Google Scholar]
- Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G.L.; Thornton, J.M. Metal ions in biological catalysis: From enzyme databases to general principles. J. Biol. Inorg. Chem. 2008, 13, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- AbouHaidar, M.G.; Ivanovb, I.G. Non-Enzymatic RNA Hydrolysis Promoted by the Combined Catalytic Activity of Buffers and Magnesium Ions. Z. Naturforsch. C 1999, 54, 542–548. [Google Scholar] [PubMed]
- Henderson, B.S.; Schimmel, P. RNA-RNA Interactions Between Oligonucleotide Substrates for Aminoacylation. Bioorg. Med. Chem. 1997, 5, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Achbergerová, L.; Nahálka, J. Polyphosphate—an ancient energy source and active metabolic regulator. Microb. Cell Fact. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, A. Inorganic Polyphosphate: Toward Making a Forgotten Polymer Unforgettable. J. Bact. 1995, 177, 491–496. [Google Scholar] [PubMed]
- Härtlein, M.; Cusack, S. Structure, Function and Evolution of Seryl-tRNA Synthetases: Implications for the Evolution of Aminoacyl-tRNA Synthetases and the Genetic Code. J. Mol. Evol. 1995, 40, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Maizels, N.; Weiner, A.M. Phylogeny from function: Evidence from the molecular fossil record that tRNA originated in replication, not translation. Proc. Natl. Acad. Sci. USA 1994, 91, 6729–6734. [Google Scholar] [CrossRef] [PubMed]
- Weiner, A.M.; Maizels, N. tRNA-like structures tag the 3' ends of genomic RNA molecules for replication: Implications for the origin of protein synthesis. Proc. Natl. Acad. Sci. USA 1987, 84, 7383–7387. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. The Place of RNA in the Origin and Early Evolution of the Genetic Machinery. Life 2014, 4, 1050–1091. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T.-F. Coevolution theory of the genetic code at age thirty. BioEssays 2005, 27, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.F.; Budin, I.; Szostak, J.W. Vesicle Extrusion Through Polycarbonate Track-etched Membranes using a Hand-held Mini-extruder. Meth. Enzymol. 2013, 533, 275–282. [Google Scholar] [PubMed]
- Perona, J.J.; Gruic-Sovulj, I. Synthetic and Editing Mechanisms of Aminoacyl-tRNA Synthetases. Top. Curr. Chem. 2013, 344, 1–41. [Google Scholar]
- Perona, J.J.; Hadd, A. Structural Diversity and Protein Engineering of the Aminoacyl-tRNA Synthetases. Biochemistry 2013, 51, 8705–8729. [Google Scholar] [CrossRef]
- Bullock, T.; Uter, N.; Nissan, T.A.; Perona, J.J. Amino Acid Discrimination by a class I aminoacyl-tRNA synthetase specified by negative determinants. J. Mol. Biol. 2003, 328, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Sakaguchi, R.; Liu, C.; Vishveshwara, S.; Hou, Y.-M. Allosteric Communication in Cysteinyl tRNA Synthetase A NETWORK OF DIRECT AND INDIRECT READOUT. J. Biol. Chem. 2011, 286, 37721–37731. [Google Scholar] [CrossRef] [PubMed]
- Marquet, R.; lsel, C.; Ehresmann, C.; Ehresmann, B. tRNAs as prirner of reverse transcriptases. Biochimie 1995, 77, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Bayes, T.; Price, R. An Essay towards solving a Problem in the Doctrine of Chance. By the late Rev. Mr. Bayes, F. R. S. communicated by Mr. Price, in a letter to John Canton, A. M. F. R. S. Philos. Trans. 1763, 53, 370–418. [Google Scholar] [CrossRef]
- Popper, K. The Logic of Scientific Discovery; Routledge: Florence, KY, USA, 1959; p. 284. [Google Scholar]
- Yarus, M. The meaning of a minuscule ribozyme. Phil. Trans. R. Soc. B 2011, 366, 2902–2909. [Google Scholar] [CrossRef] [PubMed]
- Turk, R.M.; Illangasekare, M.; Yarus, M. Catalyzed and Spontaneous Reactions on Ribozyme Ribose. J. Am. Chem. Soc. 2011, 133, 6044–6050. [Google Scholar] [CrossRef] [PubMed]
