
Integrating Full-Length and Second-Generation Transcriptomics to Reveal Differentially Expressed Genes Associated with the Development of Corydalis yanhusuo Tuber
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, Iso-Seq Library Construction, and Single-Molecular Real-Time Sequencing
2.3. Analysis of the Full-Length Transcriptome
2.4. Gene Functional Annotation
2.5. Illumina cDNA Library Construction and Second-Generation Sequencing
2.6. Analysis of Differentially Expressed Genes
2.7. GO and KEGG Pathway Enrichment Analysis of Differentially Expressed Genes
2.8. Quantitative Real-Time PCR Analysis
3. Results
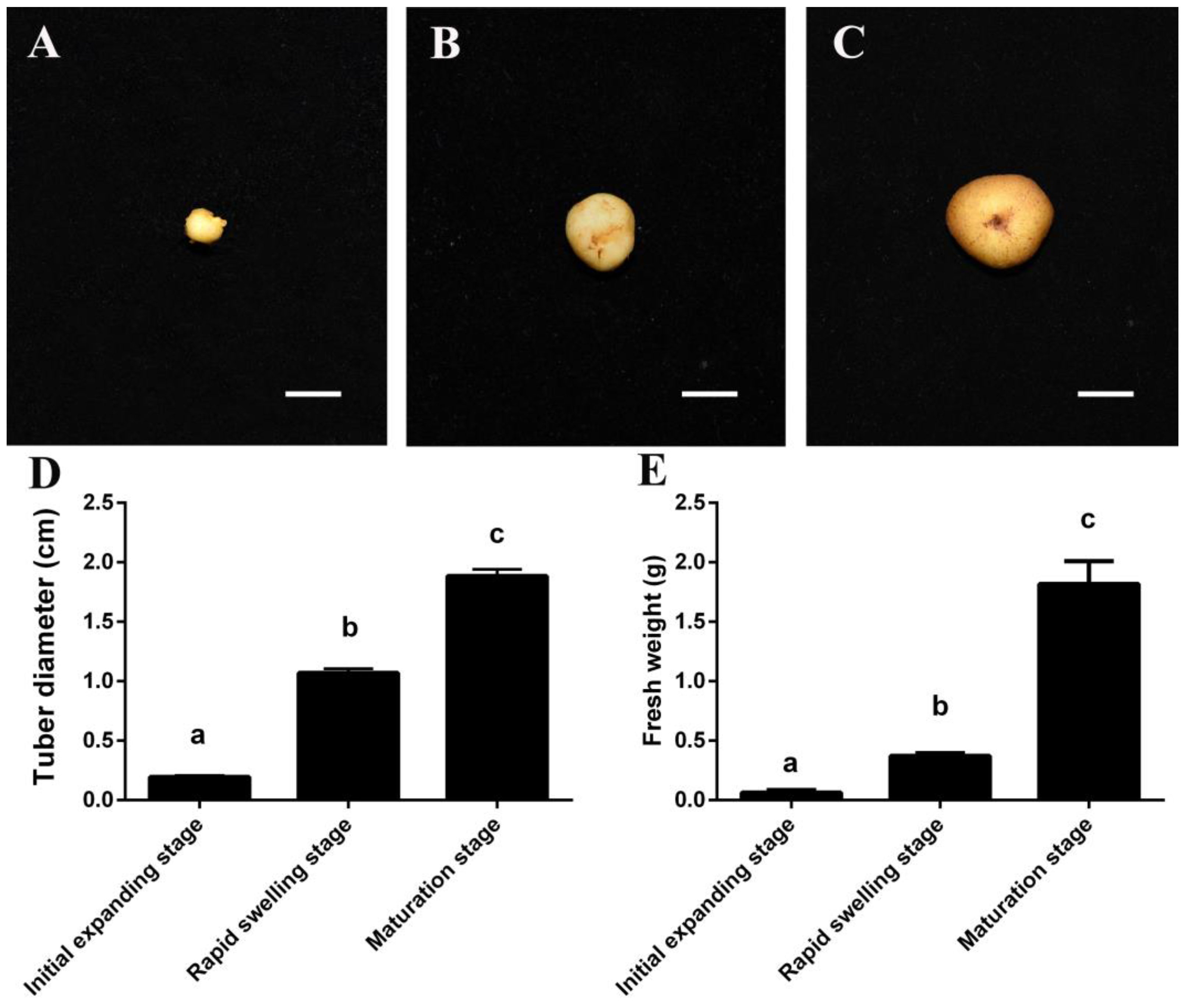
3.1. Morphological Changes during C. yanhusuo Tuber Expanding
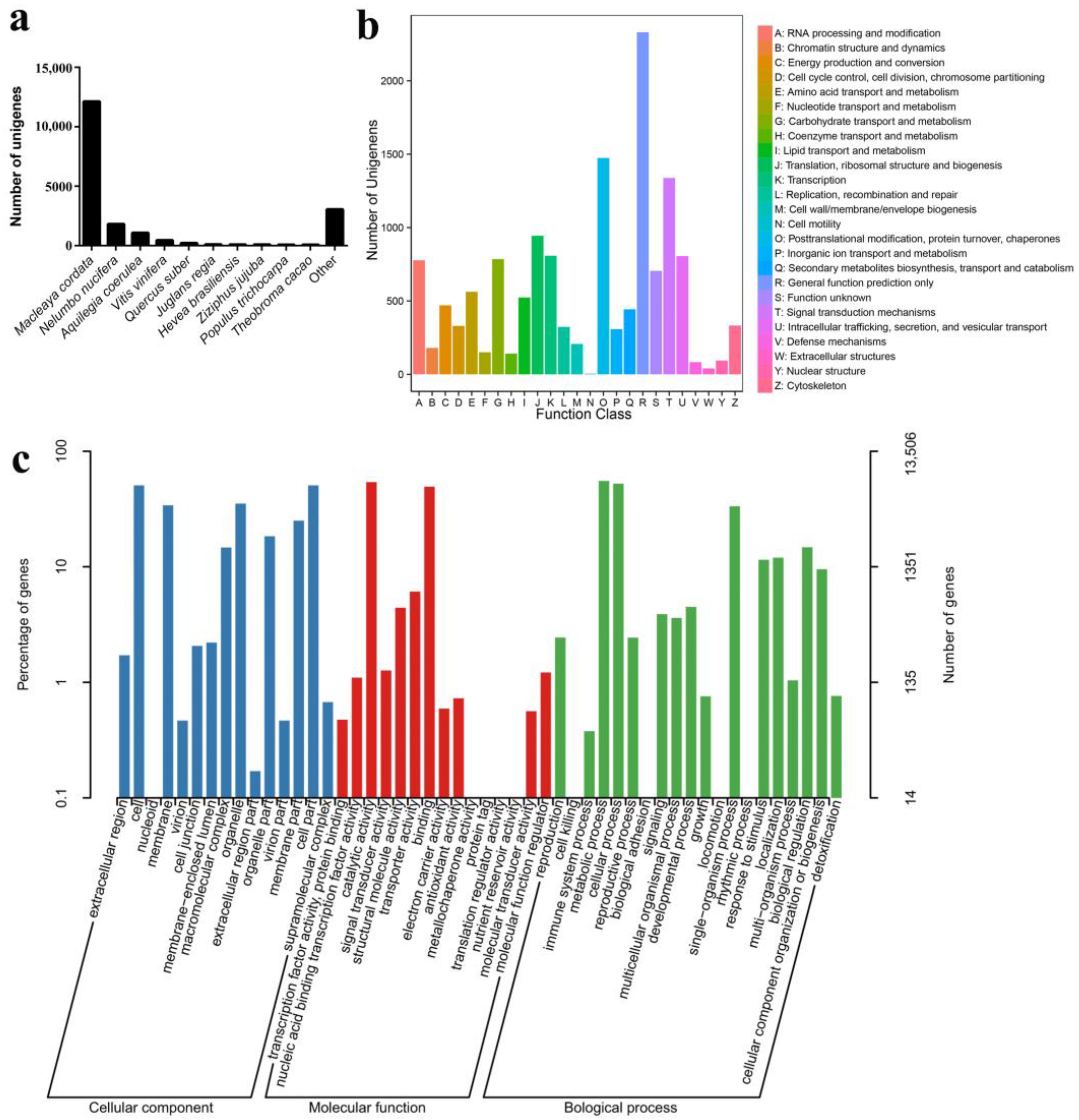
3.2. Full-Length Transcriptome Sequencing and Analysis
3.3. Function Annotation of Unigenes
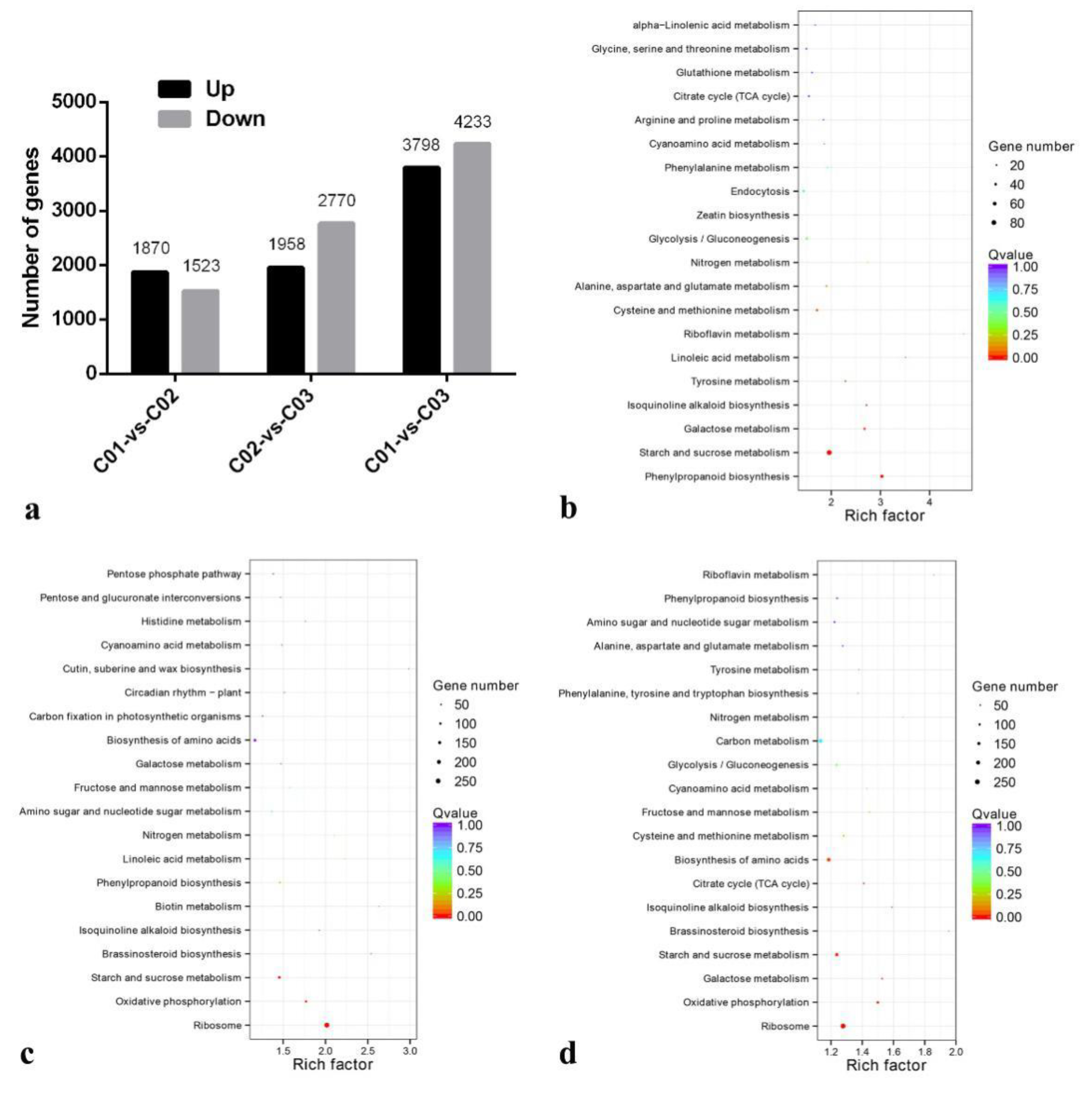
3.4. Differential Expression of Genes among C. yanhusuo Tubers at Different Developmental Stages
3.5. The Function Annotation and Enrichment Analysis of Differential Expression Genes
4. Discussion
4.1. Characterization of the C. yanhusuo Full-Length Transcriptome
4.2. Differential Expression Genes in C. yanhusuo Tuber Swelling
4.3. Differential Expression of Genes Related to Starch and Sucrose Metabolism
4.4. Differential Expression of Genes Related to Hormone Biosynthesis and Signaling Transduction
4.5. Differential Expression of Genes Related to Cell Wall Metabolism
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, X.S.; Xu, J.; Zhang, X.M.; Zhang, T.J.; Chen, C.Q. Research progress on chemical constituents and pharmacological activities of Yuanhu Zhitong Prescription. Chin. Tradit. Herb. Drugs 2015, 46, 1081–1095. [Google Scholar] [CrossRef]
- Li, Q.; Guan, H.; Wang, X.; He, Y.; Sun, H. Fingerprint-efficacy study of the quaternary alkaloids in Corydalis yanhusuo. J. Ethnopharmacol. 2017, 207, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Chinese Pharmacopeia Committee. Phamacopoeia of the People’s Republic of China; Chinese Medical Science Press: Beijing, China, 2020; p. 145. [Google Scholar]
