
Long-Term Monitoring of Cardiac Involvement under Migalastat Treatment Using Magnetic Resonance Tomography in Fabry Disease

 , ,
, , 
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Desnick, R.J.; Brady, R.; Barranger, J.; Collins, A.J.; Germain, D.P.; Goldman, M.; Grabowski, G.; Packman, S.; Wilcox, W.R. Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann. Intern. Med. 2003, 138, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J. Med. Genet. 2001, 38, 769–775. [Google Scholar] [CrossRef] [PubMed]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J. Med. Genet. 2001, 38, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, O.; Cordeiro, F.; Gago, M.F.; Miltenberger-Miltenyi, G.; Ferreira, C.; Sousa, N.; Cunha, D. Fabry Disease and the Heart: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 4434. [Google Scholar] [CrossRef]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A., III; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme Replacement Therapy in Fabry Disease. JAMA 2001, 285, 2743. [Google Scholar] [CrossRef]
- Banikazemi, M.; Bultas, J.; Waldek, S.; Wilcox, W.R.; Whitley, C.B.; McDonald, M.; Finkel, R.; Packman, S.; Bichet, D.G.; Warnock, D.G.; et al. Agalsidase-beta therapy for advanced Fabry disease: A randomized trial. Ann. Intern. Med. 2007, 146, 77–86. [Google Scholar] [CrossRef]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef]
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Eveslage, M.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Müntze, J.; et al. Treatment of Fabry Disease management with migalastat—Outcome from a prospective 24 months observational multicenter study (FAMOUS). Eur. Heart J. Cardiovasc. Pharmacother. 2021, 8, 272–281. [Google Scholar] [CrossRef]
- Bichet, D.G.; Torra, R.; Wallace, E.; Hughes, D.; Giugliani, R.; Skuban, N.; Krusinska, E.; Feldt-Rasmussen, U.; Schiffmann, R.; Nicholls, K.; et al. Long-term follow-up of renal function in patients treated with migalastat for Fabry disease. Mol. Genet. Metab. Rep. 2021, 28, 100786. [Google Scholar] [PubMed]
