
Brain Region-Specific Differences in Amyloid-β Plaque Composition in 5XFAD Mice

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Transgenic Mice
2.3. Immunohistochemistry on Paraffin Sections
2.4. Quantification of Aβ Load
2.5. Statistical Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Sirkis, D.W.; Bonham, L.W.; Johnson, T.P.; La Joie, R.; Yokoyama, J.S. Dissecting the clinical heterogeneity of early-onset Alzheimer’s disease. Mol. Psychiatry 2022, 27, 2674–2688. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Selkoe, D.J. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998, 8, 447–453. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19. [Google Scholar] [CrossRef]
- Reinert, J.; Richard, B.C.; Klafki, H.W.; Friedrich, B.; Bayer, T.A.; Wiltfang, J.; Kovacs, G.G.; Ingelsson, M.; Lannfelt, L.; Paetau, A.; et al. Deposition of C-terminally truncated Aβ species Aβ37 and Aβ39 in Alzheimer’s disease and transgenic mouse models. Acta Neuropathol. Commun. 2016, 4, 24. [Google Scholar] [CrossRef]
- Galanis, C.; Fellenz, M.; Becker, D.; Bold, C.; Lichtenthaler, S.F.; Müller, U.C.; Deller, T.; Vlachos, A. Amyloid-Beta Mediates Homeostatic Synaptic Plasticity. J. Neurosci. 2021, 41, 5157–5172. [Google Scholar] [CrossRef] [PubMed]
- Plant, L.D.; Boyle, J.P.; Smith, I.F.; Peers, C.; Pearson, H.A. The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J. Neurosci. 2003, 23, 5531–5535. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fa, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef] [PubMed]
- Lewczuk, P.; Riederer, P.; O’Bryant, S.E.; Verbeek, M.M.; Dubois, B.; Visser, P.J.; Jellinger, K.A.; Engelborghs, S.; Ramirez, A.; Parnetti, L.; et al. Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: An update of the Consensus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry. World J. Biol. Psychiatry 2018, 19, 244–328. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Capetillo-Zarate, E.; Tredici, K.D.; Braak, H. The Development of Amyloid Beta Protein Deposits in the Aged Brain. Sci. Aging Knowl. Environ. 2006, 2006, re1. [Google Scholar] [CrossRef]
