1. Introduction
Filamentous heterocyst-forming cyanobacteria fix atmospheric nitrogen to ammonium under oxic growth conditions. Nitrogen fixation occurs in specialized cells called heterocysts that differentiate at regular intervals in a filament in response to an environment that is deficient in fixed nitrogen [
1,
2,
3,
4]. Heterocysts, which comprise 5%–10% of the cells in a filament, have a glycolipid layer that may restrict oxygen diffusion into the cell, lack oxygen-evolving photosystem II activity, and have increased respiration, all of which serve to protect nitrogenase from oxygen [
5,
6,
7,
8,
9].
Anabaena variabilis is unusual among the heterocyst-forming cyanobacteria in that it has three nitrogenases, which are expressed in cultures grown in different environmental conditions (reviewed in [
10]). No other well-characterized cyanobacterial strain has three nitrogenases; in fact, no other characterized strain has even two. The primary nitrogenase that is expressed in cultures growing in an oxic environment that is deficient in fixed nitrogen but has adequate molybdate is the heterocyst-specific Mo-nitrogenase encoded by the
nif1 genes [
11,
12]. In an oxic environment that is low in fixed nitrogen and molybdate, but with vanadate,
A. variabilis synthesizes an alternative, heterocyst-specific V-nitrogenase, encoded by the
vnf genes [
11,
13]. The third nitrogenase, a Mo-nitrogenase, encoded by the
nif2 genes is made in vegetative cells only under anoxic growth conditions in an environment that is low in fixed nitrogen with molybdate [
14,
15,
16]. Synthesis of all three nitrogenases is repressed in cells grown with a source of fixed nitrogen.
Nitrogenase activity, which requires the expression of at least a dozen genes, is found late in the differentiation process, after the heterocyst becomes microoxic [
10]. The assembly of nitrogenase is a complex process requiring highly conserved proteins that are found in large
nif clusters in all nitrogen-fixing bacteria. NifD (α-subunit) and NifK (β-subunit) are the two subunits of dinitrogenase, forming a heterotetrameric enzyme with two FeMo-cofactors [7Fe-9S-Mo-C-homocitrate] [
17,
18,
19,
20,
21]. NifH, with a [Fe
4-S
4] cofactor, transfers electrons to the dinitrogenase [
22]. NifS transfers sulfur from cysteine to NifU [
23], which acts as a scaffolding protein for [Fe-S] cluster assembly [
19,
24]. The [Fe-S] clusters are transferred to NifB to make NifB-co, a [Fe
6-S
9] cluster that serves as the precursor to FeMo-cofactor [
25,
26]. NifE and NifN, a heterotetrameric complex with some similarity to NifD and NifK, respectively, function as a scaffold for FeMo-cofactor assembly, prior to its transfer to apo-nitrogenase [
19,
27]). NifW is thought to bind MoFe protein and to help with homocitrate processing [
28]. NifX serves as a transient reservoir of FeMo-cofactor [
29]. NifZ aids in P-cluster assembly [
30,
31] while NifV makes homocitrate, a component of FeMo-cofactor [
19]. Missing in cyanobacteria are the genes for NifQ, the Mo donor to FeMo-cofactor [
32], NifM, which stabilizes NifH [
33,
34], and NafY, which stabilizes the open conformation of apo-MoFe protein prior to the insertion of FeMo-cofactor [
35,
36]. NifP is a serine acetyltransferase that is thought to aid in expression of nitrogenase activity [
37]. NifT/FixU is a very small, conserved protein that is found in
nif clusters; however, its function is unknown [
38,
39]. In
Anabaena sp. PCC 7120, NifJ, pyruvate-flavodoxin dehydrogenase is required for nitrogen fixation under iron-limiting conditions [
40].
The alternative V-nitrogenase comprises two VnfD (α-subunit),
two VnfK (β-subunit) and four δ-subunits, VnfG, forming a heterooctomeric enzyme with two FeV-cofactors [
41,
42,
43]. Like NifH, VnfH, with a [Fe
4-S
4] cofactor transfers electrons to dinitrogenase. The V-nitrogenase shows different efficiency in substrate interactions than the Mo-nitrogenase; it is relatively inefficient in reducing dinitrogen and thus produces more hydrogen than the Mo-nitrogenase and, unlike the Mo-nitrogenase, it can reduce ethylene to ethane [
44]. Because it is an inefficient nitrogenase and produces hydrogen, the V-nitrogenase of
A. variabilis has been used to produce hydrogen in an outdoor bioreactor [
45].
In the Proteobacteria, the
nif genes are structured into multiple operons, including
nifHDK, encoding the structural proteins of nitrogenase,
nifBQ, producing the proteins required for FeMo-cofactor assembly,
nifUVSM, whose products are needed for Fe-S cluster formation, and
nifENX, encoding scaffolding proteins for the assembly of the nitrogenase complex [
46,
47]. In the Proteobacteria,
nif genes are under the control of the NtrBC nitrogen regulatory system, which controls synthesis of the regulatory proteins, NifA and NifL [
48]. Activation of
nif genes in the absence of oxygen and fixed nitrogen requires NifA, as well as the alternative σ
54 RNA polymerase [
48]. Similarly, the
vnf genes of
Azotobacter vinelandii are controlled by the activator VnfA [
49,
50].
In nitrogen-fixing cyanobacteria there are no homologues of NtrBC, NifA or NifL and there is no homologue of VnfA in
A. variabilis. The global nitrogen regulatory protein, NtcA, is required for nitrogen fixation in heterocyst-forming cyanobacteria; however, it is also required for heterocyst formation so its role in activation of nitrogen fixation genes is not yet known [
4,
51,
52]. While no sigma factor specifically associated with nitrogen regulation, like the σ
54 factor in Proteobacteria, has been identified in cyanobacteria, the sigma factor encoded by
sigE is important, but not essential, for expression of the
nif genes in
Anabaena sp. PCC 7120 [
53].
In
A. variabilis, and in most nitrogen-fixing cyanobacteria whose genomes have been sequenced, the
nif gene clusters comprise, in the same order,
nifB,
fdxN,
nifS,
nifU,
nifH,
nifD,
nifK,
nifE,
nifN,
nifX, and
nifW. All
nif clusters also have
hesAB and
fdxH as well as several conserved unidentified ORFs [
54]. In
A. variabilis and in
Anabaena sp. PCC 7120, the
nifD gene is interrupted by an 11-kb element that is removed from the chromosome of heterocysts by an excisase, XisA, late in heterocyst differentiation [
55,
56,
57]. Transcription of the
nif genes was first reported over 30 years ago [
58,
59]; however, little progress has been made in identifying key regulatory mechanisms. It has been assumed, based on Northern blot results and the assumed similarity to Proteobacteria, that the large
nif cluster in cyanobacteria comprises several distinct operons:
nifB-fdxN-nifS-nifU [
58,
60],
nifHDK [
59,
61,
62], as well as
hesAB [
63],
fdxH [
64] and, by default,
nifENXW. In addition to the large conserved cluster,
nifP is located just upstream of the
nifVZT operon [
39]. In
A. variabilis,
nifP is located about 11 kb downstream from the 3' end of the large
nif1 cluster. The best evidence for
nif promoters in
Anabaena are for those genes in which the apparent transcription start sites have been mapped. These include
nifB [
58,
60,
65,
66,
67],
nifH [
58,
68],
hesA [
63,
67], and
fdxH [
64]. Recent work from our lab that is described in more detail here has shown that there is a strong promoter driving
nifB1 and a separate promoter for
hesA1 in
A. variabilis, but there is no promoter for
nifH1 or
fdxH1 [
65,
69]. While most of the expression of the large cluster of
nif1 genes in
A. variabilis is driven by the
nifB1 promoter, there are additional weak promoters, including one in the
nifU1 gene and in the
nifE1 gene, that supplement transcription from
nifB1 [
65,
69]. While neither
nifH1 nor
fdxH1 has a promoter, the “transcription start sites” that were mapped upstream of these two genes are actually processed 5' transcript ends, not 5' primary transcription start sites [
65,
69].
2. Organization and Evolution of nif/vnf Gene Clusters in A. variabilis
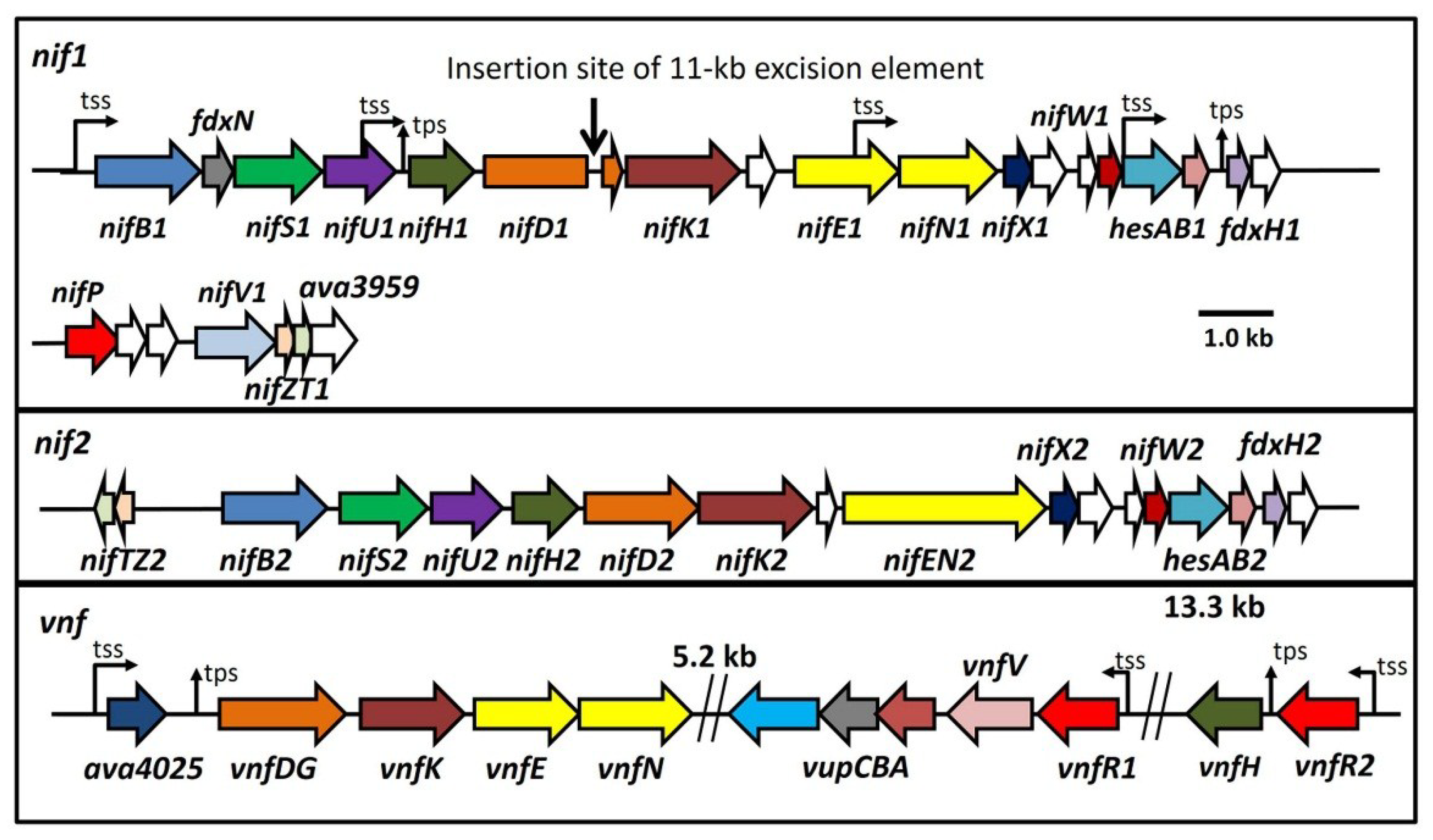
The organization of the three nitrogenase gene clusters of
A. variabilis is shown in
Figure 1 [
11,
16,
70]. The nearest relative of the
nif1 cluster of
A. variabilis is the sole
nif cluster in
Anabaena sp. PCC 7120; however, the
nif cluster in
Anabaena sp. strain PCC 7120 has a 55-kb excision element in
fdxN that is not present in
A. variabilis [
71,
72]. The
nif1 cluster is also very similar to the
nif clusters in other heterocyst-forming cyanobacteria. In
Anabaena spp., the nitrogenase encoded by these
nif1-type genes is expressed only in heterocysts, even under anoxic growth conditions [
16,
73]. The similarly organized
nif2 cluster in
A. variabilis is most similar in overall gene organization and gene similarity to the sole
nif cluster of
Chroococcidiopsis thermalis PCC 7203, a strain that belongs to a group of unicellular non-heterocystous cyanobacteria that grow in extreme environments and fix nitrogen only under anoxic conditions [
74]. The similarity of the
nif2 gene cluster to the
nif cluster in
Chroococcidiopsis thermalis PCC 7203 is interesting, since the
Chroococcidiopsis group is the closest relative of the heterocyst-forming cyanobacteria, based on 16S rRNA phylogeny [
75]. In particular, the unusual fusion of the
nifE and
nifN genes into a single gene in the
nif2 cluster and in the
nif cluster of
C. thermalis suggests that these genes have a common ancestor. Another major difference between the
nif1 and
nif2 clusters in
A. variabilis is the presence of an excision element only in the
nifD1 gene. That excision element is present in most, but not all, of the
nif clusters in the genomes of sequenced heterocyst-forming cyanobacteria. Although the size of the element and the genes present in these excision elements varies among strains, all of them have a conserved excisase gene that removes the element during heterocyst differentiation, thereby restoring a complete
nifD gene to produce the β-subunit of nitrogenase [
55,
72].