- Xu, Y.; Sun, J.; Li, W.; Zhang, S.; Yang, L.; Teng, Y.; Lv, K.; Liu, Y.; Su, Y.; Zhang, J.; et al. Analgesic effect of the main components of Corydalis yanhusuo (yanhusuo in Chinese) is caused by inhibition of voltage gated sodium channels. J. Ethnopharmacol. 2021, 280, 114457. [Google Scholar] [CrossRef] [PubMed]
- Mathur, V. Social mechanisms of psychophysical pain disparities. J. Pain 2019, 20, S1. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Wang, Z.; Gong, N.; Kweon, T.D.; Vo, B.; Wang, C.; Zhang, X.; Chung, J.Y.; Alachkar, A.; et al. The antinociceptive properties of the Corydalis yanhusuo extract. PLoS ONE 2016, 11, e0162875. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, X.; Fu, S.; Bao, J.L.; Dang, Y.; Huang, M.; Chen, L.; Wang, Y. Dehydrocorydaline inhibits breast cancer cells proliferation by inducing apoptosis in MCF-7 cells. Am. J. Chin. Med. 2012, 40, 177–185. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, W.; Tang, Y.; Bai, W.L.; Yang, F.; Xie, L.P.; Li, X.Z.; Zhou, S.M.; Pan, S.Y.; Chen, Q.; et al. l-tetrahydropalmatine, an active component of Corydalis yanhusuo W.T. Wang, protects against myocardial ischaemia reperfusion injury in rats. PLoS ONE 2012, 7, e38627. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Ryu, Y.B.; Lee, W.S.; Kim, Y.H. Neuraminidase inhibitory activities of quaternary isoquinoline alkaloids from Corydalis turtschaninovii rhizome. Bioorg. Med. Chem. 2014, 22, 6047–6052. [Google Scholar] [CrossRef]
- Chen, C.; Wang, F.Q.; Xiao, W.; Xia, Z.N.; Hu, G.; Wan, J.B.; Yang, F.Q. Effect on platelet aggregation activity: Extracts from 31 traditional chinese medicines with the property of activating blood and resolving stasis. J. Tradit. Chin. Med. 2017, 37, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Tian, M.; Huang, S.M. Advances in phytochemical and modern pharmacological research of Rhizoma corydalis. Pharm. Biol. 2020, 58, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Aksenova, N.P.; Konstantinova, T.N.; Golyanovskaya, S.A.; Sergeeva, L.I.; Romanov, G.A. Hormonal regulation of tuber formation in potato plants. Russ. J. Plant Physiol. 2012, 59, 451–466. [Google Scholar] [CrossRef]