- Fiala, J.C. Mechanisms of amyloid plaque pathogenesis. Acta Neuropathol. 2007, 114, 551–571. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Mehraein, P. Reanalysis of the first case of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249 (Suppl. S3), 10–13. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42 3 Pt 1, 631–639. [Google Scholar] [CrossRef]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimers Res. Ther. 2014, 6, 28. [Google Scholar] [CrossRef]
- Zampar, S.; Klafki, H.W.; Sritharen, K.; Bayer, T.A.; Wiltfang, J.; Rostagno, A.; Ghiso, J.; Miles, L.A.; Wirths, O. N-terminal heterogeneity of parenchymal and vascular amyloid-β deposits in Alzheimer‘s disease. Neuropathol. Appl. Neurobiol. 2020, 46, 673–685. [Google Scholar] [CrossRef]
- Portelius, E.; Bogdanovic, N.; Gustavsson, M.K.; Volkmann, I.; Brinkmalm, G.; Zetterberg, H.; Winblad, B.; Blennow, K. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010, 120, 185–193. [Google Scholar] [CrossRef]
- Bien, J.; Jefferson, T.; Čaušević, M.; Jumpertz, T.; Munter, L.; Multhaup, G.; Weggen, S.; Becker-Pauly, C.; Pietrzik, C.U. The Metalloprotease Meprin β Generates Amino Terminal-truncated Amyloid β Peptide Species. J. Biol. Chem. 2012, 287, 33304–33313. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Jumpertz, T.; Hüttenrauch, M.; Ogorek, I.; Gerber, H.; Storck, S.E.; Zampar, S.; Dimitrov, M.; Lehmann, S.; Lepka, K.; et al. The metalloprotease ADAMTS4 generates N-truncated Aβ4–x species and marks oligodendrocytes as a source of amyloidogenic peptides in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J.; Overman, M.J.; Cotman, C.W. Amino-terminal deletions enhance aggregation of beta-amyloid peptides in vitro. J. Biol. Chem. 1995, 270, 23895–23898. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Lauber, T.; Schaupp, M.; Manhart, S.; Scheel, E.; Bohm, G.; Demuth, H.U. On the seeding and oligomerization of pGlu-amyloid peptides (in vitro). Biochemistry 2006, 45, 12393–12399. [Google Scholar] [CrossRef]