- Feldt-Rasmussen, U.; Hughes, D.; Sunder-Plassmann, G.; Shankar, S.; Nedd, K.; Olivotto, I.; Ortiz, D.; Ohashi, T.; Hamazaki, T.; Skuban, N.; et al. Long-term efficacy and safety of migalastat treatment in Fabry disease: 30-month results from the open-label extension of the randomized, phase 3 ATTRACT study. Mol. Genet. Metab. 2020, 131, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat-amenable variants: Data from the phase 3 randomized, multicenter, double-blind clinical trial and extension study. Genet. Med. 2019, 21, 1987–1997. [Google Scholar] [PubMed]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Grothues, F.; Smith, G.C.; Moon, J.C.C.; Bellenger, N.G.; Collins, P.; Klein, H.U.; Pennell, D.J. Comparison of interstudy reproducibility of cardiovascular magnetic resonance with two-dimensional echocardiography in normal subjects and in patients with heart failure or left ventricular hypertrophy. Am. J. Cardiol. 2002, 90, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Bellenger, N.G.; Davies, L.C.; Francis, J.M.; Coats, A.J.; Pennell, D.J. Reduction in sample size for studies of remodeling in heart failure by the use of cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reason. 2000, 2, 271–278. [Google Scholar]
- Bottini, P.B.; Carr, A.A.; Prisant, L.M.; Flickinger, F.W.; Allison, J.D.; Gottdiener, J.S. Magnetic resonance imaging compared to echocardiography to assess left ventricular mass in the hypertensive patient. Am. J. Hypertens. 1995, 8, 221–228. [Google Scholar] [CrossRef]
- Camporeale, A.; Bandera, F.; Pieroni, M.; Pieruzzi, F.; Spada, M.; Bersano, A.; Econimo, L.; Lanzillo, C.; Rubino, M.; Mignani, R.; et al. Effect of Migalastat on cArdiac Involvement in FabRry Disease: MAIORA study. J. Med. Genet. 2023. [Google Scholar] [CrossRef]
- Messroghli, D.R.; Moon, J.C.; Ferreira, V.M.; Grosse-Wortmann, L.; He, T.; Kellman, P.; Mascherbauer, J.; Nezafat, R.; Salerno, M.; Schelbert, E.B.; et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: A consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imagi. J. Cardiovasc. Magn. Reson. 2017, 19, 75. [Google Scholar]
- Schulz-Menger, J.; Bluemke, D.A.; Bremerich, J.; Flamm, S.D.; Fogel, M.A.; Friedrich, M.G.; Kim, R.J.; Von Knobelsdorff-Brenkenhoff, F.; Kramer, C.M.; Pennell, D.J.; et al. Standardized image interpretation and post-processing in cardiovascular magnetic resonance—2020 update. J. Cardiovasc. Magn. Reson. 2020, 22, 87. [Google Scholar] [CrossRef]
