
Transcriptome Analyses Indicate Significant Association of Increased Non-Additive and Allele-Specific Gene Expression with Hybrid Weakness in Rice (Oryza sativa L.)

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Calculation of Mid-Parent Heterosis (MPH) Index
2.3. RNA Isolation, Library Construction, and Sequencing
2.4. Analysis of RNA-Seq Data
2.5. Identification of NAE Genes
2.6. Identification of ASE Genes
2.6.1. Whole Genome Re-Sequencing of Parental Lines
2.6.2. DNA Sequencing Read Alignment and Variant Calling, Filtering, and Annotation
2.6.3. Detection of Exonic SNP and InDel Markers
2.6.4. Detection of Expressed SNP and InDel Markers
2.6.5. Analysis of Allelic Imbalance and Detection of ASE Genes
3. Results
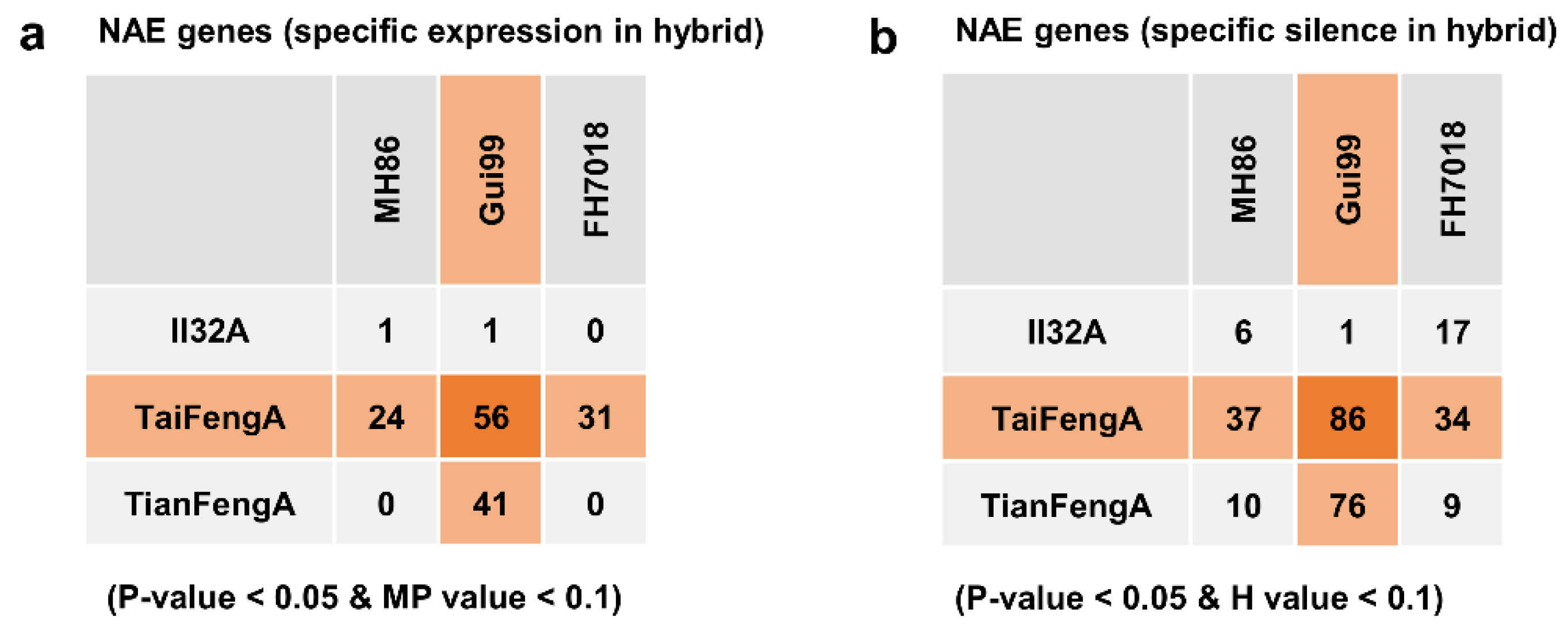
3.1. NAE Was Increased in the MPHW Hybrids
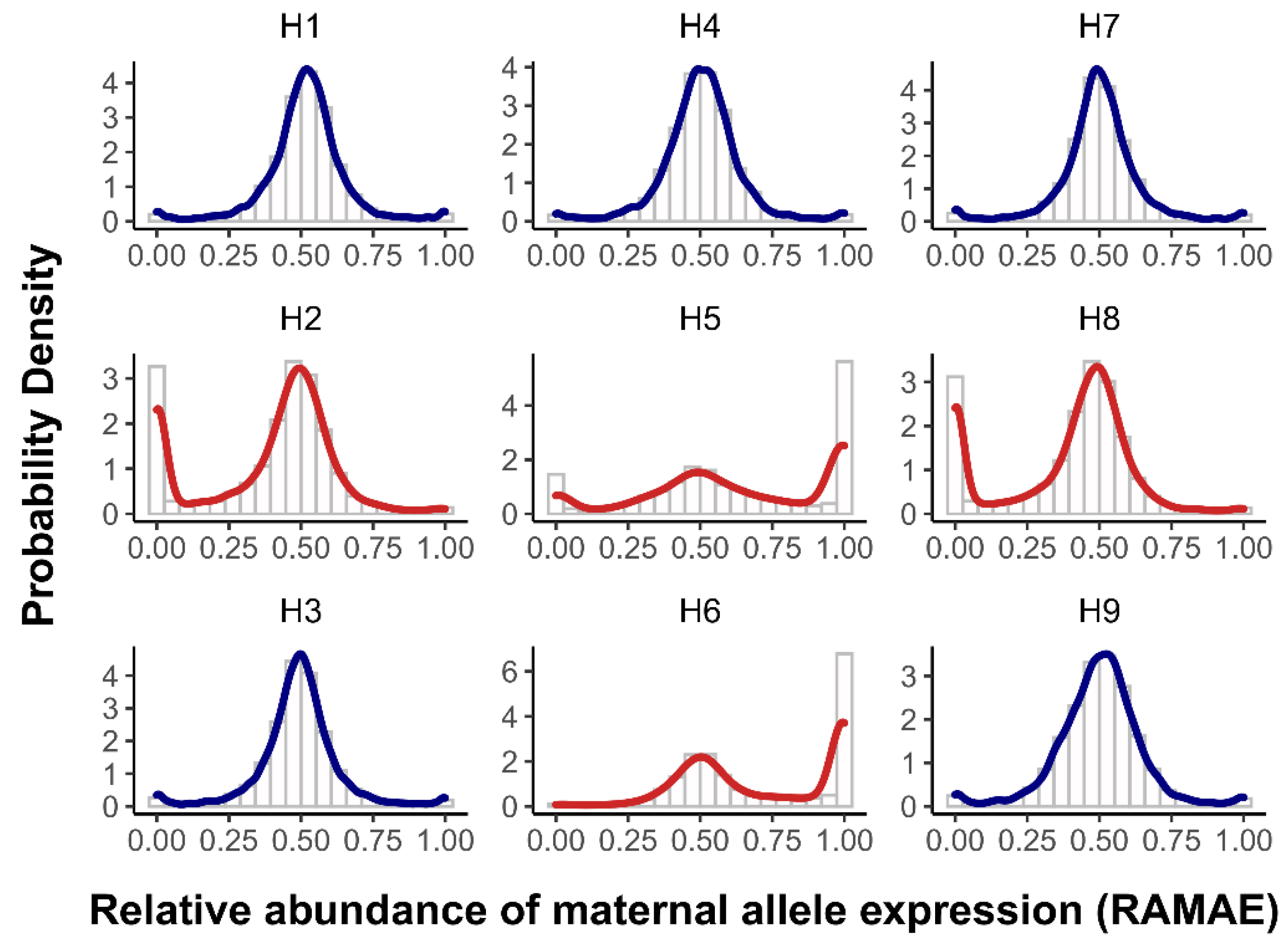
3.2. ASE Was Increased in the MPHW Hybrids
3.3. The Enhancements of NAE and ASE in the MPHW Hybrids Were Correlated
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheng, S.; Zhuang, J.; Fan, Y.; Du, J.; Cao, L. Progress in Research and Development on Hybrid Rice: A Super-Domesticate in China. Ann. Bot. 2007, 100, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Springer, N.M.; Stupar, R.M. Allelic Variation and Heterosis in Maize: How Do Two Halves Make More than a Whole? Genome Res. 2007, 17, 264–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, C.; Wang, F.; Sun, B.; Liu, W.; Li, J.; Deng, R.; Liu, D.; Liu, Z.; Zhu, M.; Liao, Y.; et al. Genetic and Cytological Analysis of a Novel Type of Low Temperature-Dependent Intrasubspecific Hybrid Weakness in Rice. PLoS ONE 2013, 8, e73886. [Google Scholar] [CrossRef] [PubMed]
- Yoneya, Y.; Wakabayashi, T.; Kato, K. The Temperature Sensitive Hybrid Breakdown 1 Induces Low Temperature-Dependent Intrasubspecific Hybrid Breakdown in Rice. Breed. Sci. 2021, 71, 20129. [Google Scholar] [CrossRef] [PubMed]