- Bouter, Y.; Dietrich, K.; Wittnam, J.L.; Rezaei-Ghaleh, N.; Pillot, T.; Papot-Couturier, S.; Lefebvre, T.; Sprenger, F.; Wirths, O.; Zweckstetter, M.; et al. N-truncated amyloid β (Aβ) 4-42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 2013, 126, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, E.; Mathews, P.; Mezhericher, E.; Beach, T.G.; Deng, J.; Neubert, T.A.; Rostagno, A.; Ghiso, J. Aβ truncated species: Implications for brain clearance mechanisms and amyloid plaque deposition. Biochim. Biophys. Acta 2018, 1864, 208–225. [Google Scholar] [CrossRef]
- Wirths, O.; Walter, S.; Kraus, I.; Klafki, H.W.; Stazi, M.; Oberstein, T.J.; Ghiso, J.; Wiltfang, J.; Bayer, T.A.; Weggen, S. N-truncated Aβ4–x peptides in sporadic Alzheimer’s disease cases and transgenic Alzheimer mouse models. Alzheimers Res. Ther. 2017, 9, 80. [Google Scholar] [CrossRef]
- Cai, W.; Wu, T.; Chen, N. The Amyloid-Beta Clearance: From Molecular Targets to Glial and Neural Cells. Biomolecules 2023, 13, 313. [Google Scholar] [CrossRef]
- Loeffler, D.A. Experimental approaches for altering the expression of Abeta-degrading enzymes. J. Neurochem. 2023, 164, 725–763. [Google Scholar] [CrossRef]
- Lao, K.; Zhang, R.; Luan, J.; Zhang, Y.; Gou, X. Therapeutic Strategies Targeting Amyloid-β Receptors and Transporters in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 79, 1429–1442. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Forner, S.; Kawauchi, S.; Balderrama-Gutierrez, G.; Kramár, E.A.; Matheos, D.P.; Phan, J.; Javonillo, D.I.; Tran, K.M.; Hingco, E.; da Cunha, C.; et al. Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer’s disease. Sci. Data 2021, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Lemere, C.A.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D.J. The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1995, 1, 1291–1296. [Google Scholar] [CrossRef]