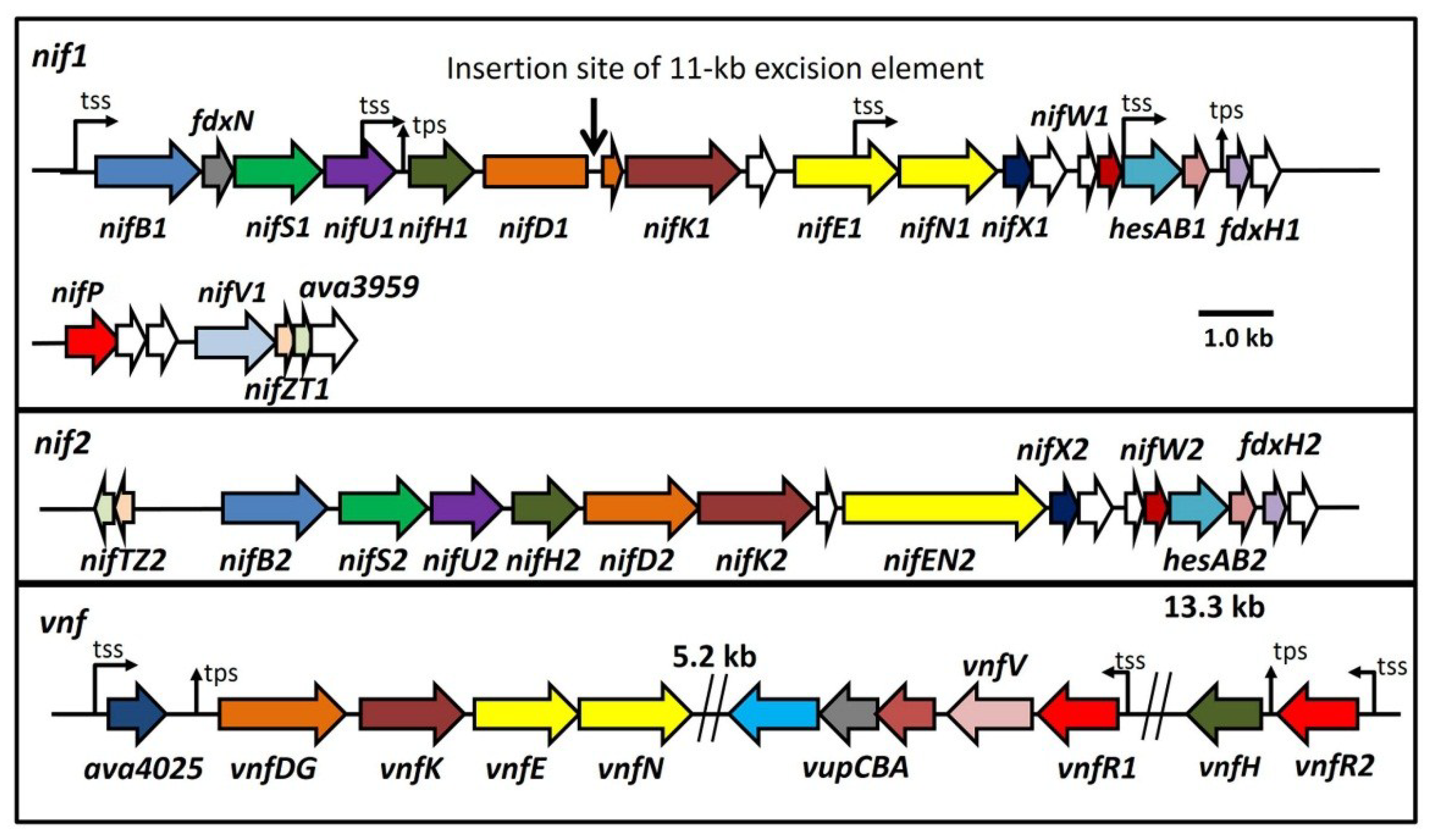
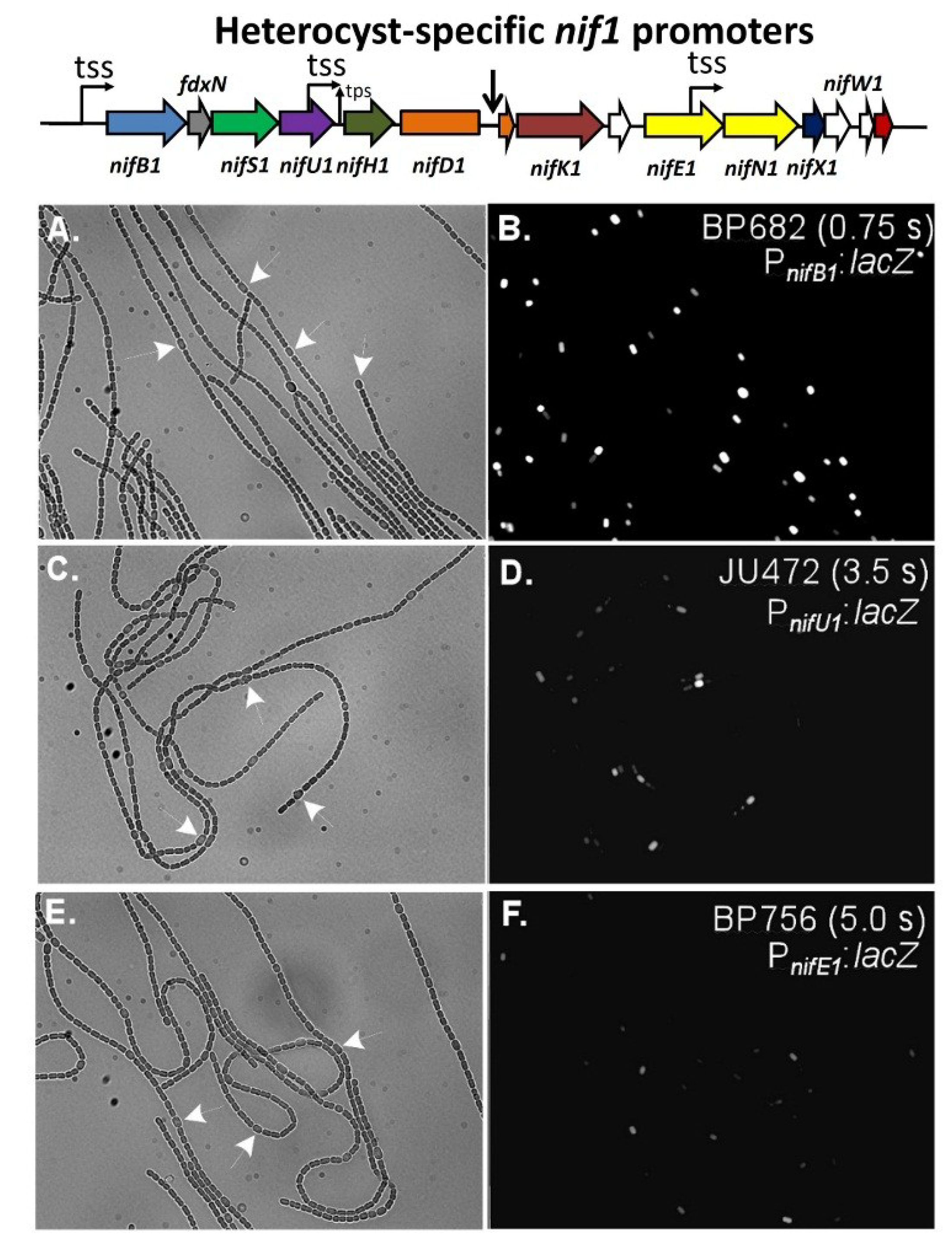
Figure 1.
Maps of the three major nitrogenase gene clusters in A. variabilis. The 11-kb excision element in nifD1 is not shown. tss, transcription start site; tps, transcriptional processing site. White ORFs indicate proteins of unknown function.
Figure 1.
Maps of the three major nitrogenase gene clusters in A. variabilis. The 11-kb excision element in nifD1 is not shown. tss, transcription start site; tps, transcriptional processing site. White ORFs indicate proteins of unknown function.
In contrast, the organization of the
vnf genes that encode an alternative V-nitrogenase [
11] is different from the two
nif clusters, in part because synthesis and assembly of the V-nitrogenase depends on the products of some of the genes that make the Mo-nitrogenase, notably NifB, NifS, NifU and possibly several of the small proteins such as NifW, HesA, HesB and ferredoxins [
76]. The
vnf genes comprise
vnfDG, a fusion of
vnfD and
vnfG, as well as genes
vnfK,
vnfE and
vnfN. Unlike the
vnf gene cluster in the Proteobacteria, there is no
vnfH near the
vnfDGKEN cluster, and in
A. variabilis,
vnfH is over 20 kb downstream from
vnfN, with
vupABC between
vnfH and the other structural genes (
Figure 1). Complete cyanobacterial genome sequences [
77] have revealed strains that have genes very similar to the
vnf genes of
A. variabilis [
70].
Fremyella diplosiphon UTEX 481 and
Fischerella muscicola PCC 7414 have orthologs of
vnfDG,
vnfK,
vnfE and
vnfN as well as the vanadate transport genes, while
Fischerella sp. PCC 9339 has orthologs of
vnfDG,
vnfK,
vnfE and
vnfN but is missing most of the vanadate transport genes. In contrast,
Chlorogleopsis sp. PCC 7702 has orthologs for the vanadate transport genes, and has most of the structural genes for the V-nitrogenase; however, the fused
vnfDG gene is missing the
vnfD portion that encodes the α-subunit of the enzyme, which is essential for dinitrogenase activity. The presence of V-nitrogenase activity has not been confirmed in any of these strains.
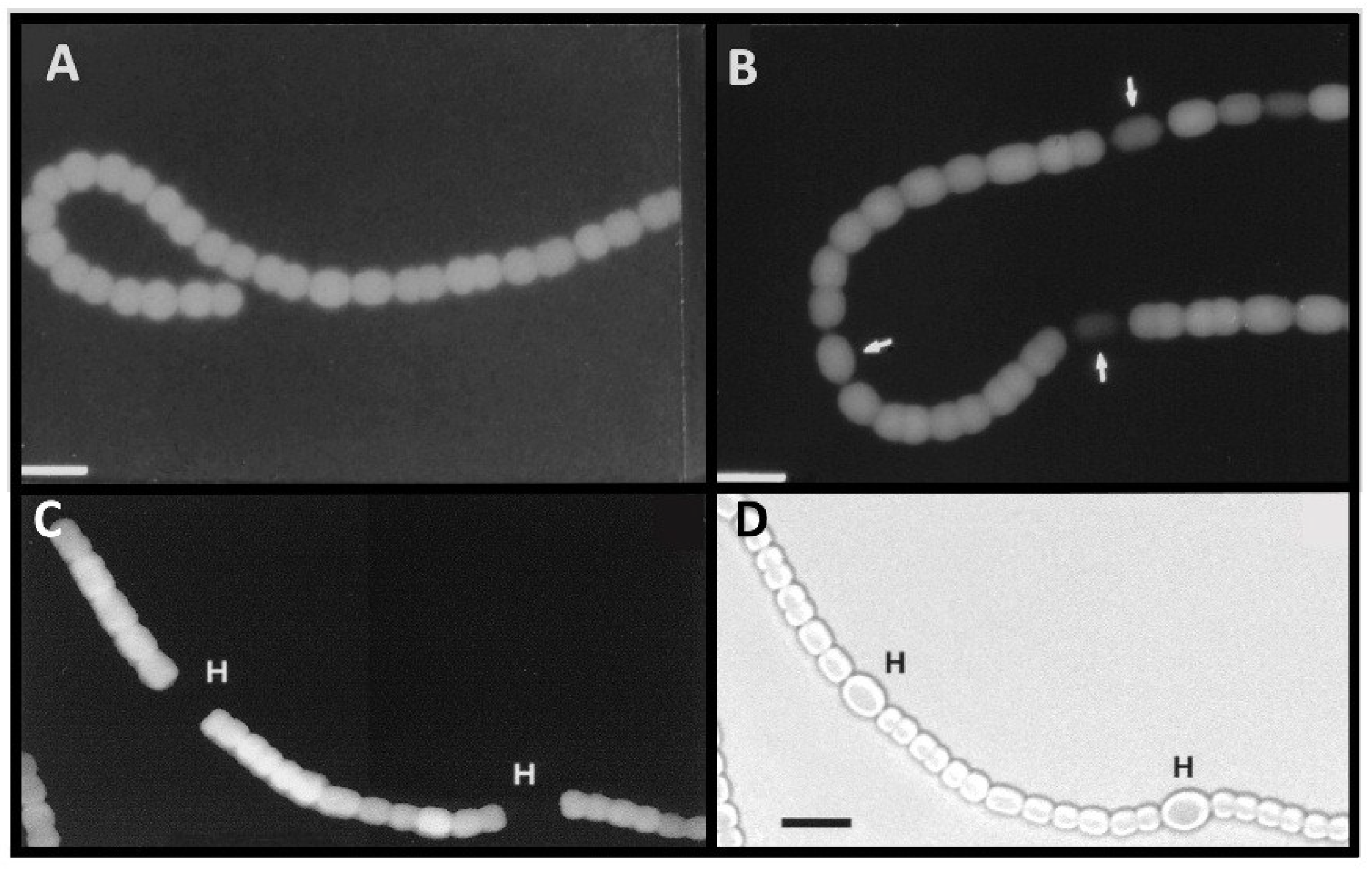
3. Cell-Type Specific Expression of the Three Nitrogenases in A. variabilis
In cyanobacteria, the best evidence for cell-type specific gene expression comes from imaging of cells expressing reporter genes such a
gfp,
luxAB or
lacZ fused to cyanobacterial promoters. In our research, we have often used promoter:
lacZ fusions because it is easy to assay β-galactosidase in the same cultures that are used for imaging and because there is no concern that the microoxic conditions in a mature heterocyst may affect the reporter protein, which might affect levels of GFP [
78]. It was first shown in
Anabaena sp. PCC 7120, using a Lux reporter, that the nitrogenase genes were expressed only in heterocysts even under anoxic conditions [
73]. Similarly, in
A. variabilis expression of
nifD1:
lacZ is confined to heterocysts, whether the cells are grown under oxic or anoxic conditions (
Figure 2), indicating that some aspect of heterocyst development, possibly a heterocyst-specific activator, is required for
nif1 gene expression [
16].