- Kawel-Boehm, N.; Hetzel, S.J.; Ambale-Venkatesh, B.; Captur, G.; Francois, C.J.; Jerosch-Herold, M.; Salerno, M.; Teague, S.D.; Valsangiacomo-Buechel, E.; van der Geest, R.J.; et al. Reference ranges (“normal values”) for cardiovascular magnetic resonance (CMR) in adults and children: 2020 update. J. Cardiovasc. Magn. Reason. 2020, 22, 87. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.L.; Castro, A.F., 3rd; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef]
- Patel, M.R.; Cecchi, F.; Cizmarik, M.; Kantola, I.; Linhart, A.; Nicholls, K.; Strotmann, J.; Tallaj, J.; Tran, T.C.; West, M.L.; et al. Cardiovascular Events in Patients with Fabry Disease. J. Am. Coll. Cardiol. 2011, 57, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Riccio, E.; Zanfardino, M.; Ferreri, L.; Santoro, C.; Cocozza, S.; Capuano, I.; Imbriaco, M.; Feriozzi, S.; Pisani, A.; Pisani, A.; et al. Switch from enzyme replacement therapy to oral chaperone migalastat for treating fabry disease: Real-life data. Eur. J. Hum. Genet. 2020, 28, 1662–1668. [Google Scholar] [CrossRef]
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Muntze, J.; Uceyler, N.; et al. Treatment of Fabry’s Disease With Migalastat: Outcome From a Prospective Observational Multicenter Study (FAMOUS). Clin. Pharmacol. Ther. 2020, 108, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Perretta, F.; Jaurretche, S. Fabry Disease: Switch from Enzyme Replacement Therapy to Oral Chaperone Migalastat: What Do We Know Today? Healthcare 2023, 11, 449. [Google Scholar] [CrossRef] [PubMed]
- Raman, B.; Ariga, R.; Spartera, M.; Sivalokanathan, S.; Chan, K.; Dass, S.; Petersen, S.E.; Daniels, M.J.; Francis, J.; Smillie, R.; et al. Progression of myocardial fibrosis in hypertrophic cardiomyopathy: Mechanisms and clinical implications. Eur. Heart J.–Cardiovasc. Imaging 2018, 20, 157–167. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac Involvement in Fabry Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 922–936. [Google Scholar] [CrossRef]
- Ponsiglione, A.; Gambardella, M.; Green, R.; Cantoni, V.; Nappi, C.; Ascione, R.; De Giorgi, M.; Cuocolo, R.; Pisani, A.; Petretta, M.; et al. Cardiovascular magnetic resonance native T1 mapping in Anderson-Fabry disease: A systematic review and meta-analysis. J. Cardiovasc. Magn. Reson. 2022, 24, 31. [Google Scholar] [CrossRef]