- Richard, B.C.; Kurdakova, A.; Baches, S.; Bayer, T.A.; Weggen, S.; Wirths, O. Gene Dosage Dependent Aggravation of the Neurological Phenotype in the 5XFAD Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2015, 45, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Oblak, A.L.; Lin, P.B.; Kotredes, K.P.; Pandey, R.S.; Garceau, D.; Williams, H.M.; Uyar, A.; O’Rourke, R.; O’Rourke, S.; Ingraham, C.; et al. Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: A MODEL-AD Study. Front. Aging Neurosci. 2021, 13, 713726. [Google Scholar] [CrossRef]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Marzloff, K.; Hyman, B.T. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology 1992, 42, 1681–1688. [Google Scholar] [CrossRef]
- Wirths, O.; Multhaup, G.; Czech, C.; Feldmann, N.; Blanchard, V.; Tremp, G.; Beyreuther, K.; Pradier, L.; Bayer, T.A. Intraneuronal APP/A beta trafficking and plaque formation in beta-amyloid precursor protein and presenilin-1 transgenic mice. Brain Pathol. 2002, 12, 275–286. [Google Scholar] [CrossRef]
- Moore, B.D.; Chakrabarty, P.; Levites, Y.; Kukar, T.L.; Baine, A.M.; Moroni, T.; Ladd, T.B.; Das, P.; Dickson, D.W.; Golde, T.E. Overlapping profiles of abeta peptides in the Alzheimer’s disease and pathological aging brains. Alzheimers Res. Ther. 2012, 4, 18. [Google Scholar] [CrossRef]
- Wildburger, N.C.; Esparza, T.J.; LeDuc, R.D.; Fellers, R.T.; Thomas, P.M.; Cairns, N.J.; Kelleher, N.L.; Bateman, R.J.; Brody, D.L. Diversity of Amyloid-beta Proteoforms in the Alzheimer’s Disease Brain. Sci. Rep. 2017, 7, 9520. [Google Scholar] [CrossRef] [PubMed]
- Casas, C.; Sergeant, N.; Itier, J.M.; Blanchard, V.; Wirths, O.; van der Kolk, N.; Vingtdeux, V.; van de Steeg, E.; Ret, G.; Canton, T.; et al. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 2004, 165, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Papayannopoulos, I.A.; Styles, J.; Bobin, S.A.; Lin, Y.Y.; Biemann, K.; Iqbal, K. Peptide Compositions of the Cerebrovascular and Senile Plaque Core Amyloid Deposits of Alzheimer’s Disease. Arch. Biochem. Biophys. 1993, 301, 41–52. [Google Scholar] [CrossRef]