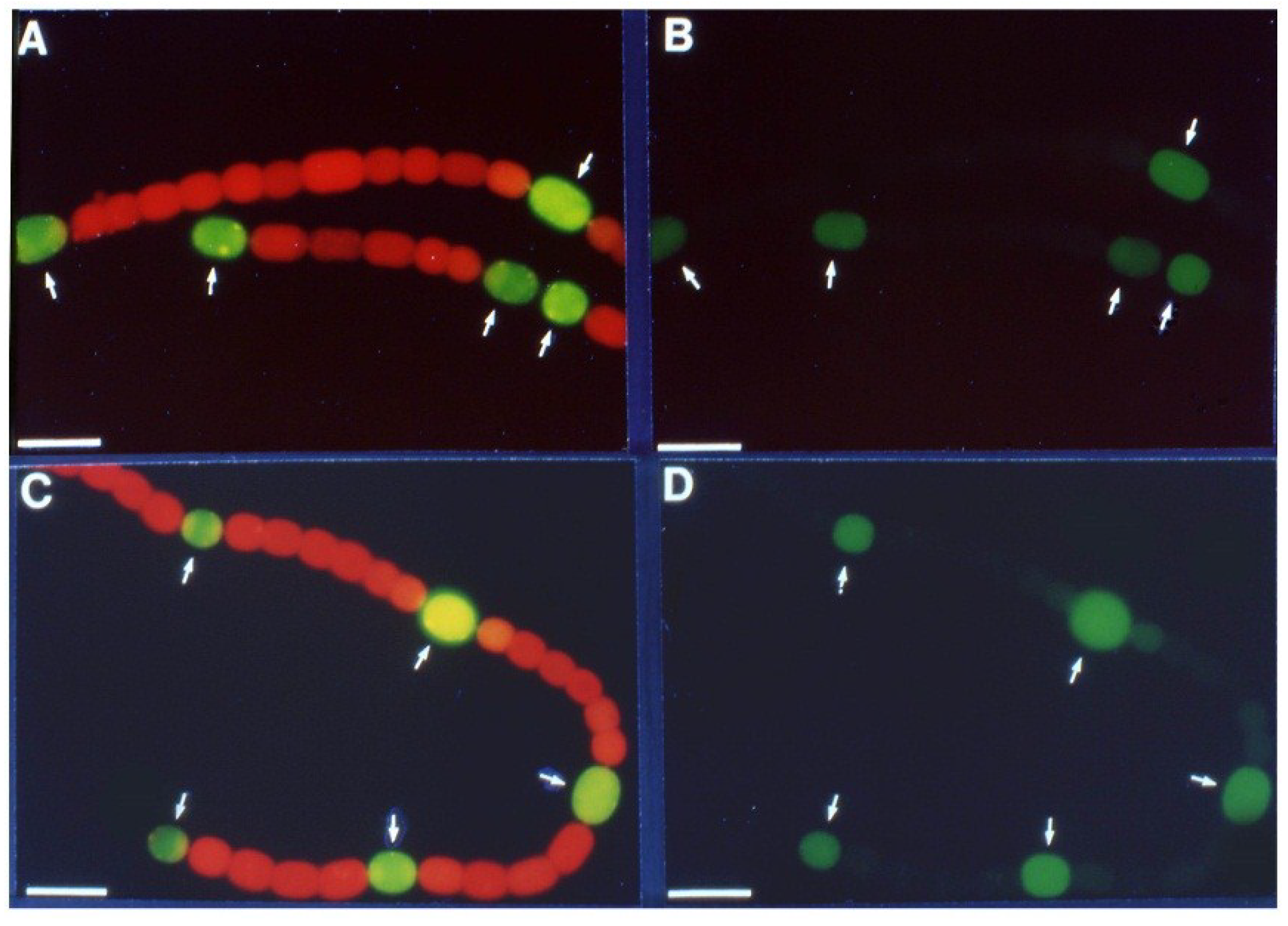
In contrast, the
nif2 genes of
A. variabilis are expressed only in cells grown under anoxic conditions and expression is evident within 4–6 h after nitrogen deprivation (
Figure 3, panels A, C) [
14,
16]. The
nif2 genes are poorly expressed in the heterocysts that form under anoxic growth conditions, and the β-galactosidase activity seen in the heterocysts (
Figure 3B) may reflect enzyme that was made in the vegetative cell prior to differentiation, rather than
de novo synthesis in heterocysts. In support of this hypothesis, we observe that expression of the
nif2 genes is restricted to vegetative cells in filaments that are first grown under oxic conditions, to allow heterocysts to form, and then switched to anoxic conditions (
Figure 3, panels C and D) [
15].
Expression of the
vnf genes, like the
nif1 genes, is restricted to heterocysts in cells grown under oxic or anoxic conditions (
Figure 4) [
13], suggesting that, like the
nif1 cluster, expression of the
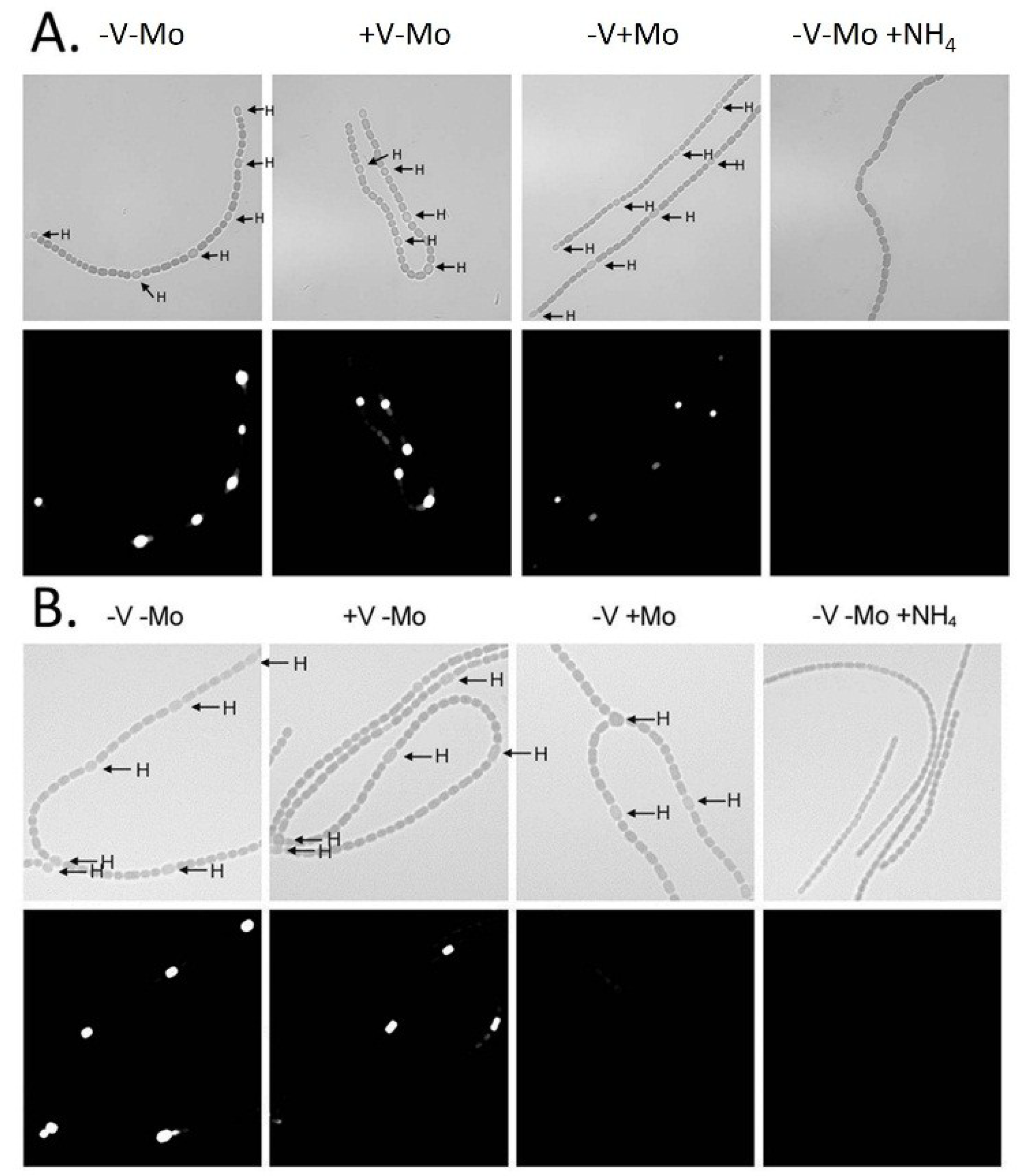
vnf genes depends on a signal that is induced during heterocyst development; however, the
vnf genes are not expressed unless the cells are starved for molybdate (
Figure 5 and
Figure 6) [
79]. The fact that the V-nitrogenase requires NifB1 and possibly other gene products in the
nif1 cluster [
76] is also consistent with the heterocyst-specific expression of the
vnf genes.
Figure 2.
Oxic
vs. anoxic expression of β-galactosidase in a
nifD1:lacZ fusion strain. Cells grown in the absence of fixed N with oxygen; fluorescence from cleavage of fluorescein-β-
d-galactopyranoside photographed without red cut-off filter (
A); or with red cut-off filter (
B); Cells grown in the absence of fixed N without oxygen; fluorescence from cleavage of fluorescein-β-
d-galactopyranoside photographed without red cut-off filter (
C); or with red cut-off filter (
D). Arrows indicate heterocysts identified by bright field microscopy. Bar = 10 μM. Reproduced from [
16] with permission.
Figure 2.
Oxic
vs. anoxic expression of β-galactosidase in a
nifD1:lacZ fusion strain. Cells grown in the absence of fixed N with oxygen; fluorescence from cleavage of fluorescein-β-
d-galactopyranoside photographed without red cut-off filter (
A); or with red cut-off filter (
B); Cells grown in the absence of fixed N without oxygen; fluorescence from cleavage of fluorescein-β-
d-galactopyranoside photographed without red cut-off filter (
C); or with red cut-off filter (
D). Arrows indicate heterocysts identified by bright field microscopy. Bar = 10 μM. Reproduced from [
16] with permission.
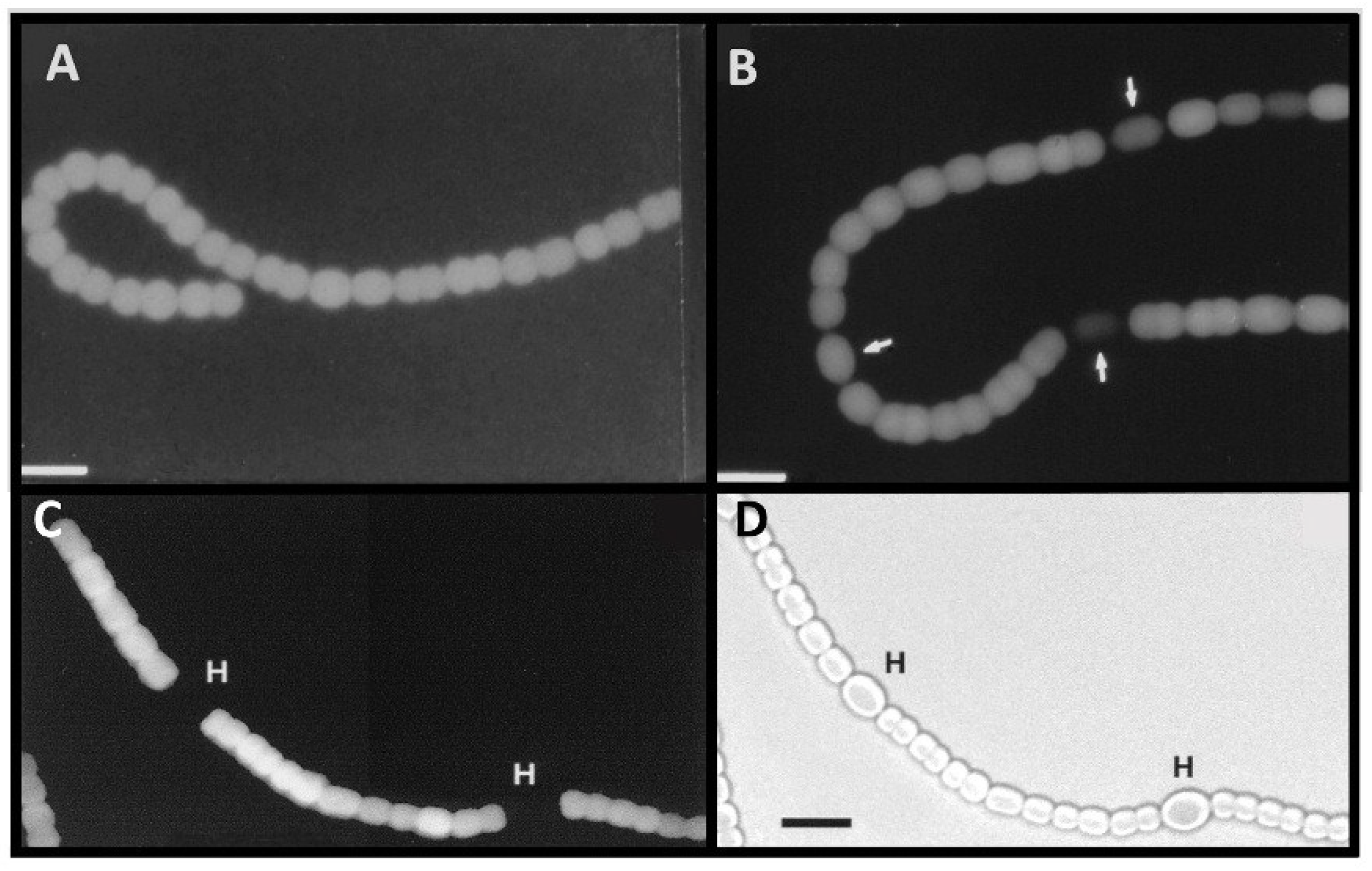
Figure 3.
Anoxic β-galactosidase expression in a
nif2:lacZ fusion strain. Panels (
A) and (
B): Cells with a
nifD2:lacZ fusion were grown in the absence of fixed N, without oxygen; fluorescence from cleavage of fluorescein-β-
d-galactopyranoside was photographed with red cut-off filter 6 h (A), or 24 h (B) after removal of fixed N. Arrows indicate heterocysts identified by bright field microscopy. Panels (
C) and (
D): Cells with a
nifD2:lacZ fusion, grown for 48 h under oxic conditions without fixed N (to induce expression of the Nif1 nitrogenase) were then shifted to anoxic conditions for 4 h to induce expression of
nifD2. Fluorescence from cleavage of fluorescein-β-
d-galactopyranoside was photographed with a red cut-off filter (A). Light micrograph (D). Bar = 10 μM. Panels (A) and (B) are reproduced from [
16] and panels (C) and (D) are reproduced from [
15], with permissions.
Figure 3.
Anoxic β-galactosidase expression in a
nif2:lacZ fusion strain. Panels (
A) and (
B): Cells with a
nifD2:lacZ fusion were grown in the absence of fixed N, without oxygen; fluorescence from cleavage of fluorescein-β-
d-galactopyranoside was photographed with red cut-off filter 6 h (A), or 24 h (B) after removal of fixed N. Arrows indicate heterocysts identified by bright field microscopy. Panels (
C) and (
D): Cells with a
nifD2:lacZ fusion, grown for 48 h under oxic conditions without fixed N (to induce expression of the Nif1 nitrogenase) were then shifted to anoxic conditions for 4 h to induce expression of
nifD2. Fluorescence from cleavage of fluorescein-β-
d-galactopyranoside was photographed with a red cut-off filter (A). Light micrograph (D). Bar = 10 μM. Panels (A) and (B) are reproduced from [
16] and panels (C) and (D) are reproduced from [
15], with permissions.
