RNA Catalysis, Thermodynamics and the Origin of Life

Abstract
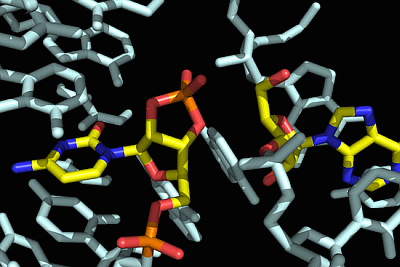
:
{kind=link}
{kind=link}
{kind=link}
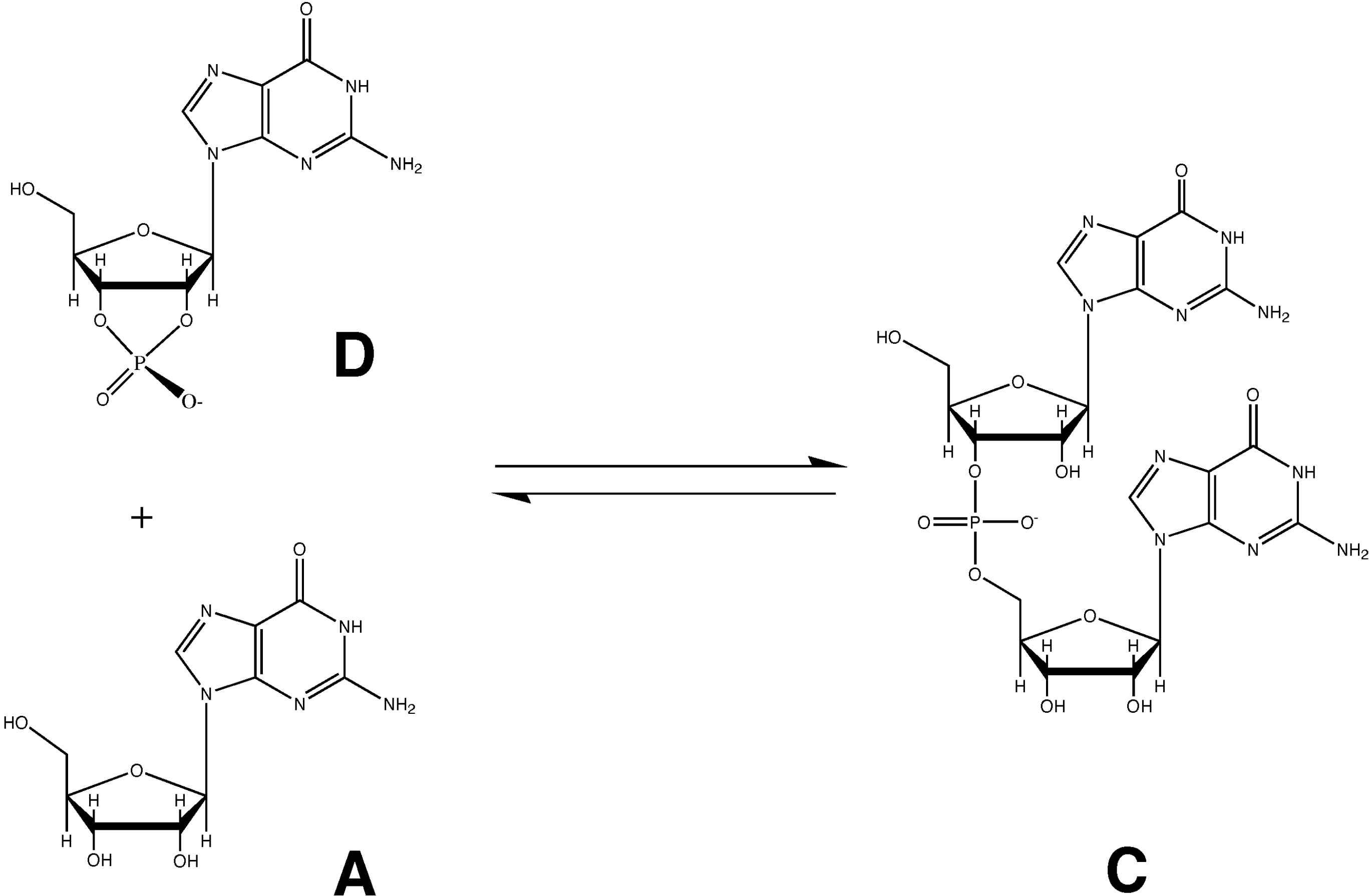{kind=link}
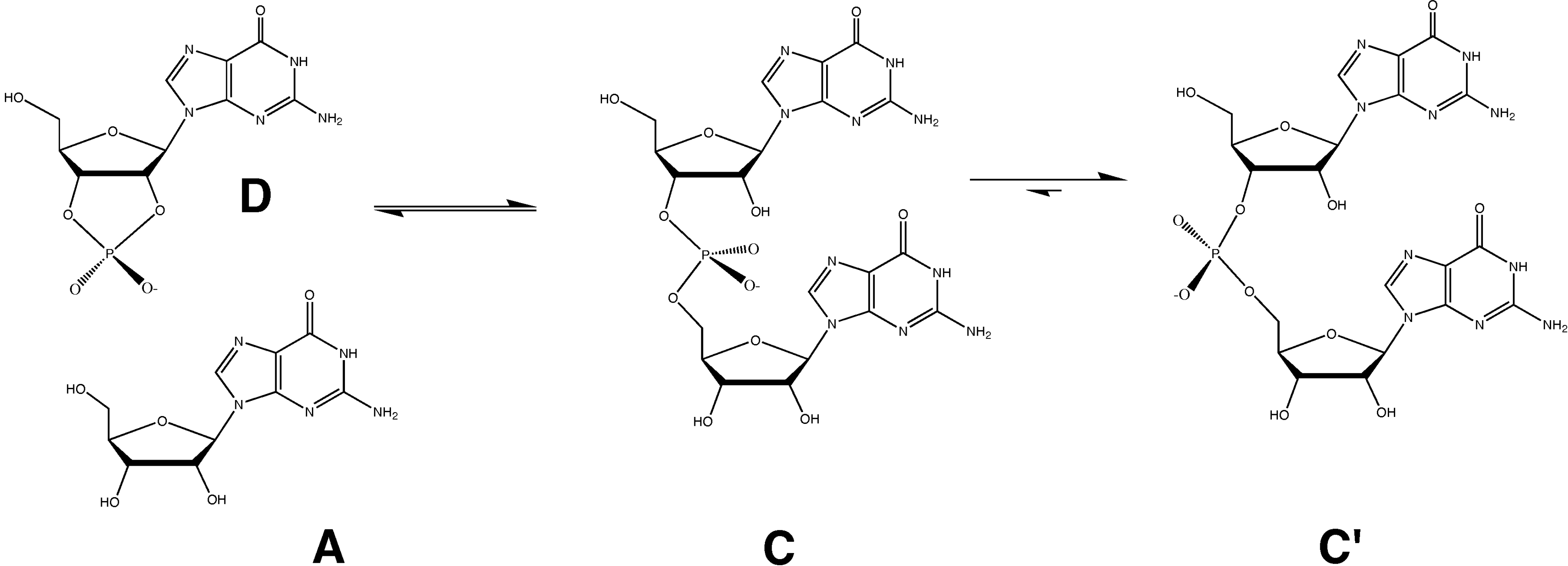{kind=link}
1. Introduction
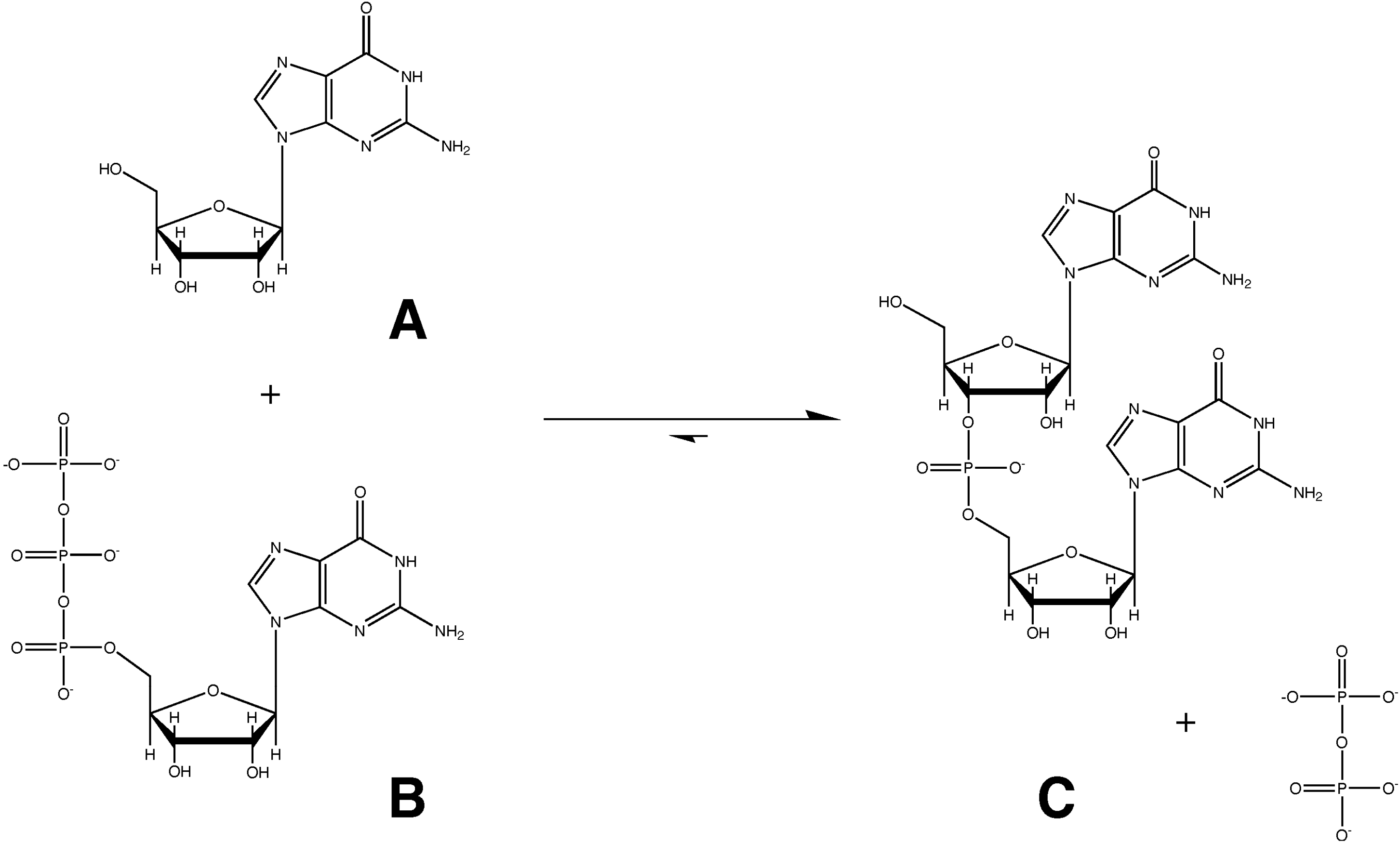

2. Formulation of Our Hypothesis