- Ma, H.; Li, C.; Yang, S.; Zhang, Y. Glutamate promotes sweet potato storage root swelling by enhancing starch accumulation. Acta Physiol. Plant 2020, 42, 58. [Google Scholar] [CrossRef]
- Hu, K.; Wei, J. Observation of growth process of Corydalis yanhusuo tuber. J. Anhui Univ. Chin. Med. 2014, 33, 78–80. [Google Scholar]
- Au, K.F.; Underwood, J.G.; Lee, L.; Wong, W.H. Improving PacBio long read accuracy by short read alignment. PLoS ONE 2012, 7, e46679. [Google Scholar] [CrossRef]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Chen, J.; Tang, X.; Ren, C.; Wei, B.; Wu, Y.; Wu, Q.; Pei, J. Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genom. 2018, 19, 548. [Google Scholar] [CrossRef]
- Zhong, F.; Huang, L.; Qi, L.; Ma, Y.; Yan, Z. Full-length transcriptome analysis of Coptis deltoidea and identification of putative genes involved in benzylisoquinoline alkaloids biosynthesis based on combined sequencing platforms. Plant Mol. Biol. 2020, 102, 477–499. [Google Scholar] [CrossRef]
- Zhong, Y.; Hu, K.; Peng, H.; Peng, D. Tuber tissue structure of wild rhizome Corydalis and histochemical localization of alkaloid. J. Anhui Univ. Chin. Med. 2018, 37, 75–78. [Google Scholar] [CrossRef]
- Liao, D.Q.; Wang, P.F.; Jia, C.; Sun, P.; Qi, J.J.; Zhou, L.L.; Li, X.E. Identification and developmental expression profiling of putative alkaloid biosynthetic genes in Corydalis yanhusuo bulbs. Sci. Rep. 2016, 6, 19460. [Google Scholar] [CrossRef]
- Xu, D.; Lin, H.; Tang, Y.; Huang, L.; Xu, J.; Nian, S.; Zhao, Y. Integration of full-length transcriptomics and targeted metabolomics to identify benzylisoquinoline alkaloid biosynthetic genes in Corydalis yanhusuo. Hortic. Res. 2021, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.H.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein databases search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.; He, F.C. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. Available online: http://genomebiology.com/2004/5/2/R7 (accessed on 10 July 2020). [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.Z.; Lopez, R.; Magrane, M.