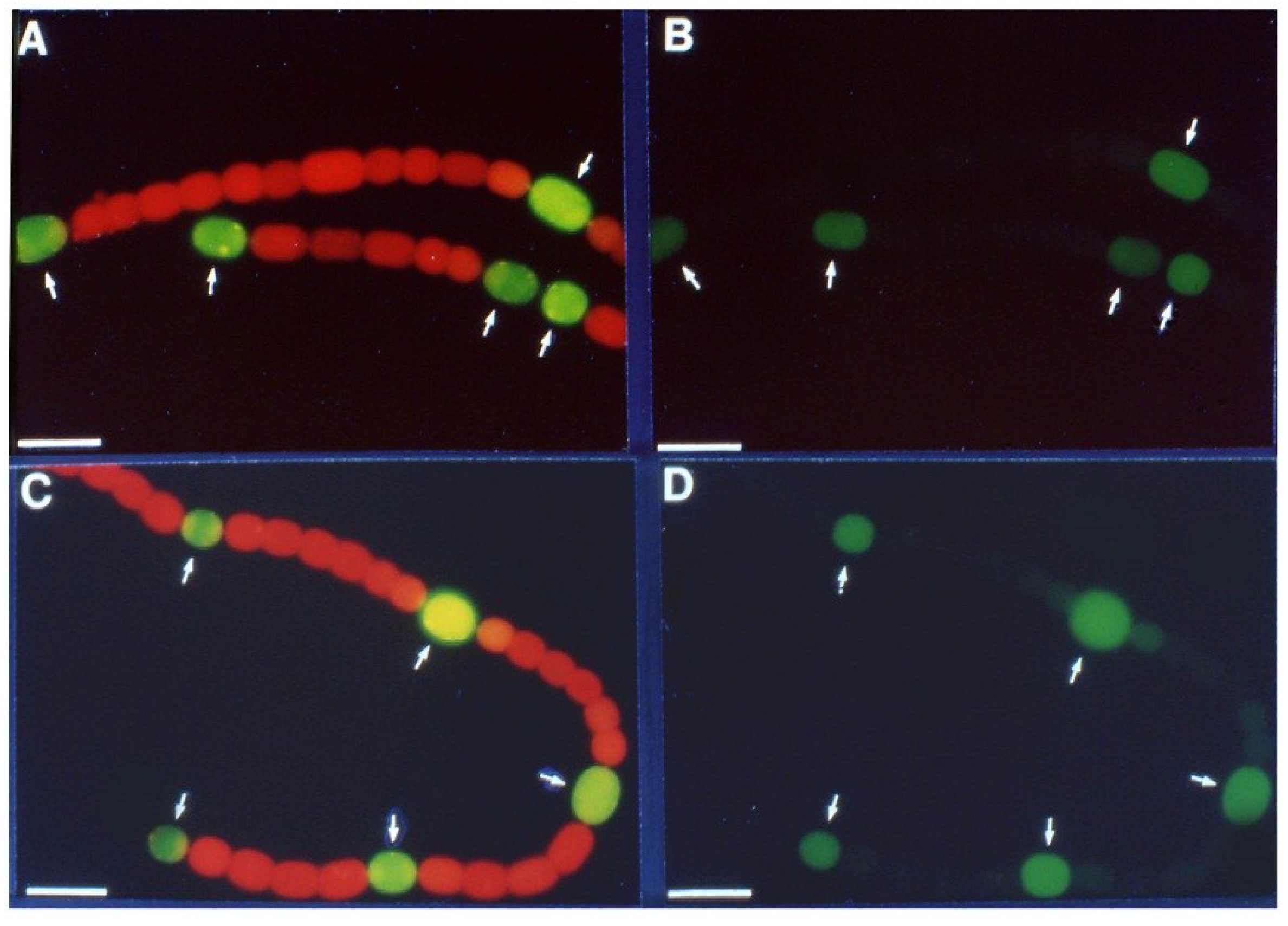
Figure 4.
In situ localization of expression of
lacZ under the control of the
vnfH promoter. Strain BP272 (
vnfH:lacZ fusion) was grown with fructose, in the absence of molybdate, with vanadate, under oxic (+O
2) or anoxic (−O
2) conditions. (
A) Light micrographs; (
B) Fluorescence from cleavage of fluorescein-β-
d-galactopyranoside was photographed with a red cut-off filter. H = heterocysts. Bar = 10 µM. Reproduced from [
15] with permission.
Figure 4.
In situ localization of expression of
lacZ under the control of the
vnfH promoter. Strain BP272 (
vnfH:lacZ fusion) was grown with fructose, in the absence of molybdate, with vanadate, under oxic (+O
2) or anoxic (−O
2) conditions. (
A) Light micrographs; (
B) Fluorescence from cleavage of fluorescein-β-
d-galactopyranoside was photographed with a red cut-off filter. H = heterocysts. Bar = 10 µM. Reproduced from [
15] with permission.
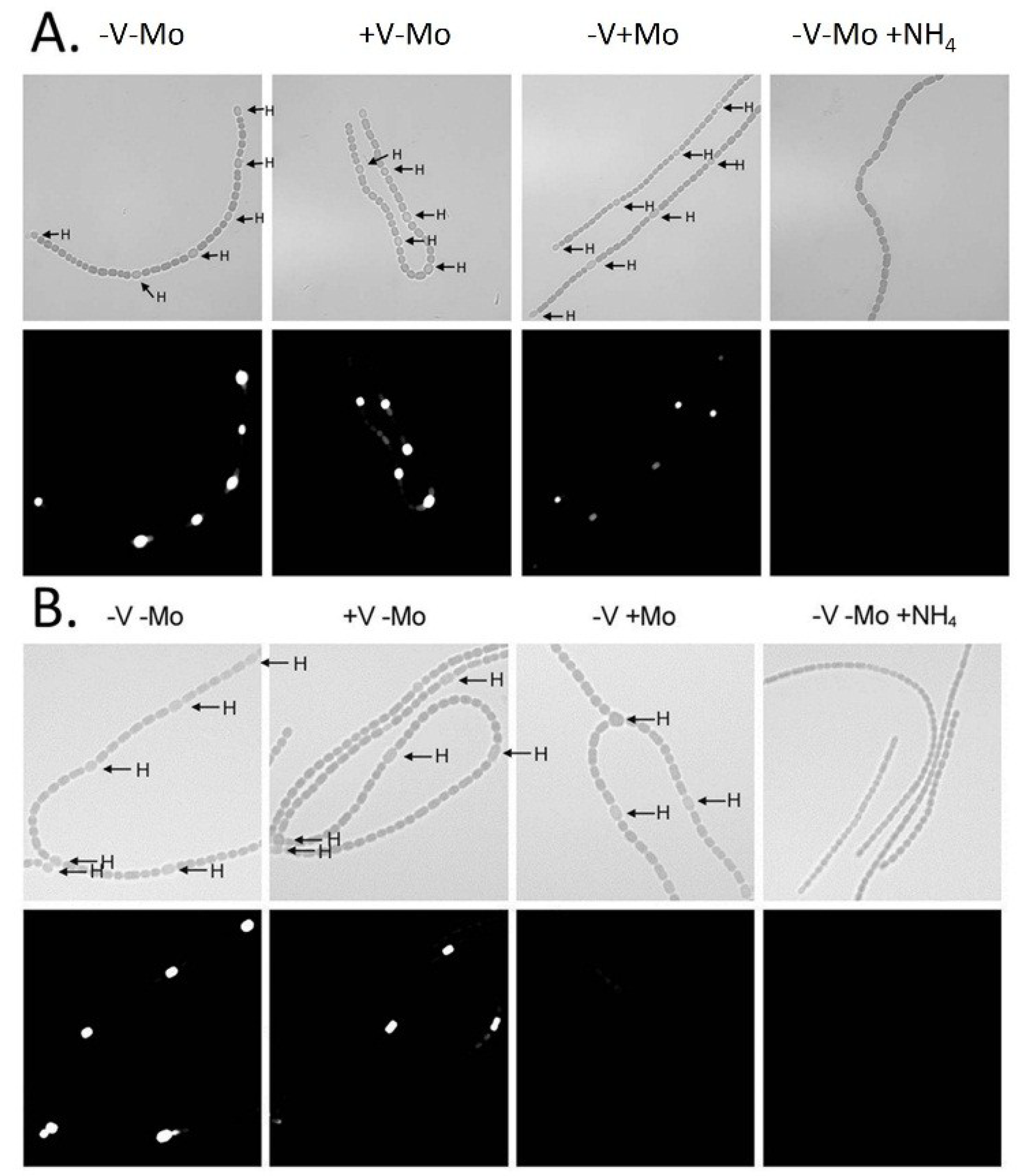
Figure 5.
In situ localization of (
A)
nifH1 expression and (
B)
vnfDG expression. Cells of strain BP221, with
lacZ fused to the promoter region of
nifH1 (A), or BP193, with
lacZ fused to the promoter region of
vnfDG, were grown in AA/8 medium, with or without 1.0 μM molybdate or 1.0 μM vanadate or with 5.0 mM NH
4Cl and 10 mM TES, pH 7.2. β-galactosidase activity was visualized using fluorescein-β-
d-galactopyranoside. Top panels are bright field images showing heterocysts (H). Bottom panels show fluorescein fluorescence. Reproduced from [
80] with permission.
Figure 5.
In situ localization of (
A)
nifH1 expression and (
B)
vnfDG expression. Cells of strain BP221, with
lacZ fused to the promoter region of
nifH1 (A), or BP193, with
lacZ fused to the promoter region of
vnfDG, were grown in AA/8 medium, with or without 1.0 μM molybdate or 1.0 μM vanadate or with 5.0 mM NH
4Cl and 10 mM TES, pH 7.2. β-galactosidase activity was visualized using fluorescein-β-
d-galactopyranoside. Top panels are bright field images showing heterocysts (H). Bottom panels show fluorescein fluorescence. Reproduced from [
80] with permission.
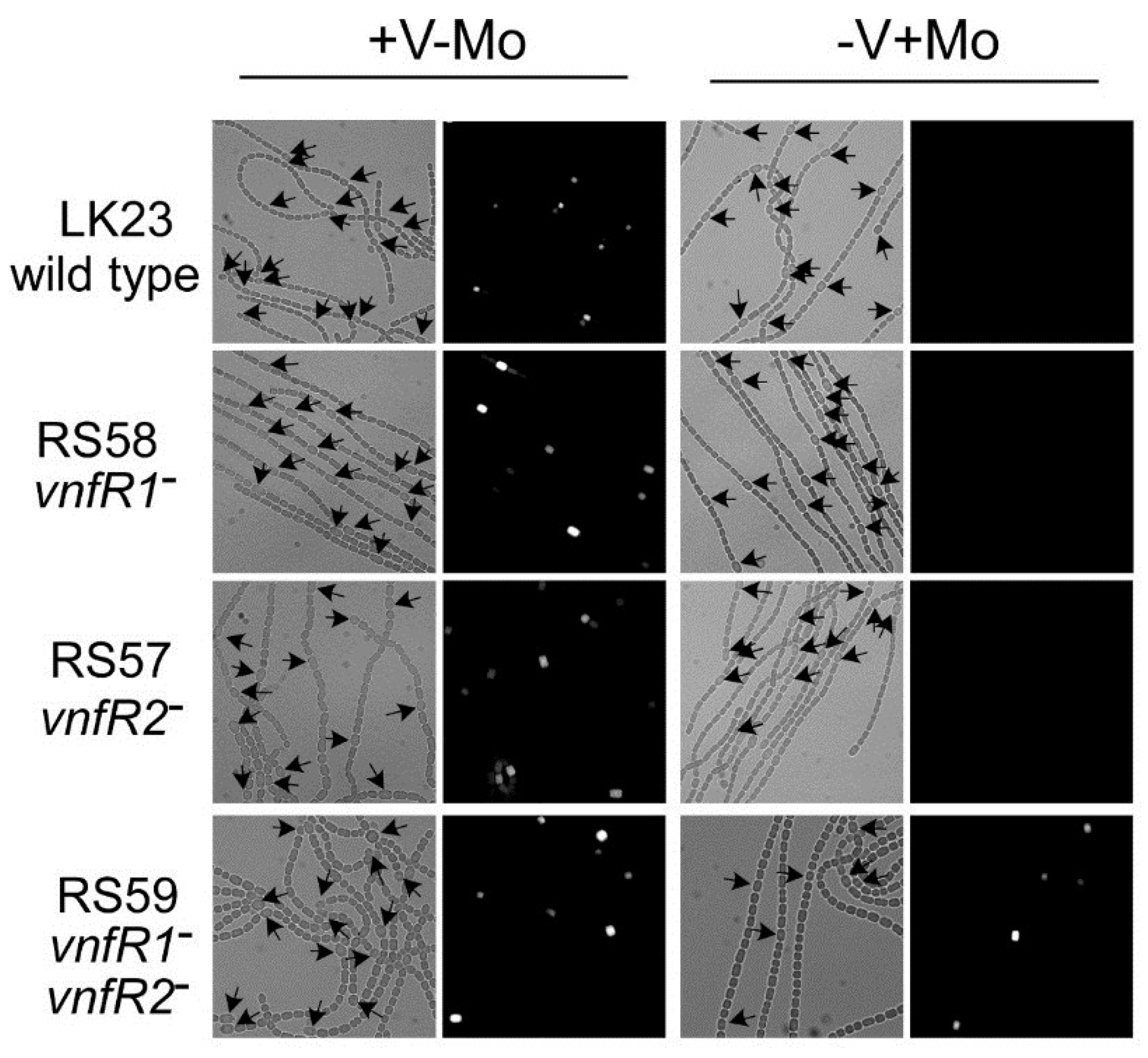
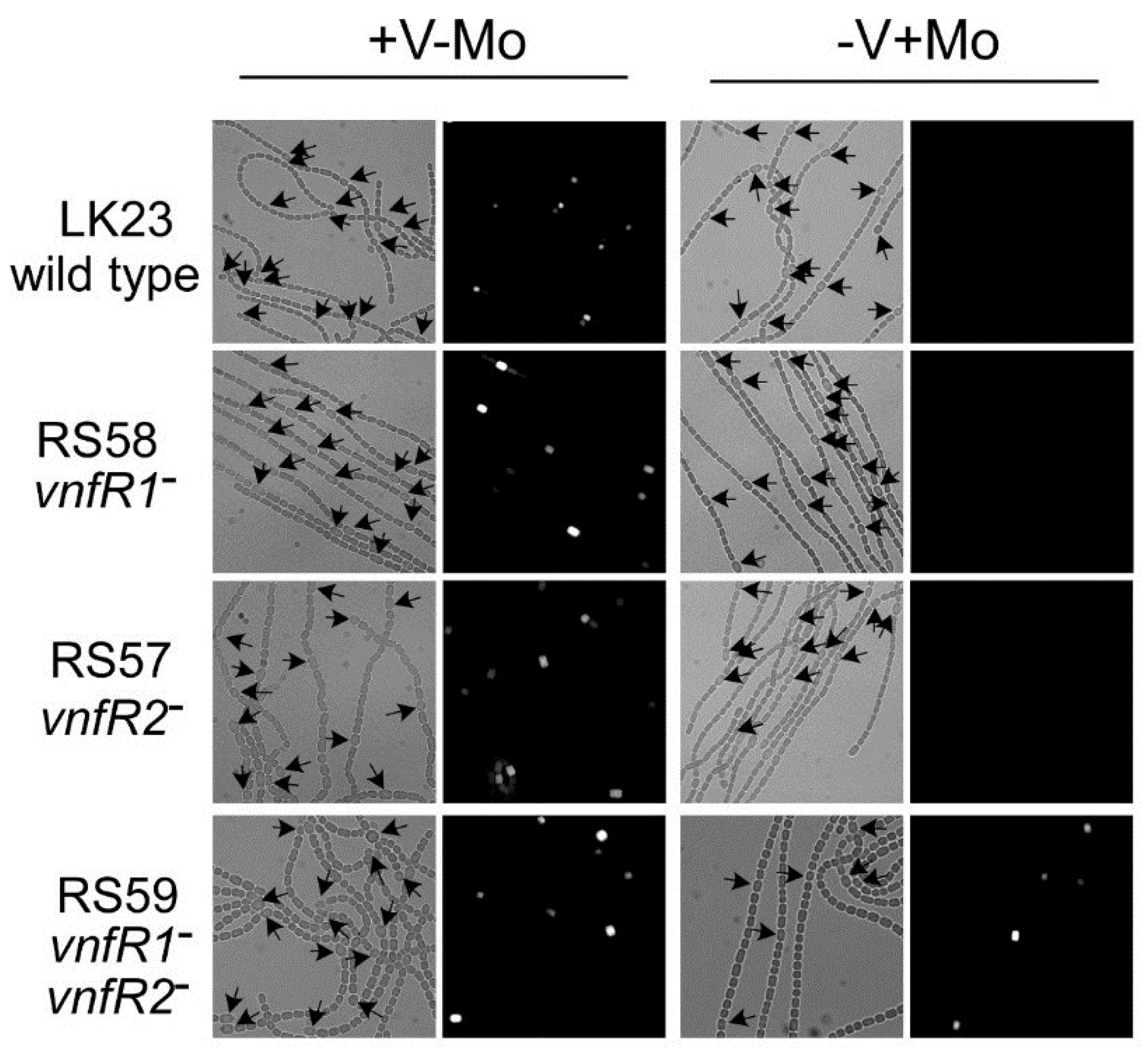
Figure 6.