- Monticelli, M.; Liguori, L.; Allocca, M.; Bosso, A.; Andreotti, G.; Lukas, J.; Monti, M.C.; Morretta, E.; Cubellis, M.V.; Hay Mele, B. Drug Repositioning for Fabry Disease: Acetylsalicylic Acid Potentiates the Stabilization of Lysosomal Alpha-Galactosidase by Pharmacological Chaperones. Int. J. Mol. Sci. 2022, 23, 5105. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Hay Mele, B.; Cozzolino, F.; Monaco, V.; Cimmaruta, C.; Monti, M.; Andreotti, G.; Monticelli, M. Enzyme Replacement Therapy for FABRY Disease: Possible Strategies to Improve Its Efficacy. Int. J. Mol. Sci. 2023, 24, 4548. [Google Scholar] [CrossRef] [PubMed]
- Seemann, S.; Ernst, M.; Cimmaruta, C.; Struckmann, S.; Cozma, C.; Koczan, D.; Knospe, A.M.; Haake, L.R.; Citro, V.; Bräuer, A.U.; et al. Proteostasis regulators modulate proteasomal activity and gene expression to attenuate multiple phenotypes in Fabry disease. Biochem. J. 2020, 477, 359–380. [Google Scholar] [CrossRef]
- Weidemann, F.; Jovanovic, A.; Herrmann, K.; Vardarli, I. Chaperone Therapy in Fabry Disease. Int. J. Mol. Sci. 2022, 23, 1887. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Pockrandt, A.M.; Seemann, S.; Sharif, M.; Runge, F.; Pohlers, S.; Zheng, C.; Gläser, A.; Beller, M.; Rolfs, A.; et al. Enzyme enhancers for the treatment of Fabry and Pompe disease. Mol. Ther. 2015, 23, 456–464. [Google Scholar] [CrossRef]
- Siegenthaler, M.; Huynh-Do, U.; Krayenbuehl, P.; Pollock, E.; Widmer, U.; Debaix, H.; Olinger, E.; Frank, M.; Namdar, M.; Ruschitzka, F.; et al. Impact of cardio-renal syndrome on adverse outcomes in patients with Fabry disease in a long-term follow-up. Int. J. Cardiol. 2017, 249, 261–267. [Google Scholar] [CrossRef]
- Roy, A.; Umar, H.; Ochoa-Ferraro, A.; Warfield, A.; Lewis, N.; Geberhiwot, T.; Steeds, R. Atherosclerosis in Fabry Disease—A Contemporary Review. J. Clin. Med. 2021, 10, 4422. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Overall (n = 15) | Male (n = 4) | Female (n = 11) | |
|---|---|---|---|
| Age at baseline (years) | 54 (37–59) | 55 (46–58) | 53 (32–66) |
| Follow-up time (months) | 34 (26–43) | 29.5 (26–40) | 37 (26–43) |
| Previous ERT | 3 (75) | 4 (36) | |
| Left ventricular hypertrophy | 10 (66.7%) | 4 (100%) | 6 (54.5%) |
| ACE-inhibitor/AT1-r. antagonists | 8 (53%) | 1 (25%) | 7 (64%) |
| Acetylsalicylic acid | 1 (6.67%) | 0 (0%) | 1 (9.09%) |
| Urinary protein to creatinine ratio | |||
| <100 mg/g | 10 (66.7%) | 1 (25%) | 9 (81.82%) |
| 100–1000 mg/g | 4 (26.7%) | 2 (50%) | 2 (18.18%) |
| >1000 mg/g | 1 (6.67%) | 1 (25%) | 0 (0%) |
| GLA mutations + (phenotype) | c.902G>A p.(R301Q) (late-onset) | c.902G>A p.(R301Q (late-onset) | |