- Turk, R.M.; Chumachenkob, N.V.; Yarus, M. Multiple translational products from a five-nucleotide ribozyme. Proc. Natl. Acad. Sci. USA 2010, 107, 4585–4589. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M. Life from an RNA World: The Ancestor within; Harvard University Press: Cambridge, MA, USA, 2011; p. 208. [Google Scholar]
- Yarus, M.; Widmann, J.; Knight, R. RNA-amino acid binding: A stereochemical era for the genetic code. J. Mol. Evol. 2009, 69, 406–429. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.; Majerfeld, I.; Yarus, M. 23S rRNA Similarity from Selection for Peptidyl Transferase Mimicry. Biochemistry 1997, 36, 6614–6623. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.; Chastang, J.; Yarus, M. An Inhibitor of Ribosomal Peptidyl Transferase Using Transition-State Analogy. Biochemtry 1995, 34, 385–390. [Google Scholar] [CrossRef]
- Niwa, N.; Yamagishi, Y.; Murakami, H.; Suga, H. A flexizyme that selectively charges amino acids activated by a water-friendly leaving group. Bioorg. Med. Chem. Lett. 2009, 19, 3892–3894. [Google Scholar] [CrossRef] [PubMed]
- Sczepanski, J.T.; Joyce, G.F. A cross-chiral RNA polymerase ribozyme. Nature 2014, 515, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, T.A.; Joyce, G.F. Self-Sustained Replication of an RNA Enzyme. Science 2009, 323, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Shechner, D.M.; Bartel, D.P. The structural basis of RNA-catalyzed RNA polymerization. Nat. Struct. Mol. Biol. 2011, 18, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Johnston, W.K.; Unrau, P.J.; Lawrence, M.S.; Glasner, M.E.; Bartel, D.P. RNA-Catalyzed RNA Polymerization: Accurate and General RNA-Templated Primer Extension. Science 2001, 292, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P.; Unrau, P.J. Constructing an RNA world. Trends Biochem. Sci. 1999, 24, M9–M13. [Google Scholar] [CrossRef]
- Wochner, A.; Attwater, J.; Coulson, A.; Holliger, P. Ribozyme-Catalyzed Transcription of an Active Ribozyme. Science 2011, 332, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Noller, H. The driving force for molecular evolution of translation. RNA 2004, 10, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.; Steitz, T.A. The Complete Atomic Structure of the Large Ribosomal Subunit at 2.4 Å Resolution. Science 2000, 289, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Henkin, T.M. RNA-dependent RNA switches in bacteria. Meth. Mol. Biol. 2009, 540, 207–214. [Google Scholar]
- Grundy, F.J.; Winkler, W.C.; Henkin, T.M. tRNA-mediated transcription antitermination in vitro: Codon-anticodon pairing independent of the ribosome. Proc. Natl. Acad. Sci. USA 2002, 99, 11121–11126. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L. Gas-liquid transfer data used to analyze hydrophobic hydration and find the nature of the Kauzmann-Tanford hydrophobic factor. Proc. Natl. Acad. Sci. USA 2012, 109, 7310–7313. [Google Scholar] [CrossRef] [PubMed]
- Kauzmann, W. Some Factors in the Interpretation of Protein Denaturation. Adv. Protein Chem. 1959, 14, 1–63. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carter, C.W., Jr. What RNA World? Why a Peptide/RNA Partnership Merits Renewed Experimental Attention. Life 2015, 5, 294-320. https://doi.org/10.3390/life5010294
Carter CW Jr. What RNA World? Why a Peptide/RNA Partnership Merits Renewed Experimental Attention. Life. 2015; 5(1):294-320. https://doi.org/10.3390/life5010294
Chicago/Turabian StyleCarter, Charles W., Jr. 2015. "What RNA World? Why a Peptide/RNA Partnership Merits Renewed Experimental Attention" Life 5, no. 1: 294-320. https://doi.org/10.3390/life5010294
APA StyleCarter, C. W., Jr. (2015). What RNA World? Why a Peptide/RNA Partnership Merits Renewed Experimental Attention. Life, 5(1), 294-320. https://doi.org/10.3390/life5010294