In situ localization of expression of
vnfDG. The
vnfDG gene was replaced in the chromosome by
lacZ, in a
vnfR1 mutant (RS58), in a
vnfR2 mutant (RS57), in a
vnfR1 vnfR2 double mutant (RS59) or in a wild-type background (LK23). β-galactosidase was visualized with a fluorescent substrate in cells grown with V (no Mo) or with Mo. Left panels are bright field images while the right panels are fluorescence images of the same field. Arrows indicate heterocysts. Reproduced from [
81], with permission.
Figure 6.
In situ localization of expression of
vnfDG. The
vnfDG gene was replaced in the chromosome by
lacZ, in a
vnfR1 mutant (RS58), in a
vnfR2 mutant (RS57), in a
vnfR1 vnfR2 double mutant (RS59) or in a wild-type background (LK23). β-galactosidase was visualized with a fluorescent substrate in cells grown with V (no Mo) or with Mo. Left panels are bright field images while the right panels are fluorescence images of the same field. Arrows indicate heterocysts. Reproduced from [
81], with permission.
4. Metal Transport and Its Effect on Nitrogenase Gene Expression
In many bacteria, including
Escherichia coli,
Rhodobacter capsulatus and
Azotobacter vinelandii, high-affinity molybdate transport is mediated by an ABC-type transport system encoded by
modABC genes [
82,
83,
84,
85]. ModA is the periplasmic component that binds molybdate, ModB is the transmembrane component of the permease, while ModC provides the energy from the cytoplasmic side of the membrane. Transcription of the
modABC operon of
E. coli is negatively regulated by dimers of ModE that are bound with four molecules of molybdate [
86]. A high-affinity molybdate transport system in
A. variabilis, with a
Km for transport of molybdate of about 0.3 nM, is encoded by genes
modA and fused genes
modBC [
79,
87] that are located 2.7 Mb apart in the 6.36 Mb chromosome [
70]. Mutants in this transport system cannot fix nitrogen unless molybdate is supplied at high concentrations (about 1 mM) or unless vanadate is supplied to allow assembly of the V-nitrogenase [
79]. Cells starved for molybdate and vanadate express the
nif1 and
vnf nitrogenase genes in heterocysts (
Figure 5); however, because they cannot make nitrogenase, these nitrogen-starved cells produce a very high frequency of heterocysts and overexpress the nitrogenase genes. The addition of vanadate to Mo-starved cells has no effect on the expression of the
nif1 genes, while the addition of molybdate, which allows the Mo-nitrogenase to function, turns off expression of the
vnf genes, but also decreases
nif1 gene expression and reduces heterocyst frequency compared to the Mo-starved cells (
Figure 5). In contrast, the
vnf genes are expressed only in the absence of molybdate, with or without vanadate [
13]. NifH1, which is made in cells starved for molybdate, can substitute for VnfH in a
vnfH mutant strain [
13]. Further, in a strain with a
vnfH promoter mutation that allows
vnfH to be expressed in cells grown with molybdate, VnfH can substitute for NifH1 when that strain has a
nifH1 mutation. Thus, the two dinitrogenase reductases for the Mo-nitrogenase and the V-nitrogenase in heterocysts are able to function in place of each other suggesting that they are not involved in determining the metal specificity of these two nitrogenases [
13]. This has not been shown
in vivo for any other organism; however, using the nitrogenase for
A. vinelandii, it has been shown
in vitro that VnfH can replace NifH for the synthesis of the FeMo-cofactor and for maturation of the Mo-nitrogenase [
88].
Between the
vnfDGKEN and
vnfH genes in the
A. variabilis genome are the
vupABC genes encoding the vanadate transport system that supplies vanadate for the V-nitrogenase [
89] (
Figure 1). The high-affinity vanadate transport system, with a K
m of about 3 nM is, to date, the only vanadate transporter that has been characterized. The vanadate transport genes, like the V-nitrogenase genes, are repressed by molybdate [
89]. These genes are most similar to the tungstate transport genes of
Eubacterium acidaminophilum. Similar genes are not present in the complete genomes of other bacterial strains that are known to have a V-nitrogenase, including
A. vinelandii, Rhodopseudomonas palustris, and
Methanosarcina barkeri, although the complete genome sequences of the cyanobacteria
Fremyella diplosiphon UTEX 481,
Chlorogloeopsis sp. PCC 7702, and
Fischerella muscicola PCC 7414 have orthologs of the vanadate transport genes.
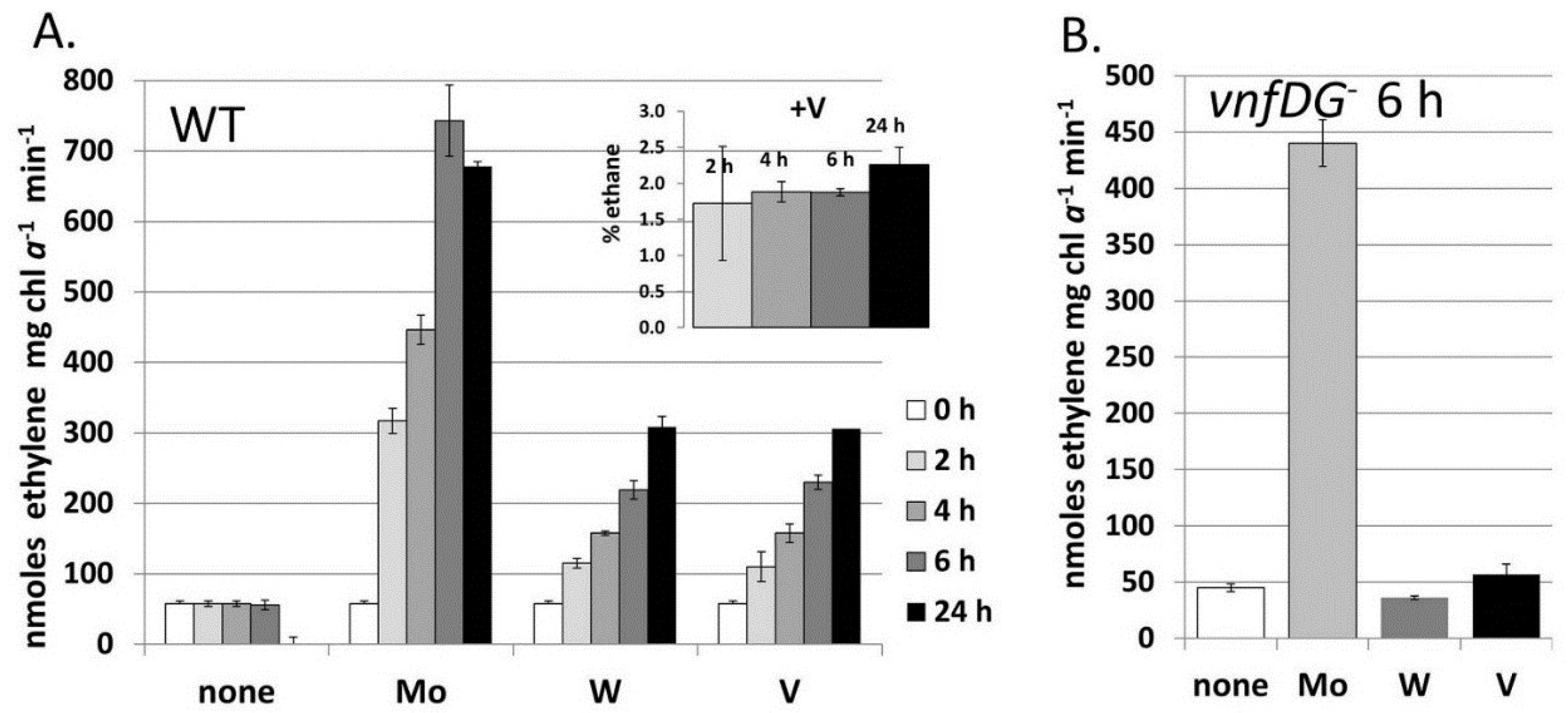
When
A. variabilis is grown in a medium without fixed nitrogen and with less than 1.0 nM Mo and V, the cells become starved for both metals; however, slow growth continues, accompanied by low levels of nitrogen fixation [
80]. This slow growth is abolished in a
nifDK1 mutant lacking the heterocyst-specific Mo-nitrogenase, but slow growth continues in a mutant lacking the V-nitrogenase, suggesting that only the Mo-nitrogenase is able to support slow growth in an environment with little molybdate or vanadate. Tungstate is transported by the molybdate transporter and could, theoretically, be incorporated into a nitrogenase [
87]. The addition of tungstate, vanadate, or molybdate to cells starved for these metals resulted in an increase in nitrogenase activity, as measured by acetylene reduction, after two hours and this increase required new protein synthesis, suggesting that new nitrogenase was being synthesized with all these metals [
80]. While tungstate functioned about as well as vanadate in supporting acetylene reduction, the cells to which tungstate was added did not grow any better with tungstate than with no added metal and did not produce ethane (
Figure 7) [
80]. A mutant lacking the V-nitrogenase showed no increase in nitrogenase activity upon addition of tungstate, suggesting that the V-nitrogenase, rather than the Mo-nitrogenase, was able to incorporate tungstate (
Figure 7). Tungstate was able to substitute for molybdate in repressing transcription of a Mo-transport gene, but not the
vnfH gene, which was, however, repressed by Mo [
80]. This suggests that the Mo-dependent regulator of the molybdate transport system, probably the product of the
modE homolog located just upstream of
modA [
79], interacts differently with molybdate/tungstate than the Mo-dependent regulators of the
vnf genes, VnfR1 and VnfR2 [
81] discussed in more detail below.
Figure 7.
Metal-induced increase in nitrogenase activity in cells starved for molybdate and vanadate.
A. variabilis strains FD (WT) and MB2 (
vnfDG mutant) were grown in Mo- and V-free medium for at least 10 generation to deplete internal stores of these metals and then Na
2MoO
4, Na
3VO
4 or Na
2WO
4 (all at 100 nM) were added to these starved cells at 0 time. Acetylene reduction was measured for strain FD at 2 h, 4 h, 6 h and 24 h after metal addition (
A) and for strain MB2 (
vnfDG mutant) at 6 h after metal addition (
B). The inset shows the ethane (% of ethylene) produced by strain FD with added Na
3VO
4, which were the only samples that produced any ethane. Reproduced from [
80] with permission.
Figure 7.
Metal-induced increase in nitrogenase activity in cells starved for molybdate and vanadate.
A. variabilis strains FD (WT) and MB2 (
vnfDG mutant) were grown in Mo- and V-free medium for at least 10 generation to deplete internal stores of these metals and then Na
2MoO
4, Na
3VO
4 or Na
2WO
4 (all at 100 nM) were added to these starved cells at 0 time. Acetylene reduction was measured for strain FD at 2 h, 4 h, 6 h and 24 h after metal addition (
A) and for strain MB2 (
vnfDG mutant) at 6 h after metal addition (
B). The inset shows the ethane (% of ethylene) produced by strain FD with added Na
3VO
4, which were the only samples that produced any ethane. Reproduced from [
80] with permission.