- Russo, C.; Salis, S.; Dolcini, V.; Venezia, V.; Song, X.H.; Teller, J.K.; Schettini, G. Identification of amino-terminally and phosphotyrosine-modified carboxy-terminal fragments of the amyloid precursor protein in Alzheimer’s disease and Down’s syndrome brain. Neurobiol. Dis. 2001, 8, 173–180. [Google Scholar] [CrossRef]
- Wirths, O.; Bethge, T.; Marcello, A.; Harmeier, A.; Jawhar, S.; Lucassen, P.J.; Multhaup, G.; Brody, D.L.; Esparza, T.; Ingelsson, M.; et al. Pyroglutamate Abeta pathology in APP/PS1KI mice, sporadic and familial Alzheimer’s disease cases. J. Neural Transm. 2010, 117, 85–96. [Google Scholar] [CrossRef]
- Hornung, K.; Zampar, S.; Engel, N.; Klafki, H.; Liepold, T.; Bayer, T.A.; Wiltfang, J.; Jahn, O.; Wirths, O. N-Terminal Truncated Abeta4-42 Is a Substrate for Neprilysin Degradation in vitro and in vivo. J. Alzheimers Dis. 2019, 67, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A. The AβCs of Aβ-cleaving Proteases. J. Biol. Chem. 2008, 283, 29645–29649. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Yuan, L.; He, W.; Yang, X. Zinc ions regulate opening of tight junction favouring efflux of macromolecules via the GSK3β/snail-mediated pathway. Metallomics 2018, 10, 169–179. [Google Scholar] [CrossRef]
- Turner, A.J.; Nalivaeva, N.N. New Insights into the Roles of Metalloproteinases in Neurodegeneration and Neuroprotection. Int. Rev. Neurobiol. 2007, 82, 113–135. [Google Scholar]
- Lyons, B.; Friedrich, M.G.; Raftery, M.J.; Truscott, R.J.W. Amyloid plaque in the human brain can decompose from Aβ1-40/1-42 by spontaneous non-enzymatic processes. Anal. Chem. 2016, 88, 2675–2684. [Google Scholar] [CrossRef]
- Bolmont, T.; Haiss, F.; Eicke, D.; Radde, R.; Mathis, C.A.; Klunk, W.E.; Kohsaka, S.; Jucker, M.; Calhoun, M.E. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 2008, 28, 4283–4292. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Saul, A.; Sprenger, F.; Bayer, T.A.; Wirths, O. Accelerated tau pathology with synaptic and neuronal loss in a novel triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2564–2573. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