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Mao, X.; Tao, C.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Liu, Y.P.; Zhang, L.Y.; Yue, D.M.; Qi, D.Y.; Liu, G.J.; Liu, S. Levo-tetrahydropalmatine attenuates bone cancer pain by inhibiting microglial cells activation. Mediat. Inflamm. 2015, 2015, 752512. [Google Scholar] [CrossRef]
- Yang, L.F.; Jin, Y.H.; Huang, W.; Sun, Q.; Liu, F.; Huang, X.Z. Full-length transcriptome sequences of ephemeral plant Arabidopsis pumila provides insight into gene expression dynamics during continuous salt stress. BMC Genom. 2018, 19, 717. [Google Scholar] [CrossRef]
- Zhang, H.L.; Liu, Z.; Hu, A.S.; Wu, H.W.; Zhu, J.F.; Wang, F.Z.; Cao, P.P.; Yang, X.Y.; Zhang, H.X. Full-Length transcriptome analysis of the halophyte Nitraria sibirica Pall. Genes 2022, 13, 661. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Yang, J.L.; Hao, B.; Lu, C.; Qian, Z.L.; Li, Y.; Ye, S.; Tang, J.R.; Chen, M.; Long, G.Q.; et al. Comparative transcriptome and metabolome analyses provide new insights into the molecular mechanisms underlying taproot thickening in Panax notoginseng. BMC Plant Biol. 2019, 19, 451. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.L.; Zhong, Q.; Tian, M.Y.; Han, R.; Ren, Y.J. Comparative transcriptome analysis reveals differentially expressed genes associated with the development of Jerusalem artichoke tuber (Helianthus tuberosus L.). Ind. Crop. Prod. 2020, 151, 112455. [Google Scholar] [CrossRef]
- Abelenda, J.A.; Navarro, C.; Prat, S. From the model to the crop: Genes controlling tuber formation in potato. Curr. Opin. Biotechnol. 2011, 22, 287–292. [Google Scholar] [CrossRef]
- Yang, M.; Zhu, L.P.; Pan, C.; Xu, L.; Liu, Y.; Ke, W.; Yang, P. Transcriptomic analysis of the regulation of rhizome formation in temperate and tropical lotus (Nelumbo nucifera). Sci. Rep. 2015, 5, 13059. [Google Scholar] [CrossRef]
- Wang, X.; Chang, L.; Tong, Z.; Wang, D.; Yin, Q.; Wang, D.; Jin, X.; Yang, Q.; Wang, L.; Sun, Y.; et al. Proteomics profiling reveals carbohydrate metabolic enzymes and 14-3-3 proteins play important roles for starch accumulation during cassava root tuberization. Sci. Rep. 2016, 6, 19643. [Google Scholar] [CrossRef]