5. Transcription of Nitrogenase Genes
By analogy with the
nif operons of other nitrogen-fixing bacteria, including
Klebsiella and
Azotobacter, it has been thought that the large cluster comprising most of the
nif genes in
Anabaena could be divided into several discrete operons, including
nifBSU,
nifHDK and
nifENX. Northern blots appeared to confirm this and, in fact, putative transcription start sites were mapped for the
nifB,
nifH,
hesA and
fdxH genes in the
nif clusters of
Anabaena spp. [
58,
60,
63,
64,
65,
66,
67,
68]. We mapped what appeared to be transcription start sites for
vnfDG and
vnfH (although they are actually processing sites) and confirmed that the apparent transcription start sites for
nifB1 and
nifH1 in
A. variabilis were identical to those mapped in
Anabaena sp. PCC 7120. We also identified additional weak promoters within the coding regions of
nifU1 and
nifE1 [
65,
69]. However, when we attempted to use the
nifH1 promoter to drive expression of
lacZ, using a 300-bp promoter fragment that extended at least 150 bp upstream from the putative
nifH1 transcription start site, there was no reporter activity. A strain in which this same 300-bp fragment was used to drive expression of the
nifH1 gene failed to grow under nitrogen-fixing conditions and had no nitrogenase activity [
90]. The same problem occurred when we attempted to drive transcription of
lacZ with the putative
vnfDG promoter [
81] or with the
vnfH promoter [
65]; there was no expression using these promoter regions, although they extended well upstream from the putative transcription start sites. However, the promoters of other genes gave good activity, including
nifB1 [
65],
ava4025, the gene upstream of
vnfDG [
81], and
vnfR2, the gene upstream of
vnfH [
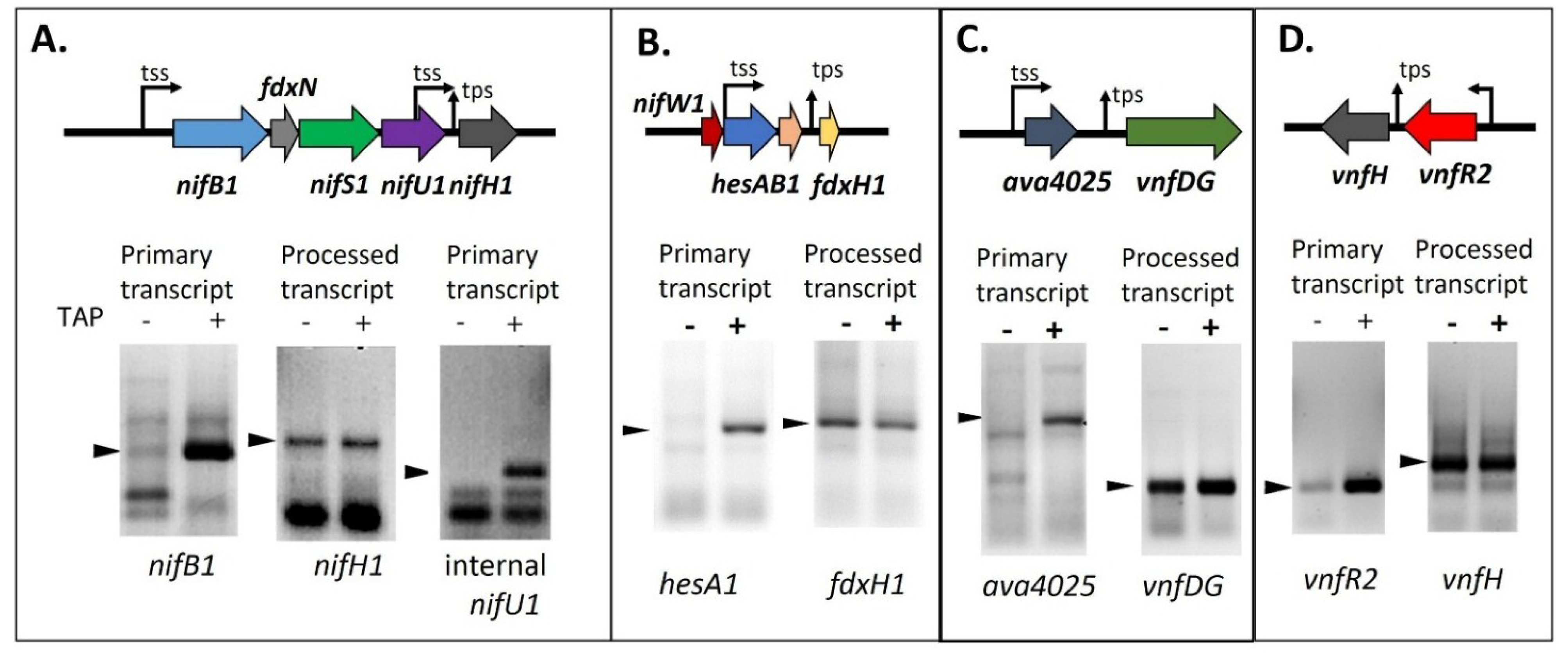
65]. The answer to this puzzle came when the 5' ends of these transcripts were characterized and
nifH1,
fdxH1,
vnfH, and
vnfDG were found to have the 5' monophosphate end of a processed RNA, rather than the 5' triphosphate characteristic of a primary transcript (
Figure 8) [
65,
69,
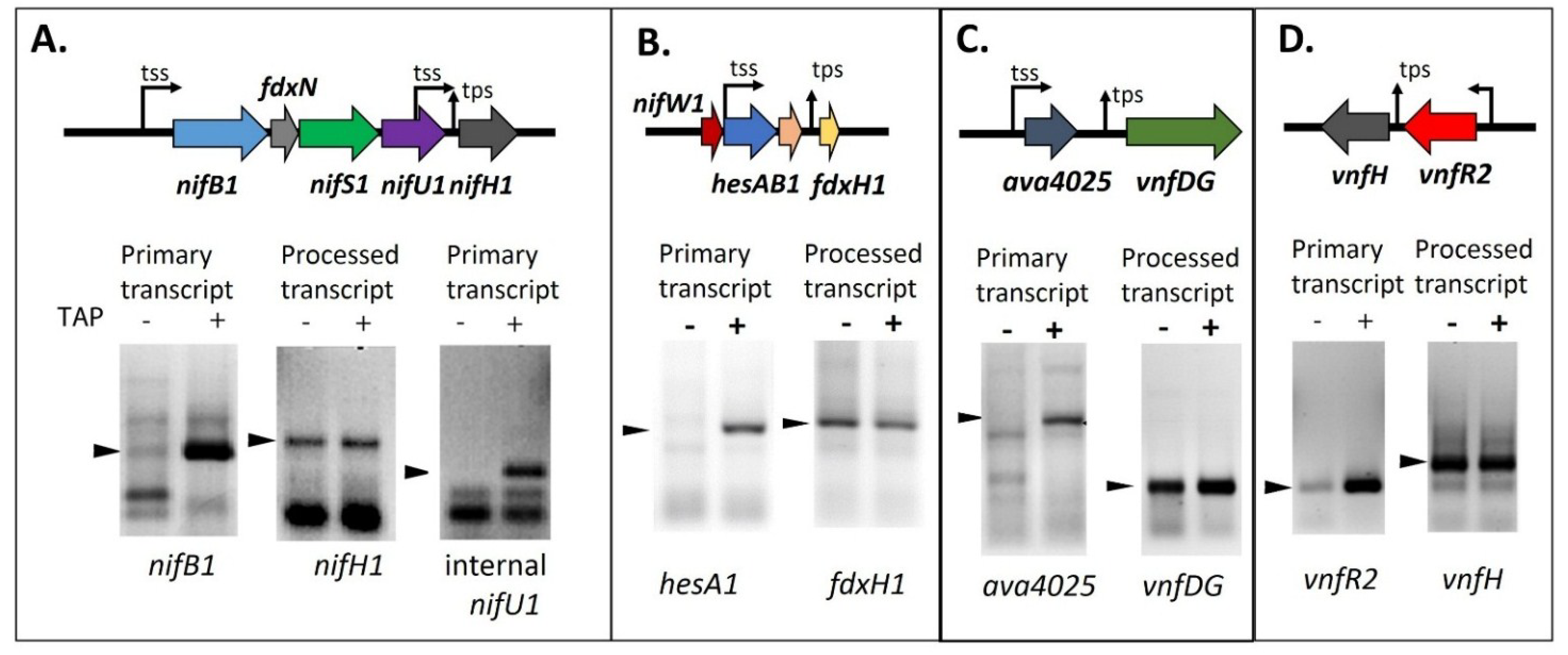
81]. The difference in the 5' end structure of the mRNA can be determined by using a technique called RNA Ligase Mediated Rapid Amplification of cDNA Ends (5' RACE) with RNA that is treated, or not treated, with tobacco acid phosphatase (TAP), which converts a 5' triphosphate end to a 5' monophosphate end in preparation for ligation of the RNA adapter to the 5' end of the transcript. If the transcript is a primary transcript, the RNA adapter cannot ligate to the 5' triphosphate of the RNA unless it is treated with TAP; however, if the transcript is processed, it already has a 5' monophosphate and does not require TAP treatment. A 5' RACE product that is made equally well with RNA treated or not treated with TAP provides good evidence that the transcript is the result of processing
in vivo. If very little 5' RACE product is made when the RNA is not treated with TAP, this indicates that the mRNA is a primary transcript. As shown in
Figure 8, TAP treatment was not required to produce strong products by 5' RACE for
nifH1,
fdxH1,
vnfDG, or
vnfH, while TAP treatment was required for 5' RACE amplification of transcripts for
nifB1,
nifU1,
hesA1,
ava4025 and
vnfR2 [
65,
69,
81].
Figure 8.
Determining the 5' ends of transcripts by 5' RACE with and without TAP treatment. (
A)
nifB1 and
nifU1, requiring TAP, are primary transcripts, but
nifH1 is a processed transcript; (
B)
hesA1, requiring TAP, is a primary transcript, but
fdxH1 is a processed transcript; (
C)
ava4025, requiring TAP, is a primary transcript, but
vnfDG is a processed transcript; (
D)
vnfR2, requiring TAP, is a primary transcript, but
vnfH is a processed transcript. tss, transcription start site; tps, transcriptional processing site. Arrows indicate the PCR products that were sequenced to determine the 5' transcript ends whose location is shown on the gene maps. Gel images in panels A and D are reproduced from [
65], in panel B from [
69], and in panel C from [
81], with permissions.
Figure 8.
Determining the 5' ends of transcripts by 5' RACE with and without TAP treatment. (
A)
nifB1 and
nifU1, requiring TAP, are primary transcripts, but
nifH1 is a processed transcript; (
B)
hesA1, requiring TAP, is a primary transcript, but
fdxH1 is a processed transcript; (
C)
ava4025, requiring TAP, is a primary transcript, but
vnfDG is a processed transcript; (
D)
vnfR2, requiring TAP, is a primary transcript, but
vnfH is a processed transcript. tss, transcription start site; tps, transcriptional processing site. Arrows indicate the PCR products that were sequenced to determine the 5' transcript ends whose location is shown on the gene maps. Gel images in panels A and D are reproduced from [
65], in panel B from [
69], and in panel C from [
81], with permissions.
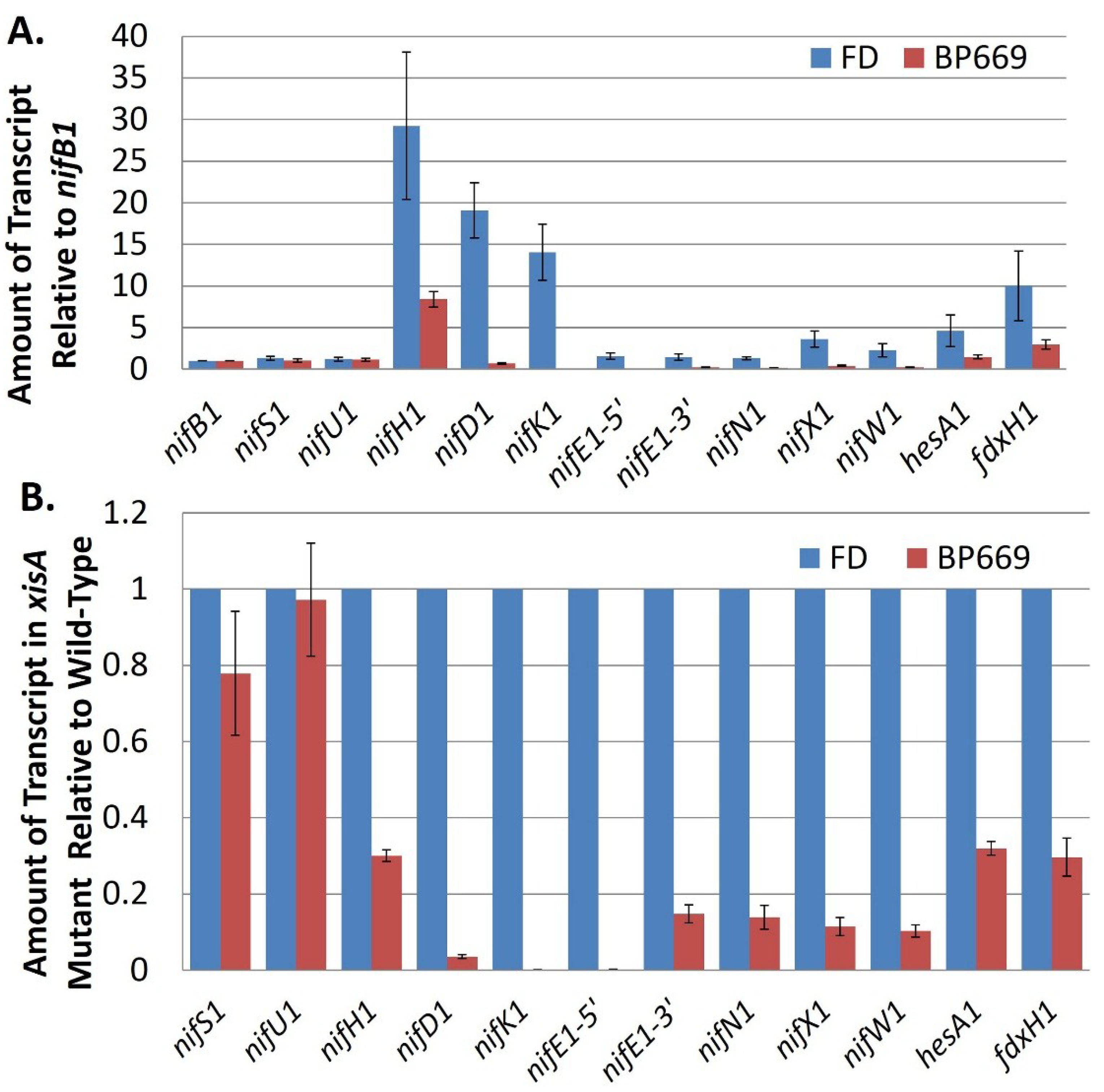
Transcription of the
nif1 gene cluster of
A. variabilis appears to depend primarily on the promoter for the first gene in the cluster,
nifB1. While there is a promoter inside
nifU1, it is very weak compared to the
nifB1 promoter and a strain in which
nifH1 is driven only by the
nifU1 promoter fixes nitrogen poorly compared to the wild-type strain [
65]. There is no promoter upstream of
nifK1 or
nifE1, so their transcription depends on the
nifB1 and
nifU1 promoters. Like the
nifU1 promoter, the promoter within
nifE1 is weak, suggesting that it serves an auxiliary rather than a primary function for gene expression [
69]. Further support for the importance of the
nifB1 promoter in expression of the far downstream genes, including
nifKENXW1 is the near loss of these transcripts in a mutant strain that lacks
xisA, the gene that makes the excisase that removes the 11-kb element from
nifD1 [
69]. In this mutant, the
nifB1 and
nifU1 promoters cannot drive expression of the genes downstream from the 11-kb element, which is not excised, and these genes are poorly transcribed (
Figure 9). Even
hesA1, which has its own promoter, shows decreased expression in the
xisA mutant, which suggests that the
nifB1 promoter is capable, at least partially, of driving transcription of a gene that is 14 kb away. Consistent with the fact that the
nif1 cluster encodes an enzyme that functions only in heterocysts, the
nifB1,
nifU1 and
nifE1 promoters showed heterocyst-specific expression (
Figure 10) [
69].
Figure 9.
Transcript abundance of
nif cluster genes in the wild-type strain compared to BP669, an
xisA mutant. (
A) The amount of transcript for genes in the
nif cluster (relative to
nifB1) was determined by RT-qPCR using RNA isolated from the wild-type strain, FD and from BP669, which cannot remove the 11-kb excision element in
nifD1. Strains were grown with ammonium and then
nif genes were induced by 24 h of starvation for fixed nitrogen; (
B) Transcript levels in BP669 are shown relative to the wild-type strain, FD, in order to more clearly visualize the low levels of transcript for
nifK1,
nifE1,
nifN1,
nifX1, and
nifW1 in the mutant. Reproduced from [
69], with permission.
Figure 9.
Transcript abundance of
nif cluster genes in the wild-type strain compared to BP669, an
xisA mutant. (
A) The amount of transcript for genes in the
nif cluster (relative to
nifB1) was determined by RT-qPCR using RNA isolated from the wild-type strain, FD and from BP669, which cannot remove the 11-kb excision element in
nifD1. Strains were grown with ammonium and then
nif genes were induced by 24 h of starvation for fixed nitrogen; (
B) Transcript levels in BP669 are shown relative to the wild-type strain, FD, in order to more clearly visualize the low levels of transcript for
nifK1,
nifE1,
nifN1,
nifX1, and
nifW1 in the mutant. Reproduced from [
69], with permission.
Figure 10.
In situ localization of expression of β-galactosidase in strains with fusions of a promoterless
lacZ to the
nifB1 promoter alone in strain BP682 (panels (
A) and (
B)), the
nifU1 promoter alone in strain JU472 (panels in (
C) and (
D)), or the
nifE1 promoter alone in strain BP756 (panels (
E) & (
F)). Panels (A), (C), and (E), are bright-field images of filaments with white arrows indicating a few representative heterocysts. Panels (B), (D) and (F) are fluorescence images showing the expression of β-galactosidase primarily in heterocysts. Exposure times in seconds for the fluorescence images are provided in panels (B), (D) and (F). Reproduced from [
69], with permission.
Figure 10.