- Shiragaki, K.; Furukawa, H.; Yokoi, S.; Tezuka, T. Temperature-Dependent Sugar Accumulation in Interspecific Capsicum F1 Plants Showing Hybrid Weakness. J. Plant Res. 2021, 134, 1199–1211. [Google Scholar] [CrossRef]
- He, G.; Zhu, X.; Elling, A.A.; Chen, L.; Wang, X.; Guo, L.; Liang, M.; He, H.; Zhang, H.; Chen, F. Global Epigenetic and Transcriptional Trends among Two Rice Subspecies and Their Reciprocal Hybrids. Plant Cell 2010, 22, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Yang, S.; Gong, J.; Zhao, Y.; Feng, Q.; Gong, H.; Li, W.; Zhan, Q.; Cheng, B.; Xia, J. Genomic Analysis of Hybrid Rice Varieties Reveals Numerous Superior Alleles That Contribute to Heterosis. Nat. Commun. 2015, 6, 6258. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Wang, X.; Ren, D.; Huang, H.; Xu, M.; He, G.; Deng, X.W. Genomic Architecture of Biomass Heterosis in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, 8101–8106. [Google Scholar] [CrossRef] [Green Version]
- Seymour, D.K.; Chae, E.; Grimm, D.G.; Pizarro, C.M.; Habring-Müller, A.; Vasseur, F.; Rakitsch, B.; Borgwardt, K.M.; Koenig, D.; Weigel, D. Genetic Architecture of Nonadditive Inheritance in Arabidopsis Thaliana Hybrids. Proc. Natl. Acad. Sci. USA 2016, 113, E7317–E7326. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.; Xing, F.; Xu, C.; Zhang, Q.; Che, J.; Wang, X.; Song, J.; Li, X.; Xiao, J.; Chen, L. Patterns of Genome-Wide Allele-Specific Expression in Hybrid Rice and the Implications on the Genetic Basis of Heterosis. Proc. Natl. Acad. Sci. USA 2019, 116, 5653–5658. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Xing, F.; Jia, Q.; Zhang, Q.; Hu, T.; Wu, B.; Shao, L.; Zhao, Y.; Zhang, Q.; Zhou, D. Parental Variation in CHG Methylation Is Associated with Allelic-Specific Expression in Elite Hybrid Rice. Plant Physiol. 