| c.902G>A p.(R301Q) (late-onset) | c.902G>A p.(R301Q) (late-onset) | ||
| c.713G>A p.(S238N) (late-onset) | c.902G>A p.(R301Q) (late-onset) | ||
| c.713G>A p.(S238N) (late-onset) | c.902G>A p.(R301Q) (late-onset) | ||
| c.772G>A p.(G258R) (late-onset) | |||
| c.1010T>C p.(F337S) (late-onset) | |||
| c.125T>C p.(M42T) (classic) | |||
| c.125T>C p.(M42T) (classic) | |||
| c.1033T>C p.(S345P) (classic) | |||
| c.581C>T p.(T194I) (classic) | |||
| c.581C>T p.(T194I) (classic) |
| Pre-Migalastat n = 8 | Baseline n = 15 | Follow-Up n = 15 | p-Value for Change between Baseline and Follow-Up | |
|---|---|---|---|---|
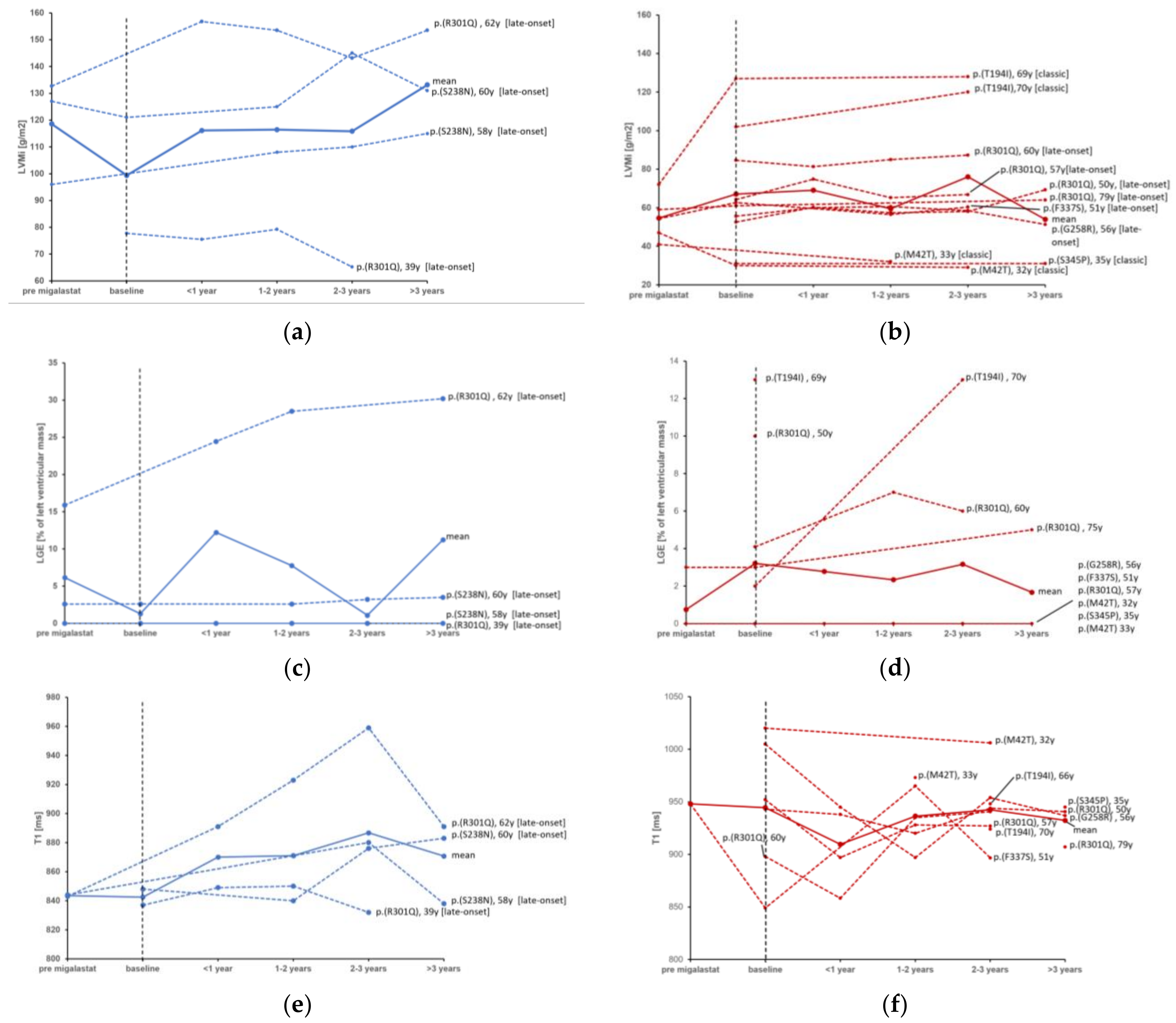
| LVMi (g/m2) | 66 (51–111) | 64.2 (52.5–108) | 66.8 (51.4–120) | 0.294 |
| IVS (mm) | 12 (7–15) | 13 (10–14) | 13 (11–15) | 0.470 |
| PWT [mm] | 8.8 (7.5–12.5) | 9 (8–11) | 10 (8–13) | 0.577 |
| LVEDV [ml] | 83 (77–107) | 96 (66–108) | 95 (76.8–121) | 0.096 |
| LVEF [%] | 62 (59–71) | 67 (60.6–76.9) | 61 (58–72) | 0.152 |
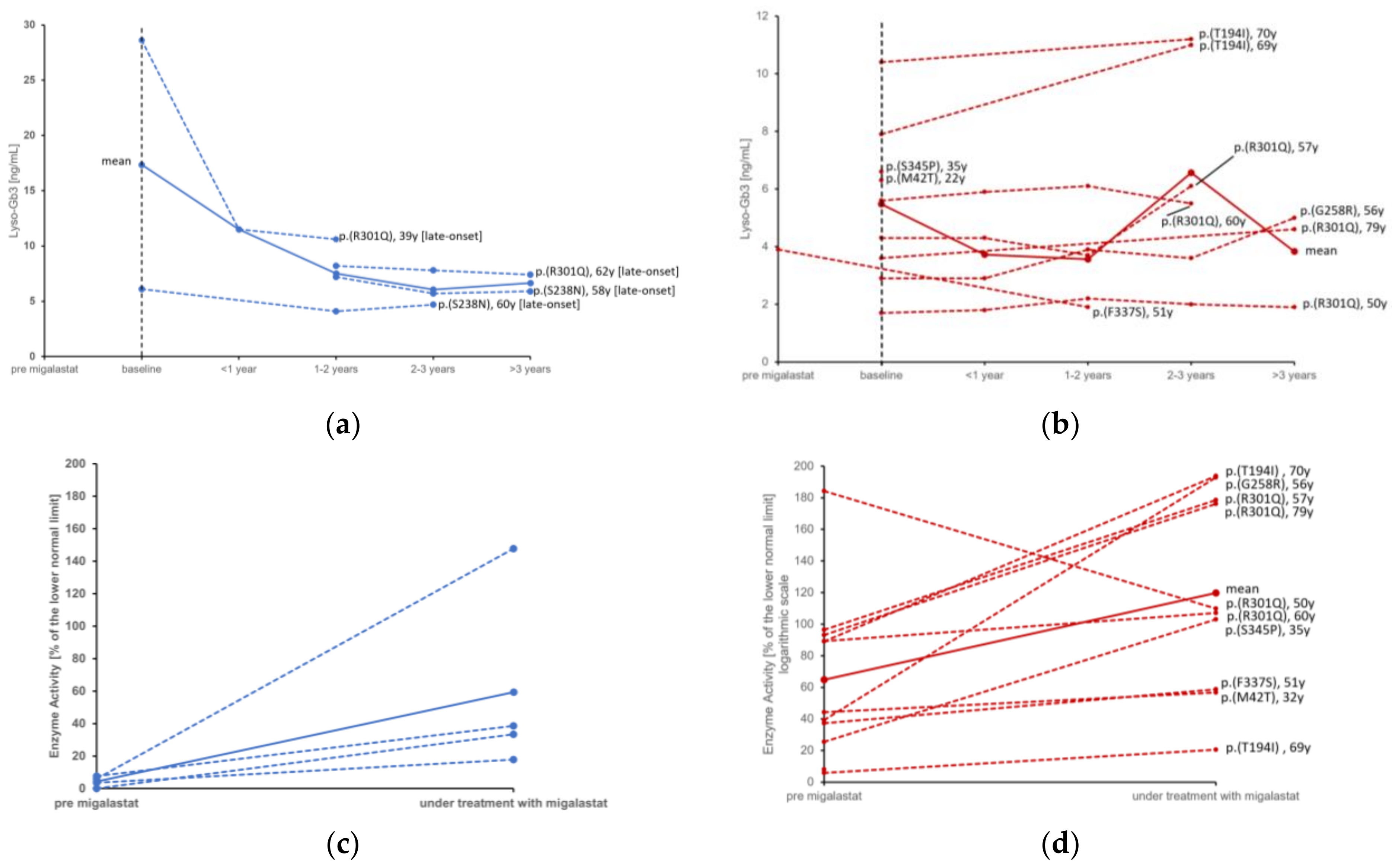
| LGE [%] | n = 7 0 (0–3) | n = 15 0 (0–4.1) | n = 13 0 (0–5.5) | 0.043 |
| T1 [ms] | n = 3 844 (843–948) | n = 14 916 (865–915) | n = 15 927 (891–945) | 0.859 |
| eGFR [ml/min/1,73 m2] | n = 9 99 (81–8-105) | n = 14 93.5 (78.7–102) | n = 15 90.48 (74–101) | 0.074 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatterer, C.; Beitzke, D.; Graf, S.; Lenz, M.; Sunder-Plassmann, G.; Mann, C.; Ponleitner, M.; Manka, R.; Fritschi, D.; Krayenbuehl, P.-A.; et al. Long-Term Monitoring of Cardiac Involvement under Migalastat Treatment Using Magnetic Resonance Tomography in Fabry Disease. Life 2023, 13, 1213. https://doi.org/10.3390/life13051213
Gatterer C, Beitzke D, Graf S, Lenz M, Sunder-Plassmann G, Mann C, Ponleitner M, Manka R, Fritschi D, Krayenbuehl P-A, et al. Long-Term Monitoring of Cardiac Involvement under Migalastat Treatment Using Magnetic Resonance Tomography in Fabry Disease. Life. 2023; 13(5):1213. https://doi.org/10.3390/life13051213
Chicago/Turabian StyleGatterer, Constantin, Dietrich Beitzke, Senta Graf, Max Lenz, Gere Sunder-Plassmann, Christopher Mann, Markus Ponleitner, Robert Manka, Daniel Fritschi, Pierre-Alexandre Krayenbuehl, and et al. 2023. "Long-Term Monitoring of Cardiac Involvement under Migalastat Treatment Using Magnetic Resonance Tomography in Fabry Disease" Life 13, no. 5: 1213. https://doi.org/10.3390/life13051213