2.1. The Previous Assumption
2.2. Our Proposed Revision
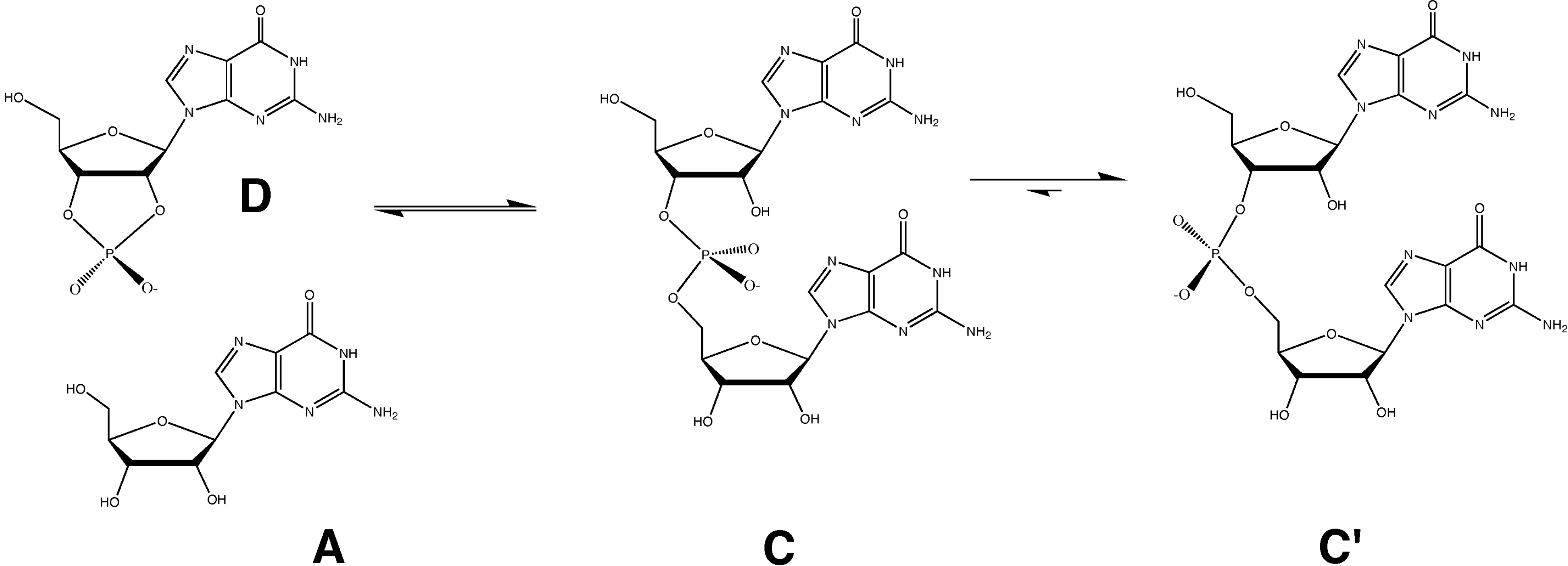
3. Results and Discussion
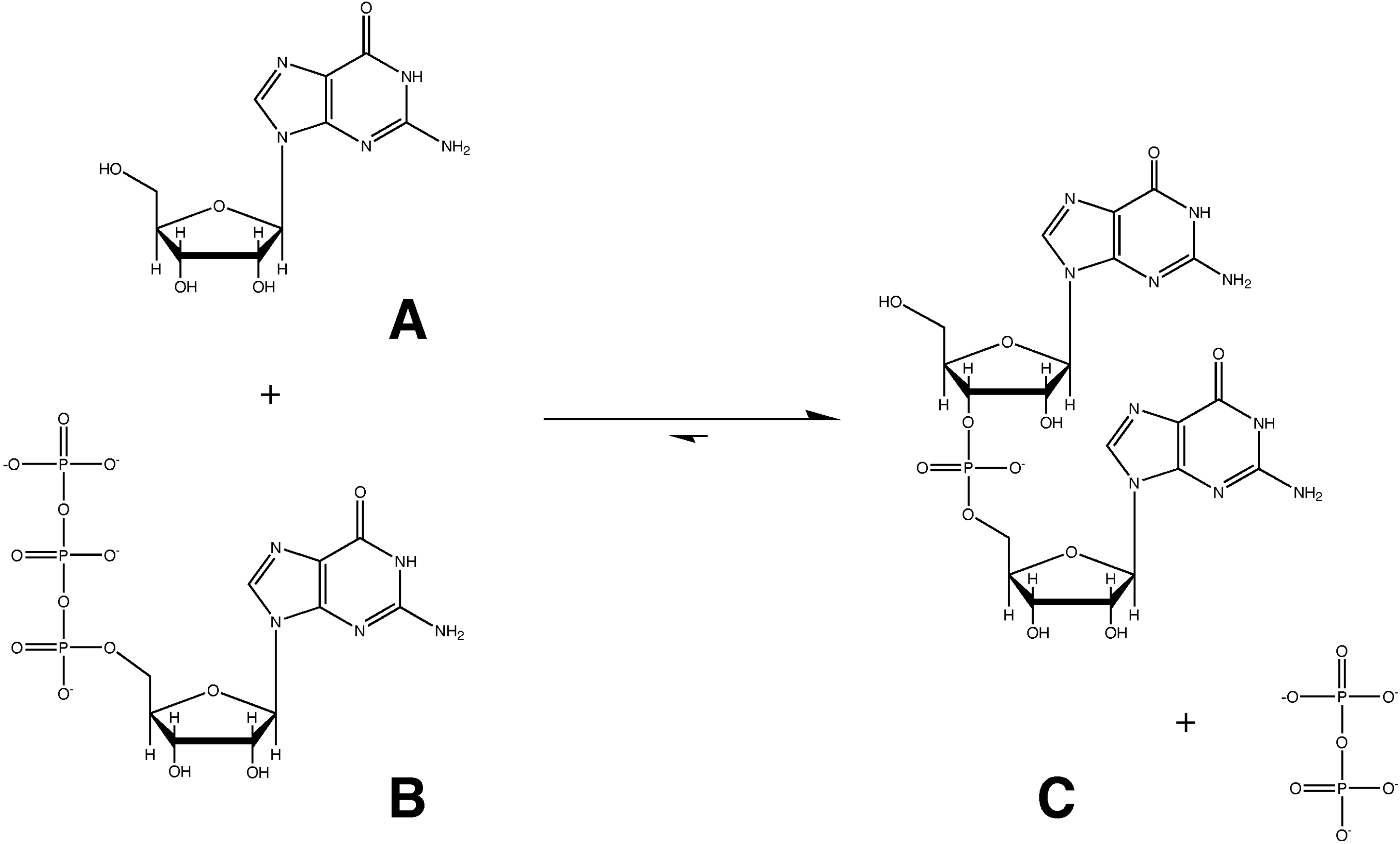
3.1. Pre-Biotic RNA Synthesis with Nucleotide 2ʹ,3ʹ-Cyclic Phosphates is in Fact Plausible
3.1.1. The Fossil Record (Ribozymes)
3.1.2. Comparative Abundance of Nucleotide 2ʹ,3ʹ-Cyclic Phosphates
3.1.3. Nucleotide 2ʹ,3ʹ-Cyclic Phosphates and Prebiotic Pyrimidine Nucleotide Synthesis
3.1.4. Simplicity of the Reaction would Allow Evolutionary Boot-Strapping
3.1.5. A Viable First RNA Editing Mechanism
3.1.6. The Advantage of RNA vs. DNA
4. Conclusions
4.1. The Origin of Catalysis and the Origin of Life
4.2. Implications for the in Vitro Evolution of an RNA Self-Replicase
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Woese, C. The Evolution of the Genetic Code. In The Genetic Code; Harper and Row: New York, NY, USA, 1967; pp. 179–195. [Google Scholar]
- Crick, F.H. The origin of the genetic code. J. Mol. Biol. 1968, 38, 367–379. [Google Scholar] [CrossRef]
- Orgel, L.E. Evolution of the genetic apparatus. J. Mol. Biol. 1968, 38, 384–393. [Google Scholar] [CrossRef]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1968, 31, 147–157. [Google Scholar]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Gesteland, R.F.; Atkins, J.F. (Eds.) The RNA World. Cold Spring Harbor Laboratory Press: New York, NY, USA, 1993.