In situ localization of expression of β-galactosidase in strains with fusions of a promoterless
lacZ to the
nifB1 promoter alone in strain BP682 (panels (
A) and (
B)), the
nifU1 promoter alone in strain JU472 (panels in (
C) and (
D)), or the
nifE1 promoter alone in strain BP756 (panels (
E) & (
F)). Panels (A), (C), and (E), are bright-field images of filaments with white arrows indicating a few representative heterocysts. Panels (B), (D) and (F) are fluorescence images showing the expression of β-galactosidase primarily in heterocysts. Exposure times in seconds for the fluorescence images are provided in panels (B), (D) and (F). Reproduced from [
69], with permission.
The primary promoter for the structural genes for the V-nitrogenase,
vnfDGKEN, is the Mo-repressible promoter for the gene upstream from this cluster,
ava4025. The predicted product of this gene shows similarity to the periplasmic component of molybdate transporters, suggesting that it may have a role in sensing molybdate in the environment; however, a mutant in
ava4025 has no apparent phenotype and the gene is not required for Mo-repression of
vnfDGKEN [
81]. Although the
ava4025 promoter controls expression of
ava4025 and
vnfDG, levels of
vnfDG transcript are about 500-fold higher than
ava4025, perhaps resulting from increased stability of the
vnfDG transcript, which is processed at the site that was initially identified as the transcription start site [
11,
81]. Expression of
vnfDG, under the control of the
ava4025 promoter is heterocyst specific (see
Figure 5 and
Figure 6) [
81]. Like
vnfDGKEN,
vnfH, encoding the dinitrogenase reductase component of the V-nitrogenase, is the result of the processing of a transcript that is made from the promoter of the upstream gene,
vnfR1. Although we initially reported, based on Northern blots, that vanadate transport genes,
vupABC, form an operon [
89], it now seems possible that the promoter for
vnfR1, located upstream of the
vupABC cluster may control these genes as well as the gene between
vnfR1 and
vupABC, which may be
vnfV, and that the
vupABC transcripts may also result from RNA processing; however, this hypothesis awaits experimental support.
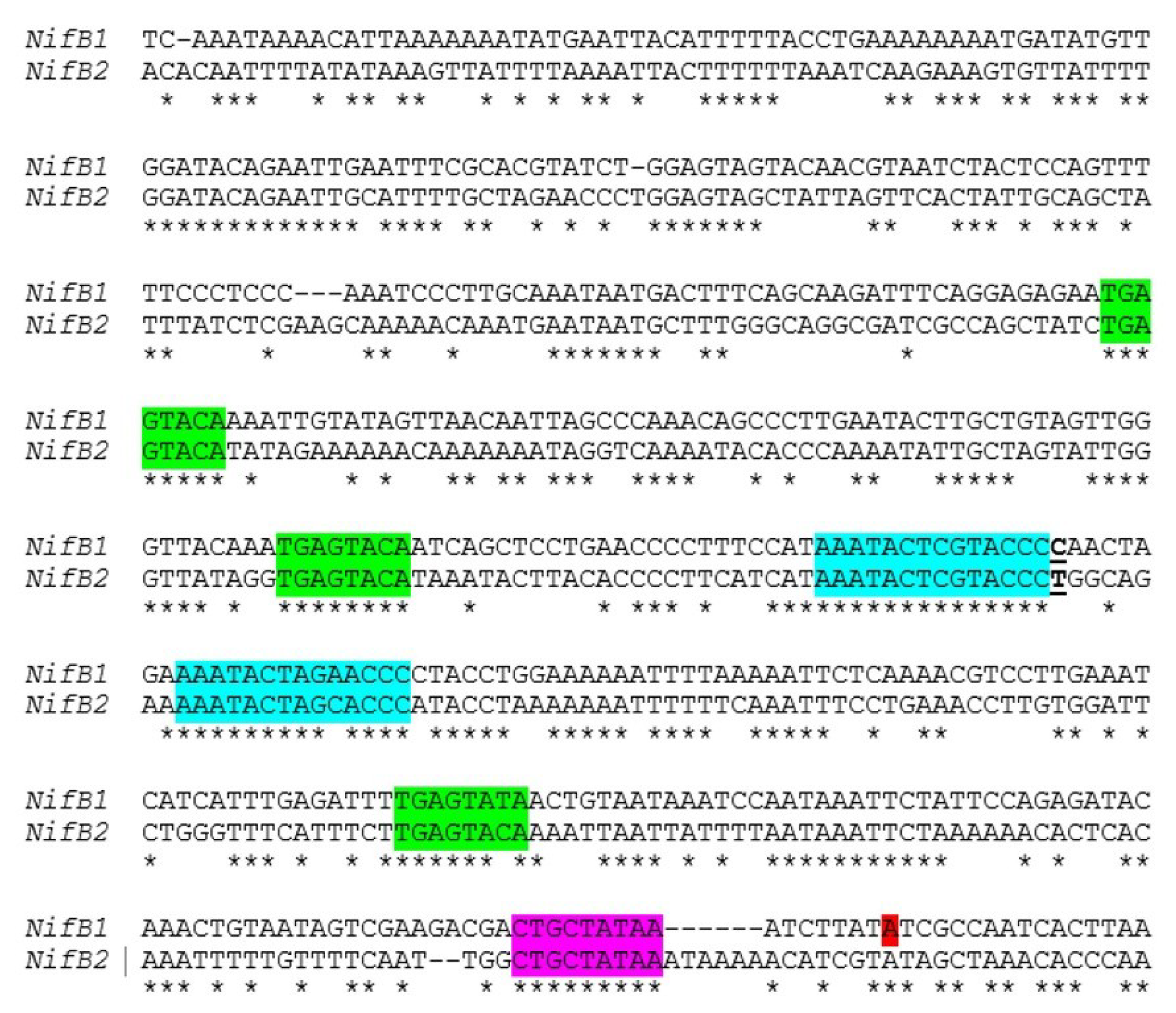
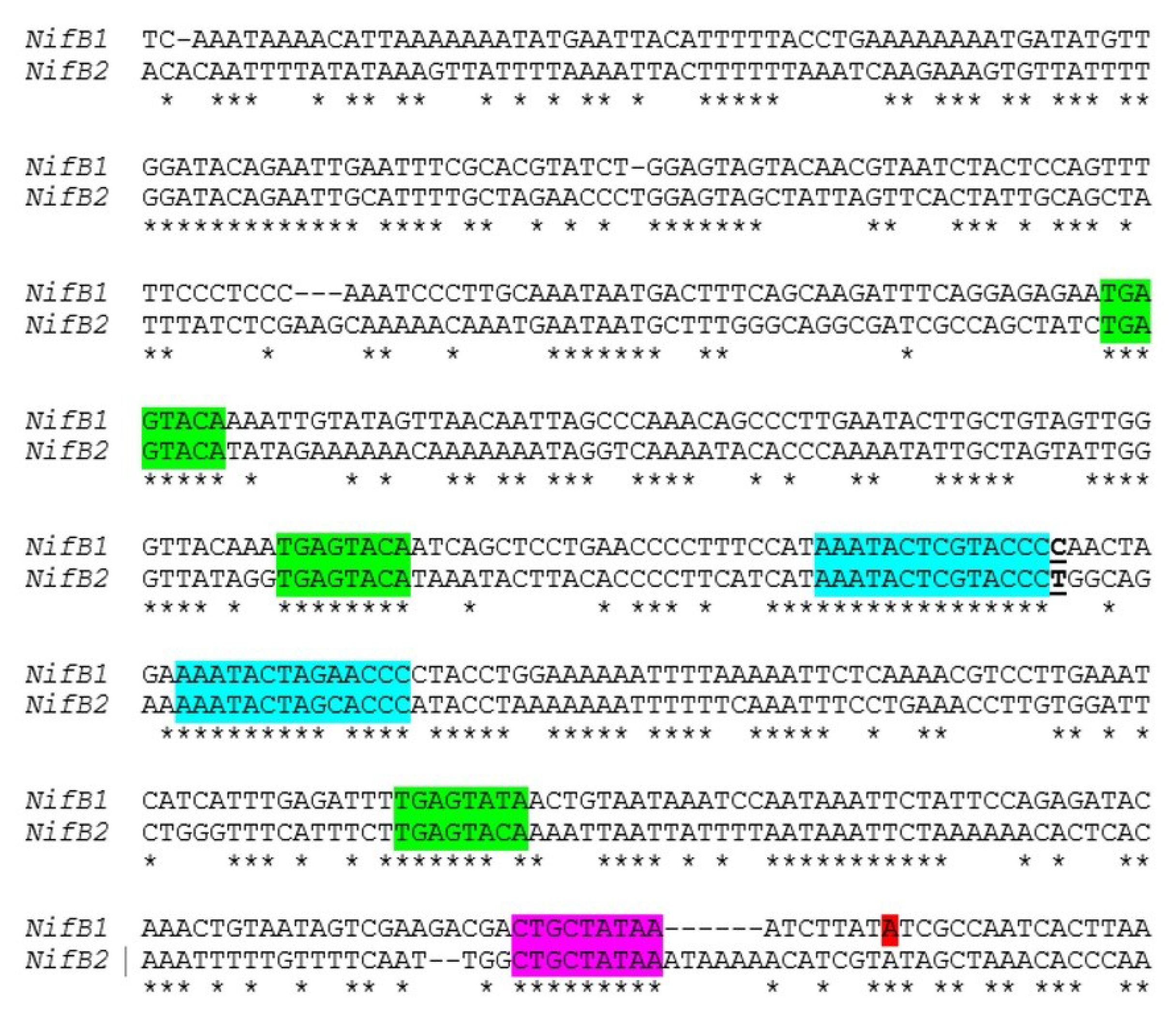
Little is known about the control of
nif genes that function under anoxic conditions, including the
nif2 cluster in
A. variabilis; however, the conservation of the organization of the entire cluster suggests that these genes may also be under the control of a single primary promoter. There are striking similarities in the sequences of
nifB1 and
nifB2 in the region upstream of the
nifB1 transcription start site, including conserved motifs, that suggest that the two
nifB genes have some aspects of regulation in common (
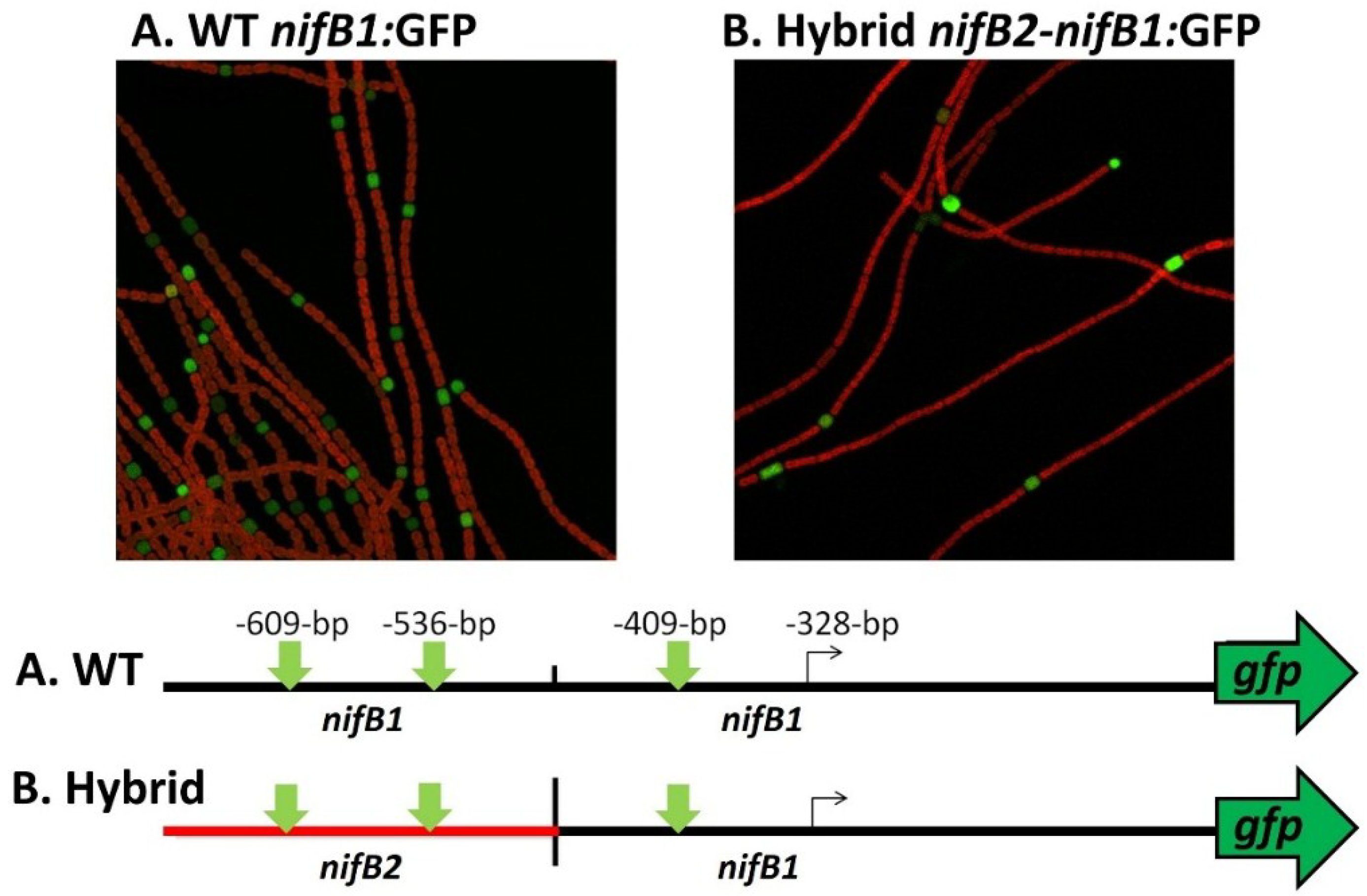
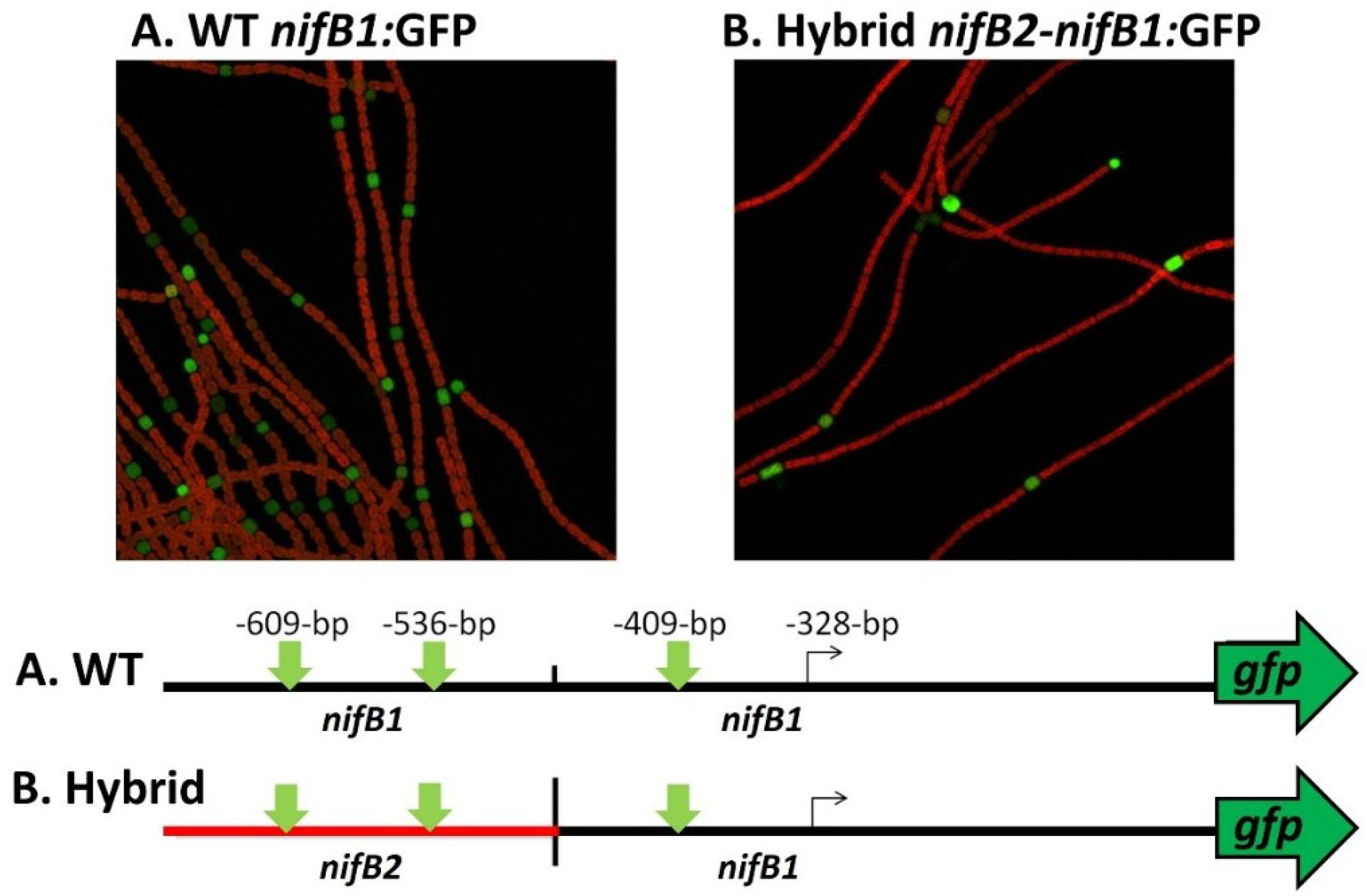
Figure 11). To test this hypothesis we created a fusion between the upstream region of
nifB2, up to and including the first conserved motif shown in turquoise (
Figure 11), to the downstream region of
nifB1 and then fused the hybrid promoter to GFP (strain JJ146). Expression of the
nifB2:
nifB1 hybrid promoter fused to GFP was localized specifically to heterocysts as was GFP expressed from the control
nifB1 promoter (strain JJ72) (unpublished data and
Figure 12). This heterocyst-specific expression of the hybrid promoter indicates that the conserved region upstream of the promoter of
nifB1 may serve in regulation that senses oxygen levels, since that signal is the primary one that induces expression of
nifB2, and that heterocyst specificity may be conferred by sequences closer to the promoter that are not shared with the
nifB2 promoter and by heterocyst-specific protein(s) that may recognize these sequences. An understanding of the roles of the various elements in the promoter and upstream regions awaits a more detailed genetic analysis of both promoters and the identification of proteins that may control cell-specific transcription of the different nitrogenase genes.