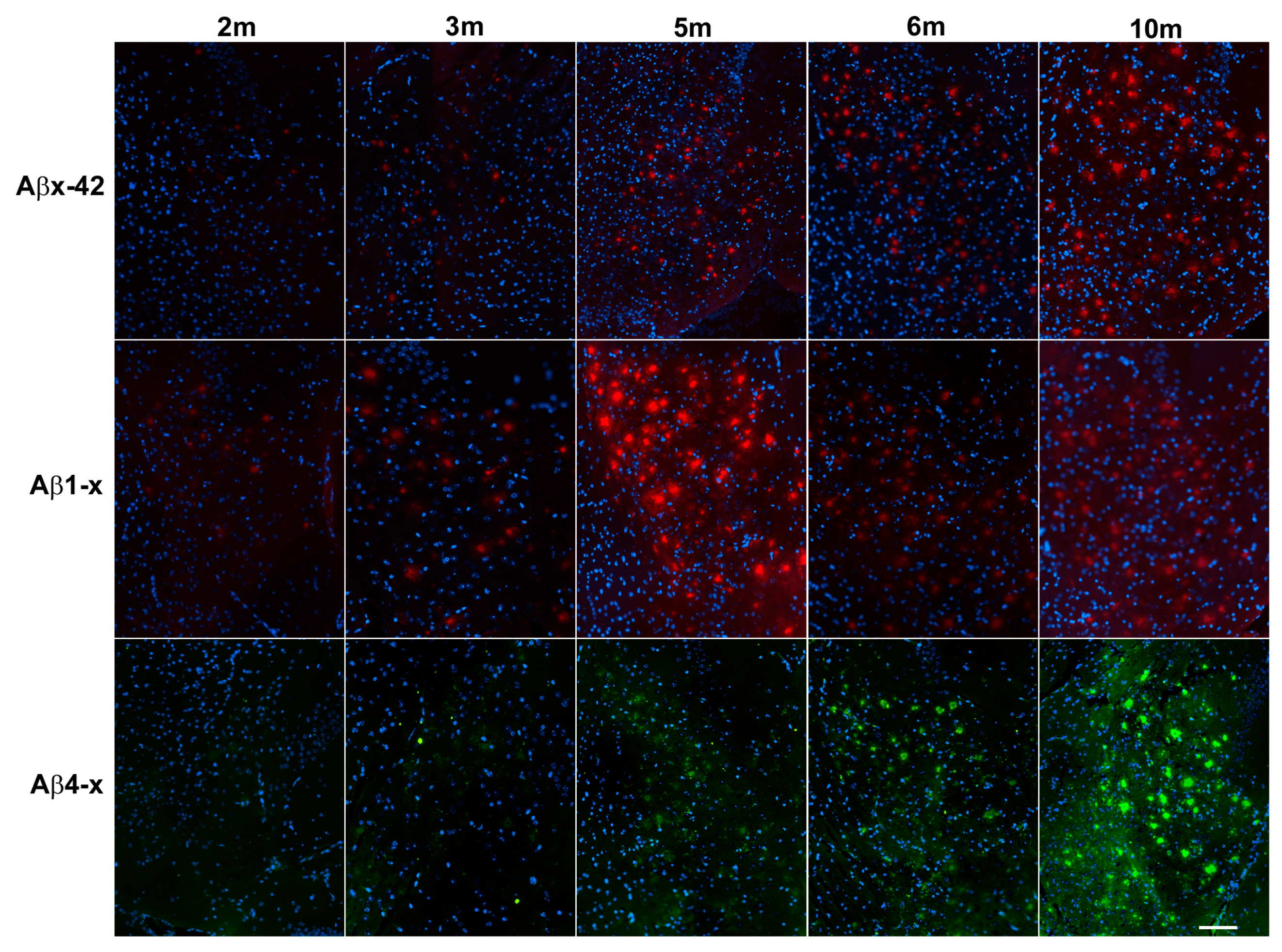{kind=link}
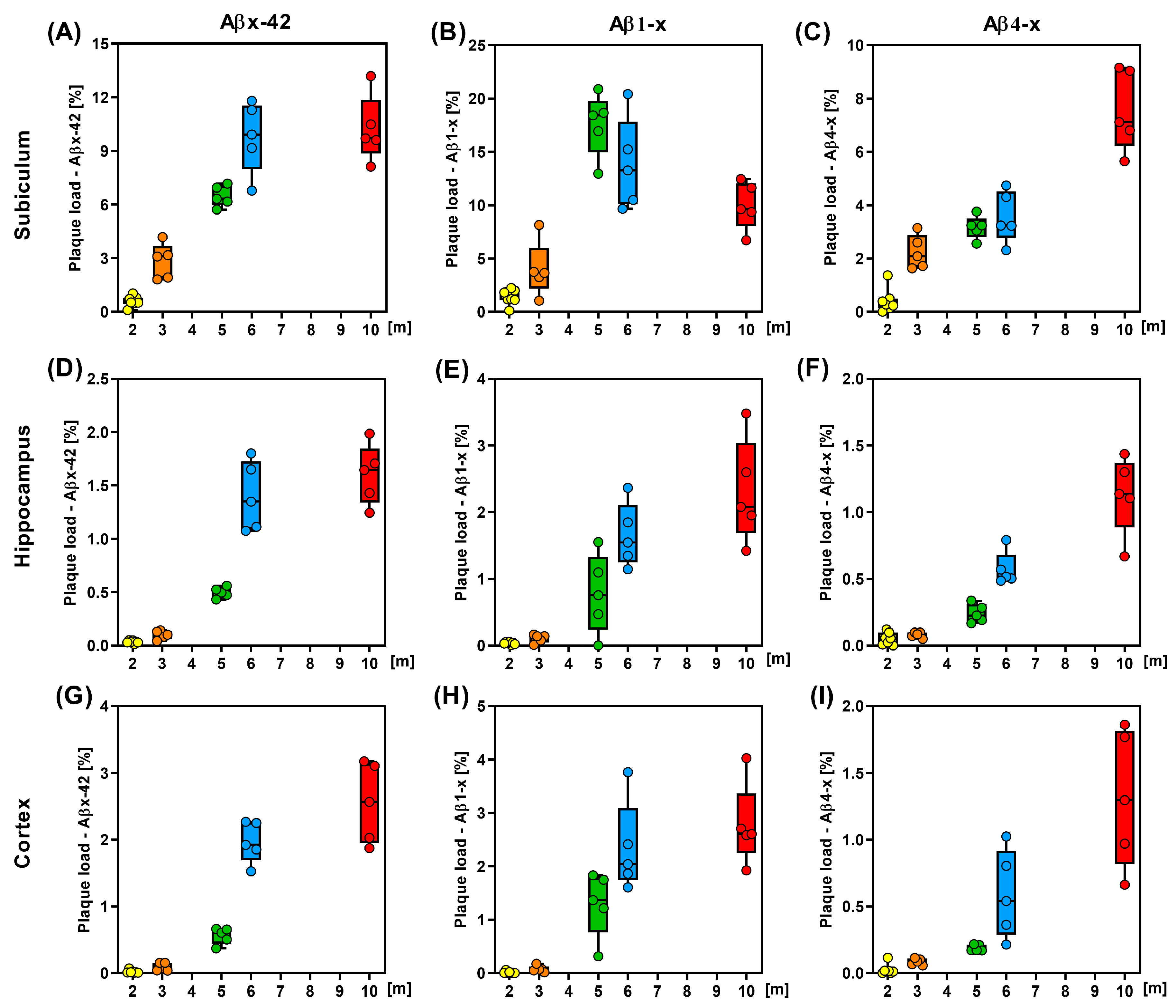{kind=link}
{kind=link}
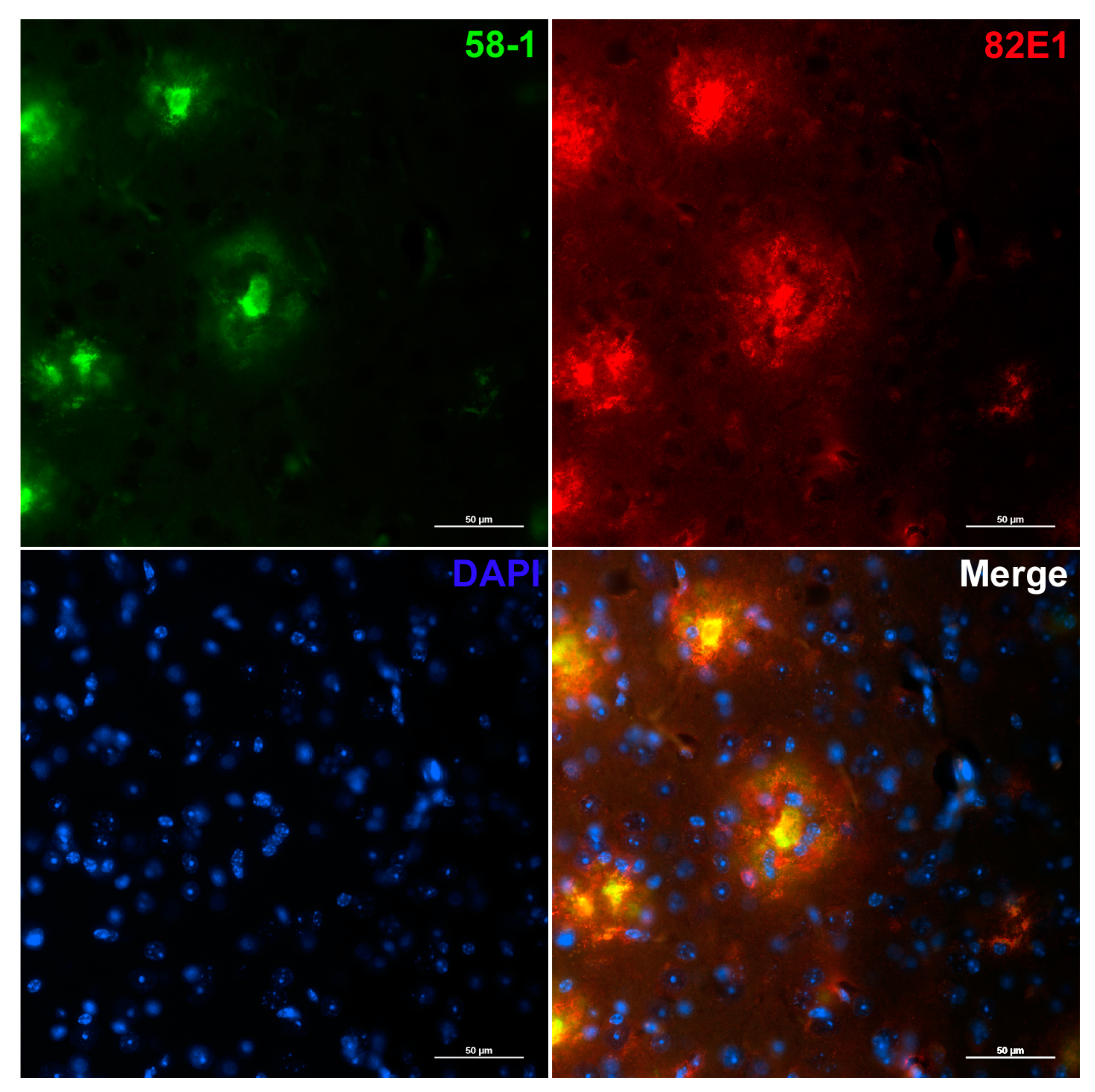{kind=link}
| Brain Area | 2 m | 3 m | 5 m | 6 m | 10 m |
|---|---|---|---|---|---|
| Subiculum | |||||
| Aβx-42 | 0.6 ± 0.29 | 2.83 ± 0.98 | 6.47 ± 0.59 | 9.79 ±1.98 | 10.22 ± 1.86 |
| Aβ1-x | 1.38 ± 0.71 | 3.97 ± 2.58 | 17.57 ± 2.94 | 13.81 ± 4.31 | 9.95 ± 2.24 |
| Aβ4-x | 0.41 ± 0.45 | 2.24 ± 0.64 | 3.17 ± 0.43 | 3.56 ± 0.97 | 7.56 ± 1.51 |
| Hippocampus | |||||
| Aβx-42 | 0.03 ± 0.013 | 0.1 ± 0.04 | 0.5 ± 0.05 | 1.4 ± 0.32 | 1.6 ± 0.28 |
| Aβ1-x | 0.04 ± 0.018 | 0.11 ± 0.06 | 0.76 ± 0.59 | 1.65 ± 0.48 | 2.31 ± 0.78 |
| Aβ4-x | 0.05 ± 0.05 | 0.08 ± 0.02 | 0.24 ± 0.07 | 0.57 ± 0.13 | 1.13 ± 0.29 |
| Cortex | |||||
| Aβx-42 | 0.02 ± 0.02 | 0.09 ± 0.06 | 0.56 ± 0.12 | 1.96 ± 0.31 | 2.55 ± 0.6 |
| Aβ1-x | 0.014 ± 0.02 | 0.08 ± 0.06 | 1.3 ± 0.61 | 2.34 ± 0.85 | 2.77 ± 0.77 |
| Aβ4-x | 0.03 ± 0.04 | 0.09 ± 0.02 | 0.19 ± 0.02 | 0.59 ± 0.33 | 1.31 ± 0.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bader, A.S.; Gnädig, M.-U.; Fricke, M.; Büschgens, L.; Berger, L.J.; Klafki, H.-W.; Meyer, T.; Jahn, O.; Weggen, S.; Wirths, O. Brain Region-Specific Differences in Amyloid-β Plaque Composition in 5XFAD Mice. Life 2023, 13, 1053. https://doi.org/10.3390/life13041053
Bader AS, Gnädig M-U, Fricke M, Büschgens L, Berger LJ, Klafki H-W, Meyer T, Jahn O, Weggen S, Wirths O. Brain Region-Specific Differences in Amyloid-β Plaque Composition in 5XFAD Mice. Life. 2023; 13(4):1053. https://doi.org/10.3390/life13041053
Chicago/Turabian StyleBader, Angelika Sabine, Marius-Uwe Gnädig, Merle Fricke, Luca Büschgens, Lena Josefine Berger, Hans-Wolfgang Klafki, Thomas Meyer, Olaf Jahn, Sascha Weggen, and Oliver Wirths. 2023. "Brain Region-Specific Differences in Amyloid-β Plaque Composition in 5XFAD Mice" Life 13, no. 4: 1053. https://doi.org/10.3390/life13041053