- Yang, J.; An, D.; Zhang, P. Expression profiling of cassava storage roots reveals an active process of glycolysis/gluconeogenesis. J. Integr. Plant Biol. 2011, 53, 193–211. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.; Willmitzer, L. Molecular and biochemical triggers of potato tuber development. Plant Physiol. 2001, 127, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.K.H.; McCouch, S. Getting to the roots of it: Genetic and hormonal control of root architecture. Front. Plant Sci. 2013, 4, 186. [Google Scholar] [CrossRef] [PubMed]
- Ljung, K. Auxin metabolism and homeostasis during plant development. Development 2013, 140, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Sojikul, P.; Saithong, T.; Kalapanulak, S.; Pisuttinusart, N.; Limsirichaikul, S.; Tanaka, M.; Utsumi, Y.; Sakurai, T.; Seki, M.; Narangajavana, J. Genome-wide analysis reveals phytohormone action during cassava storage root initiation. Plant Mol. Biol. 2015, 88, 531–543. [Google Scholar] [CrossRef]
- Clouse, S.D. Brassinosteroid signal transduction: From receptor kinase activation to transcriptional networks regulating plant development. Plant Cell 2011, 23, 1219–1230. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Bai, M.Y.; Oh, E.; Zhu, J.Y. Brassinosteroid signaling network and regulation of photomorphogenesis. Annu. Rev. Genet. 2012, 46, 701–724. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Y.; Xu, L.; Han, Z.; Yin, H.; Ai, S. Photoelectrochemical apta-biosensor for zeatin detection based on graphene quantum dots improved photoactivity of graphite-like carbon nitride and streptavidin induced signal inhibition. Sens. Actuators B-Chem. 2018, 257, 237–244. [Google Scholar] [CrossRef]