2021, 186, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, S.; Gong, J.; Zhao, Q.; Feng, Q.; Zhan, Q.; Zhao, Y.; Li, W.; Cheng, B.; Xia, J. Genomic Architecture of Heterosis for Yield Traits in Rice. Nature 2016, 537, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Shan, J.; Zhu, M.; Shi, M.; Gao, J.; Lin, H. Genetic and Physiological Analysis of a Novel Type of Interspecific Hybrid Weakness in Rice. Mol. Plant 2013, 6, 716–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Lin, Y.-S.; Shen, J.-B.; Shan, J.-X.; Qi, P.; Shi, M.; Zhu, M.-Z.; Huang, X.-H.; Feng, Q.; et al. A Two-Locus Interaction Causes Interspecific Hybrid Weakness in Rice. Nat. Commun. 2014, 5, 3357. [Google Scholar] [CrossRef] [Green Version]
- Shiragaki, K.; Iizuka, T.; Ichitani, K.; Kuboyama, T.; Morikawa, T.; Oda, M.; Tezuka, T. HWA1- and HWA2-Mediated Hybrid Weakness in Rice Involves Cell Death, Reactive Oxygen Species Accumulation, and Disease Resistance-Related Gene Upregulation. Plants 2019, 8, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichitani, K.; Namigoshi, K.; Sato, M.; Taura, S.; Aoki, M.; Matsumoto, Y.; Saitou, T.; Marubashi, W.; Kuboyama, T. Fine Mapping and Allelic Dosage Effect of Hwc1, a Complementary Hybrid Weakness Gene in Rice. Appl. Genet. 2007, 114, 1407–1415. [Google Scholar] [CrossRef]
- Nadir, S.; Li, W.; Zhu, Q.; Khan, S.; Zhang, X.; Zhang, H.; Wei, Z.; Li, M.-T.; Zhou, L.; Li, C.; et al. A Novel Discovery of a Long Terminal Repeat Retrotransposon-Induced Hybrid Weakness in Rice. J. Exp. Bot. 2019, 70, 1197–1207. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zheng, Y.; Cai, Q.; Liao, C.; Mao, X.; Xie, H.; Zhu, Y.; Lian, L.; Luo, X.; Xie, H. Population Structure and Association Analysis of Yield and Grain Quality Traits in Hybrid Rice Primal Parental Lines. Euphytica 2016, 212, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Copeland, L.O.; Elias, S.G.; Kelly, J.D. Relationship between Genetic Distance and Heterosis for Yield and Morphological Traits in Winter Canola (Brassica Napus L.). Theoret. Appl. Genet. 1995, 91, 118–121. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 9 August 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate Paired Shotgun Read Merging via Overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza Sativa Nipponbare Reference Genome Using next Generation Sequence and Optical Map Data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-Level Expression Analysis of RNA-Seq Experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Holik, A.Z.; Law, C.W.; Liu, R.; Wang, Z.; Wang, W.; Ahn, J.; Asselin-Labat, M.; Smyth, G.K.; Ritchie, M.E. RNA-Seq Mixology: Designing Realistic Control Experiments to Compare Protocols and Analysis Methods. Nucleic Acids Res 2017, 45, e30. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013. [Google Scholar] [CrossRef]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast Duplicate Marking and Structural Variant Read Extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deonovic, B.; Wang, Y.; Weirather, J.; Wang, X.-J.; Au, K.F. IDP-ASE: Haplotyping and Quantifying Allele-Specific Expression at the Gene and Gene Isoform Level by Hybrid Sequencing. Nucleic Acids Res. 2017, 45, e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid | Type | HC Genes a | NAE Genes b | ||||
|---|---|---|---|---|---|---|---|
| H > MP | % | H < MP | % | Total % | |||
| H1 | MPH | 19,962 | 24 | 0.12 | 433 | 2.17 | 2.29 |
| H2 | MPHW | 20,257 | 202 | 1.00 | 1823 | 9.00 | 10.00 |
| H3 | MPH | 20,109 | 30 | 0.15 | 726 | 3.61 | 3.76 |
| H4 | MPH | 19,574 | 9 | 0.05 | 227 | 1.16 | 1.21 |
| H5 | MPHW | 20,041 | 216 | 1.08 | 659 | 3.29 | 4.37 |
| H6 | MPHW | 20,048 | 156 | 0.78 | 439 | 2.20 | 2.98 |
| H7 | MPH | 20,490 | 64 | 0.31 | 1261 | 6.15 | 6.46 |
| H8 | MPHW | 20,631 | 167 | 0.81 | 804 | 3.90 | 4.71 |
| H9 | MPH | 20,601 | 25 | 0.12 | 591 | 2.87 | 2.99 |
| Mean ± SD | MPH | 0.15 ± 0.10 | 3.19 ± 1.88 | ||||
| MPHW | 0.92 ± 0.15 | 4.60 ± 3.02 | |||||
| p-value | 5.77 × 10−5 | 0.428 | |||||
| Crosses | SNP | InDel | Total Genes | ||