Figure 11.
Similarity of the
nifB1 and
nifB2 regions upstream from the promoter. Alignment of the region upstream of the transcription start site of
nifB1, shown in red, with a similar region upstream of
nifB2. A putative extended −10 region is highlighted in magenta, a conserved TGAGTATA motif is highlighted in green, and another conserved motif is highlighted in turquoise. The T that is underlined at the end of the first motif in turquoise indicates the fusion site of a
nifB2:
nifB1 hybrid promoter (see
Figure 12).
Figure 11.
Similarity of the
nifB1 and
nifB2 regions upstream from the promoter. Alignment of the region upstream of the transcription start site of
nifB1, shown in red, with a similar region upstream of
nifB2. A putative extended −10 region is highlighted in magenta, a conserved TGAGTATA motif is highlighted in green, and another conserved motif is highlighted in turquoise. The T that is underlined at the end of the first motif in turquoise indicates the fusion site of a
nifB2:
nifB1 hybrid promoter (see
Figure 12).
Figure 12.
Heterocyst-specific expression of the
nifB1 promoter and a
nifB2:
nifB1 hybrid promoter. The wild-type
nifB1 promoter was fused to the reporter gene,
gfp, and a hybrid promoter of
nifB2/
nifB1, fused at the first nucleotide following the first conserved motif shown in turquoise in
Figure 10, was also fused to
gfp using fusion PCR, and the construct was integrated into the
A. variabilis genome as described previously [
65,
81]. Strains were imaged by confocal microscopy 24 h after nitrogen step-down. GFP fluorescence was localized to heterocysts, which are green in the images. The map of the fusion shows the location of the tss in
nifB1 at −328 bp from the translation start site and the approximate location of the putative tss of
nifB2. The vertical green arrows on the map show the locations of the three conserved motifs that are highlighted in green in
Figure 11.
Figure 12.
Heterocyst-specific expression of the
nifB1 promoter and a
nifB2:
nifB1 hybrid promoter. The wild-type
nifB1 promoter was fused to the reporter gene,
gfp, and a hybrid promoter of
nifB2/
nifB1, fused at the first nucleotide following the first conserved motif shown in turquoise in
Figure 10, was also fused to
gfp using fusion PCR, and the construct was integrated into the
A. variabilis genome as described previously [
65,
81]. Strains were imaged by confocal microscopy 24 h after nitrogen step-down. GFP fluorescence was localized to heterocysts, which are green in the images. The map of the fusion shows the location of the tss in
nifB1 at −328 bp from the translation start site and the approximate location of the putative tss of
nifB2. The vertical green arrows on the map show the locations of the three conserved motifs that are highlighted in green in
Figure 11.
The lack of discrete operons in the
nif1 clusters is inconsistent with the differences in transcript levels for different
nif genes, especially for the highly abundant
nifH1. The
nifH1 transcript is present in much greater quantity than
nifU1, the gene directly upstream of
nifH1, and the other structural genes for nitrogenase,
nifD1 and
nifK1, also show high levels of transcription compared to
nifB1 (
Figure 9A). If a strong promoter is not directly driving transcription of
nifHDK1, then the higher levels of these transcripts may result from their stability. A striking feature of the region near the transcriptional processing sites of
nifH1,
vnfH1,
vnfDG, and
fdxH1 is the presence of conserved stem-loop structures that may play a role in stabilizing the transcript [
65,
69]. By measuring RNA by RT-qPCR at various times after the addition of rifampin, which inhibits initiation of transcription by RNA polymerase, we determined the half-lives of most of the genes in the
nif1 gene cluster [
69]. The half-life of
nifH1 is much longer than the genes upstream of the processing site and the half-lives of the transcripts downstream of
nifH1 decline with increasing distance from the processing site [
69] (
Table 1), suggesting that transcript stability plays a major role in controlling the relative amount of transcript. A mutant strain in which the stem-loop structure located at the processing site of
nifH1 is abolished shows a shorter half-life for the
nifH1 transcript than the wild-type strain and nitrogen fixation is strongly inhibited in this mutant (Thiel; unpublished data).
Table 1.
Half-life of nif1 transcripts 1.
Table 1.
Half-life of nif1 transcripts 1.
| Gene | Half-Life (min) |
|---|
| nifB1 | 12.3 ± 2.2 |
| nifS1 | 14.7 ± 4.8 |
| nifU1 | 8.6 ± 2.3 |
| nifH1 | 33.8 ± 7.8 |
| nifD1 | 22.2 ± 4.1 |
| nifK1 | 16.7 ± 3.6 |
| nifE1 | 7.4 ± 1.3 |
| nifN1 | 9.1 ± 2.5 |
| nifX1 | 12.3 ± 2.1 |
| hesA1 | 20.9 ± 4.6 |
| fdxH1 | 20.5 ± 4.0 |
6. Proteins Involved in Regulation of Nitrogenase Genes
The global regulator NtcA affects many genes that respond to nitrogen availability in the cell and its binding sites have been the subject of several studies [
52,
91,
92]. Although NtcA is a regulator in all cyanobacteria, its significance in heterocyst-forming cyanobacteria is the key role it plays in sensing nitrogen starvation and initiating the complex process of heterocyst differentiation [
3,
4]. In addition to its role in activating genes that are required for the differentiation of heterocysts, NtcA activates expression of PipX, which is thought to work in concert with NtcA to allow full expression of late heterocyst-specific genes, including
nifH,
coxB3 and
coxB2 (encoding heterocyst-specific cytochrome oxidases). A
pipX mutant shows low levels of expression of these genes and is impaired in nitrogenase activity [
93,
94]. However, since the expression of the
nif operon depends on low oxygen levels that result from high respiration that is mediated by the products of the
cox2 and
cox3 genes, the low levels of
nif gene expression may be a secondary effect of relatively high oxygen levels in the heterocyst resulting from low levels of
cox gene expression in the
pipX mutant. In
Anabaena sp. PCC 7120,
patB was shown to be important for growth in the absence of fixed nitrogen [
95] and we have found that expression of
nifB1 requires PatB1 and expression of
nifB2 requires PatB2 (Thiel; unpublished data). Similarly, in a non-heterocystous cyanobacterium a PatB homologue called CnfR has been identified as a key regulator of
nif gene expression [
96].
NtcA has been reported to bind weakly to a region upstream of
nifH in
Anabaena sp. PCC 7120 and a putative non-canonical NtcA binding site was identified [
68,
97]. However, recent ChIPSeq data for
Anabaena sp. PCC 7120 showed that NtcA did not bind to any region upstream of
nifH, but rather to a site within the coding region of
nifH in
Anabaena sp. PCC 7120 [
52]. In this study NtcA was found to bind upstream of
nifB, but the binding site was hundreds of nucleotides upstream of the transcription start site of
nifB [
52], suggesting that NtcA does not directly control expression from the
nifB promoter. Using the
nifUH1 intergenic region of
A. variabilis as the target, we were unable to detect binding of NtcA and mutations in the putative NtcA binding site in this region had no effect on expression of
nifH1 [
65]; however, this is not surprising since we could find no evidence of a promoter in the
nifUH1 intergenic region (see
Section 5 above). There may be differences in
nifHDK regulation between
Anabaena sp. PCC 7120 and
A. variabilis, and the putative NtcA binding site upstream of
nifH in
Anabaena sp. PCC 7120 is not well conserved in
A. variabilis. However, because of the high degree of overall sequence homology between the two
nif1 clusters in both strains, it seems unlikely that the same 5'
nifH transcript end, found in both strains, results from fundamentally different processes. Further, a RNAseq mapping technique that identified transcription start sites (and excluded processed sites) for the genome of
Anabaena sp. PCC 7120 found the anticipated
nifB transcription start site at the published site, but failed to identify the putative
nifH transcription start site, even though it is found at levels of at least 20-fold higher that
nifB [
67]. It is clear that NtcA is important for expression of the
nif genes, but its effect is likely to be indirect, reflecting the fact that it may be essential for the expression of other genes whose products may act more directly to regulate expression of the
nif genes.
The role of NtcA in expression of the
nif2 cluster is also not clear. An
ntcA mutant of
A. variabilis failed to fix nitrogen using the
nif2-encoded Mo-nitrogenase, indicating that NtcA has an important role in expression of this enzyme; however, there is no canonical NtcA binding site, GTAN
8TAC, anywhere in the region that is likely to have the promoter [
15]. Because of the diversity of NtcA binding sites and of their locations relative to the start of genes [
52] as well as the global effect of NtcA regulation it may be difficult to assign a specific role for NtcA in expression of these nitrogenase genes.
There are at least two proteins that are repressors of
vnfDG, VnfR1 and VnfR2. VnfR1 is encoded by
ava4042, located upstream of the vanadate transport genes,
vupABC (
Figure 1) [
89]. VnfR2 is encoded by
ava4055, upstream from
vnfH, which encodes the dinitrogenase reductase for the V-nitrogenase. The promoter for
vnfR2 serves both
vnfR2 and
vnfH (
Figure 8) [
65]. These proteins with a conserved
N-terminal helix-turn-helix motif show 73% protein identity and act as Mo-dependent repressors that independently repress transcription of
ava4025-
vnfDG in cells grown with molybdate [
81]. Although each protein can repress expression of
ava4025-
vnfDG, only VnfR1 binds specifically,
in vitro, to a region upstream of the
ava4025 promoter. Cells lacking either
vnfR1 or
vnfR2 still show heterocyst-specific, Mo-repressed expression of
ava4025-
vnfDG. A mutant lacking both
vnfR1 and
vnfR2 expresses
ava4025-
vnfDG in the presence of Mo and expression is heterocyst specific (
Figure 6), indicating that other factors activate expression of this promoter in heterocysts.
7. Conclusions
Although there are three nitrogenases in
A. variabilis, and two of them are Mo-nitrogenases, the tight control of expression of the
nif1,
nif2 and
vnf genes ensures that the cell makes the correct enzyme for the environment in which it is growing. This regulation includes differential expression of nitrogenases in response to cell differentiation, oxic
versus anoxic growth conditions, and for environments with or without molybdate. The
nif2 cluster has not been found in any other well-characterized heterocyst-forming cyanobacterium and it shares an evolutionary origin with
nif genes from non-heterocystous cyanobacteria especially with the primitive cyanobacterium
Chroococcidiopsis thermalis [
75,
98], suggesting that an ancestor of this unicellular cyanobacterium may represent the evolutionary origin of the
nif2 genes. The
vnf genes are not present in well-studied cyanobacteria; however, these genes have recently been found in cyanobacteria in a lichen symbiosis [
99], suggesting that they may be represented in symbiotic interactions. The culturable cyanobionts from the water fern
Azolla filiculoides,
Anabaena spp. [
58,
100], are virtually indistinguishable morphologically and physiologically from
A. variabilis and also have
vnf genes [
11] and
nif2 genes [
16]; however, the non-culturable
Azolla symbiont,
Nostoc azollae 0708, shows a degraded genome incapable of supporting independent growth and this strain lacks both
vnf and
nif2 genes [
101].
While we understand at an environmental-response level and even at a whole-cell level how these three nitrogenases are regulated, information at the molecular level is still lacking. The fact that at least two of the three gene clusters employ RNA processing, and its associated transcript stability, as a regulatory mechanism suggests that this may be a more general mechanism of cyanobacterial gene regulation; however, that needs to be tested experimentally. While several promoters that show late heterocyst-specific gene expression have been identified, including a number that are described here, we still do not understand how those genes are activated late in heterocyst development. Proteins NtcA [
4] and SigE [
53] are important for expression of late heterocyst genes, but their specific function in controlling these genes is not known. Research is still needed to understand how the environmentally important process of nitrogen fixation and the synthesis of associated essential proteins, such as the uptake hydrogenase, ferredoxins, and cytochrome oxidases, are regulated.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}