- Wen, J.; Zhao, G.; Yang, Y.; Chen, R.; Cai, H. Analysis of comparative transcriptome in the development of Tetrastigma hemsleyanum tuber. Mol. Plant Breed. 2019, 17, 5660–5667. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Corinne, R.; Thomas, B.; Vincent, L.; Bernard, H. The carbohydrate-active enzymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Fabiana, D.G.; Bermúdez, L.; Silvestre, L.B.; Pereira, D.S.A.; Paula, E.; Demarco, D.; Alseekh, S.; Insani, M.; Buckeridge, M.; Almeida, J.; et al. Galacturonosyltransferase 4 silencing alters pectin composition and carbon partitioning in tomato. J. Exp. Bot. 2013, 64, 2449–2466. [Google Scholar] [CrossRef]
- Mohnen, D. Pectin structure and biosynthesis. Curr. Opin. Plant Biol. 2008, 11, 266–277. [Google Scholar] [CrossRef]
- Wang, L.; Wang, W.; Wang, Y.Q.; Liu, Y.Y.; Wang, J.X.; Zhang, X.Q.; Ye, D.; Chen, L.Q. Arabidopsis galacturonosyltransferase GAUT13 and GAUT14 have redundant functions in pollen tube growth. Mol. Plant 2013, 6, 1131–1148. [Google Scholar] [CrossRef] [PubMed]
- Amoah, J.N.; Ko, C.S.; Yoon, J.S.; Weon, S.Y. Effect of drought acclimation on oxidative stress and transcript expression in wheat (Triticum aestivum L.). J. Plant Interact. 2019, 14, 492–505. [Google Scholar] [CrossRef]
- Martínez-Cortés, T.; Pomar, F.; Espiñeira, J.M.; Merino, F.; Novo-Uzal, E. Purification and kinetic characterization of two peroxidases of Selaginella martensii Spring. involved in lignification. Plant Physiol. Biochem. 2012, 52, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Passardi, F.; Tognolli, M.; De Meyer, M.; Penel, C.; Dunand, C. Two cell wall associated peroxidases from Arabidopsis influence root elongation. Planta 2006, 223, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Si, J.J.; Miao, Y.; Peng, Y.; Wang, L.; Cai, X. Comparative proteomics of Euphorbia kansui Liou milky sap at two different developmental stages. Plant Physiol. Biochem. 2014, 79, 60–65. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCS Number | Read Bases of CCS | Mean Read Length of CCS | FLNC Reads Number | Consensus Isoforms Number | Average Consensus Isoforms Length | Polished High-Quality Isoforms Number |
|---|---|---|---|---|---|---|
| 136,469 | 288,681,328 | 2115 | 90,496 | 36,347 | 1795 | 35,513 |
| Database | Number of Unigenes | 300 ≤ Length < 1000 | Length ≥ 1000 |
|---|---|---|---|