- Bernfield, M.R. Ribonuclease and oigoribonucleotide synthesis. II. Synthesis of oligonucleotides of specific sequence. J. Biol. Chem. 1966, 241, 2014–2023. [Google Scholar]
- Bernfield, M.R. Ribonuclease and oligoribonucleotide synthesis. I. Synthetic activity of bovine pancreatic ribonuclease derivatives. J. Biol. Chem. 1965, 240, 4753–4762. [Google Scholar]
- Kierzek, R.; Caruthers, M.H.; Longfellow, C.E.; Swinton, D.; Turner, D.H.; Freier, S.M. Polymer-supported RNA synthesis and its application to test the nearest-neighbor model for duplex stability. Biochemistry 1986, 25, 7840–7846. [Google Scholar]
- Freier, S.M.; Kierzek, R.; Jaeger, J.A.; Sugimoto, N.; Caruthers, M.H.; Neilson, T.; Turner, D.H. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl. Acad. Sci. USA 1986, 83, 9373–9377. [Google Scholar] [CrossRef]
- Loverix, S.; Laus, G.; Martins, J.C.; Wyns, L.; Steyaert, J. Reconsidering the energetics of ribonuclease catalysed RNA hydrolysis. Eur. J. Biochem. 1998, 257, 286–290. [Google Scholar]
- Mohr, S.C.; Thach, R.E. Application of ribonuclease T1 to the synthesis of oligoribonucleotides of defined base sequence. J. Biol. Chem. 1969, 244, 6566–6576. [Google Scholar]
- Erman, J.E.; Hammes, G.G. Relaxation spectra of ribonuclease. IV. The interaction of ribonuclease with cytidine 2':3'-cyclic phosphate. J. Am. Chem. Soc. 1966, 88, 5607–5614. [Google Scholar] [CrossRef]
- Joyce, G.F.; Orgel, L.E. Prospects for Understanding the Origin of the RNA World. In The RNA World; Gesteland, R.F., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1993; pp. 1–25. [Google Scholar]
- Govil, G. Conformational structure of polynucleotides around the O-P bonds: Refined parameters for CPF calculations. Biopolymers 1976, 15, 2303–2307. [Google Scholar] [CrossRef]
- Noller, H.F. The driving force for molecular evolution of translation. RNA 2004, 10, 1833–1837. [Google Scholar] [CrossRef]
- Ferré-D’Amaré, A.R.; Scott, W.G. Small Self-cleaving Ribozymes. Cold Spring Harb. Perspect Biol. 2010, 2, a003574. [Google Scholar]
- Robertson, M.P.; Scott, W.G. The structural basis of ribozyme-catalyzed RNA assembly. Science 2007, 315, 1549–1553. [Google Scholar] [CrossRef]
- Fuller, W.D.; Sanchez, R.A.; Orgel, L.E. Studies in prebiotic synthesis VI. Synthesis of purine nucleosides. J. Mol. Biol. 1972, 67, 25–33. [Google Scholar] [CrossRef]
- Robertson, M.P.; Miller, S.L. An efficient prebiotic synthesis of cytosine and uracil. Nature 1995, 373, 772–774. [Google Scholar] [CrossRef]
- Powner, M.W.; Gerland, B.; Sutherland, J.D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 2009, 459, 239–242. [Google Scholar] [CrossRef]
- Murray, J.B.; Seyhan, A.A.; Walter, N.G.; Burke, J.M.; Scott, W.G. The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem. Biol. 1998, 5, 587–595. [Google Scholar] [CrossRef]
- Breaker, R.R.; Joyce, G.F. A DNA enzyme with Mg2+-dependent RNA phosphoesterase activity. Chem. Biol. 1995, 2, 655–660. [Google Scholar] [CrossRef]
- Schrodinger, E. What is Life? Cambridge University Press: Cambridge, UK, 1944. [Google Scholar]
- Szoke, A.; Scott, W.G.; Hajdu, J. Catalysis, evolution and life. FEBS Lett. 2003, 553, 18–20. [Google Scholar] [CrossRef]
- Mutschler, H.J.; Holliger, P. Non-canonical 3'-5' extension of RNA with prebiotically plausible ribonucleoside 2', 3'-cyclic phosphates. J. Am. Chem. Soc. 2014. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Scott, W.G.; Szöke, A.; Blaustein, J.; O'Rourke, S.M.; Robertson, M.P. RNA Catalysis, Thermodynamics and the Origin of Life. Life 2014, 4, 131-141. https://doi.org/10.3390/life4020131
Scott WG, Szöke A, Blaustein J, O'Rourke SM, Robertson MP. RNA Catalysis, Thermodynamics and the Origin of Life. Life. 2014; 4(2):131-141. https://doi.org/10.3390/life4020131
Chicago/Turabian StyleScott, William G., Abraham Szöke, Josh Blaustein, Sara M. O'Rourke, and Michael P. Robertson. 2014. "RNA Catalysis, Thermodynamics and the Origin of Life" Life 4, no. 2: 131-141. https://doi.org/10.3390/life4020131
APA StyleScott, W. G., Szöke, A., Blaustein, J., O'Rourke, S. M., & Robertson, M. P. (2014). RNA Catalysis, Thermodynamics and the Origin of Life. Life, 4(2), 131-141. https://doi.org/10.3390/life4020131