| Nr | 19,252 | 3055 | 16,103 |
| Swiss-prot | 14,427 | 2090 | 12,278 |
| Pfam | 15,860 | 2054 | 13,798 |
| GO | 13,506 | 2165 | 11,301 |
| COG | 8172 | 945 | 7219 |
| KOG | 12,663 | 1827 | 10,797 |
| KEGG | 9324 | 1520 | 7764 |
| All | 19,341 | 3095 | 16,143 |
| Sample | Read Number | Base Number | GC Content (%) | Q30 Percentage (%) |
|---|---|---|---|---|
| C01-1 | 20,187,733 | 6,047,288,820 | 43.90 | 93.06 |
| C01-2 | 22,295,014 | 6,674,832,134 | 43.90 | 93.20 |
| C01-3 | 24,651,686 | 7,382,052,386 | 43.96 | 93.09 |
| C02-1 | 19,850,543 | 5,946,722,130 | 43.43 | 92.87 |
| C02-2 | 19,876,319 | 5,954,700,720 | 43.44 | 93.49 |
| C02-3 | 20,915,770 | 6,264,762,952 | 43.43 | 93.28 |
| C03-1 | 20,088,356 | 6,020,004,766 | 43.44 | 92.72 |
| C03-2 | 21,393,807 | 6,411,557,256 | 43.38 | 93.24 |
| C03-3 | 21,930,778 | 6,570,423,706 | 43.43 | 92.34 |
| Sample Pairs | Annotated | COG | GO | KEGG | KOG | Pfam | Swiss-Prot | eggNOG | Nr |
|---|---|---|---|---|---|---|---|---|---|
| C01 vs. C02 | 3223 | 1511 | 2271 | 1321 | 1805 | 2748 | 2539 | 3134 | 3208 |
| C01 vs. C03 | 4453 | 2122 | 3240 | 2168 | 2743 | 3733 | 3539 | 4322 | 4430 |
| C02 vs. C03 | 7649 | 3487 | 5519 | 3634 | 821 | 6466 | 5938 | 7441 | 7620 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Wang, L.; Zhou, Y.; Wang, Q.; Wang, F.; Li, Y. Integrating Full-Length and Second-Generation Transcriptomics to Reveal Differentially Expressed Genes Associated with the Development of Corydalis yanhusuo Tuber. Life 2023, 13, 2207. https://doi.org/10.3390/life13112207
Zhao X, Wang L, Zhou Y, Wang Q, Wang F, Li Y. Integrating Full-Length and Second-Generation Transcriptomics to Reveal Differentially Expressed Genes Associated with the Development of Corydalis yanhusuo Tuber. Life. 2023; 13(11):2207. https://doi.org/10.3390/life13112207
Chicago/Turabian StyleZhao, Xueyan, Li Wang, Yafu Zhou, Qing Wang, Fangyuan Wang, and Yan Li. 2023. "Integrating Full-Length and Second-Generation Transcriptomics to Reveal Differentially Expressed Genes Associated with the Development of Corydalis yanhusuo Tuber" Life 13, no. 11: 2207. https://doi.org/10.3390/life13112207