|---|---|---|---|---|---|
| Variants | Genes | Variants | Genes | ||
| H1 | 32,325 | 7122 | 3312 | 2195 | 7366 |
| H2 | 25,535 | 5570 | 2596 | 1715 | 5773 |
| H3 | 27,753 | 6433 | 2847 | 1875 | 6648 |
| H4 | 34,962 | 7111 | 3479 | 2210 | 7308 |
| H5 | 32,953 | 6472 | 3363 | 2108 | 6657 |
| H6 | 33,052 | 6807 | 3380 | 2148 | 7015 |
| H7 | 38,979 | 7557 | 3932 | 2474 | 7788 |
| H8 | 32,465 | 6385 | 3200 | 2043 | 6574 |
| H9 | 35,434 | 7233 | 3543 | 2259 | 7449 |
| Crosses | Het. Genes | Maternal-ASE Genes | Paternal-ASE Genes | ASE Genes | ||
|---|---|---|---|---|---|---|
| Number | Rate | Number | Rate | |||
| H1 | 7366 | 205 | 2.78% | 188 | 2.55% | 393 |
| H2 | 5773 | 116 | 2.01% | 1233 | 21.36% | 1349 |
| H3 | 6648 | 165 | 2.48% | 213 | 3.20% | 378 |
| H4 | 7308 | 178 | 2.44% | 173 | 2.37% | 351 |
| H5 | 6657 | 2367 | 35.56% | 693 | 10.41% | 3060 |
| H6 | 7015 | 2984 | 42.54% | 111 | 1.58% | 3095 |
| H7 | 7788 | 194 | 2.49% | 232 | 2.98% | 426 |
| H8 | 6574 | 141 | 2.14% | 1348 | 20.51% | 1489 |
| H9 | 7449 | 182 | 2.44% | 222 | 2.98% | 404 |
| Crosses | ASE | Non-Additive Expressed | p-Value | |
|---|---|---|---|---|
| TRUE | FALSE | |||
| H1 | TRUE | 14 | 317 | 0.000526 |
| FALSE | 98 | 6836 | ||
| H2 | TRUE | 140 | 1157 | 1.56 × 10−8 |
| FALSE | 262 | 4122 | ||
| H3 | TRUE | 6 | 316 | 0.585 |
| FALSE | 161 | 6056 | ||
| H4 | TRUE | 5 | 302 | 0.029 |
| FALSE | 36 | 6877 | ||
| H5 | TRUE | 119 | 2814 | 7.10 × 10−8 |
| FALSE | 66 | 3557 | ||
| H6 | TRUE | 100 | 2855 | 1.10 × 10−10 |
| FALSE | 44 | 3897 | ||
| H7 | TRUE | 30 | 322 | 0.0008 |
| FALSE | 313 | 6986 | ||
| H8 | TRUE | 86 | 1322 | 1.20 × 10−10 |
| FALSE | 122 | 4917 | ||
| H9 | TRUE | 14 | 330 | 0.0023 |
| FALSE | 112 | 6857 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Xia, J.; Huang, L.; Lin, Q.; Cai, Q.; Xie, H.; He, W.; Wei, Y.; Xie, H.; Tang, W.; et al. Transcriptome Analyses Indicate Significant Association of Increased Non-Additive and Allele-Specific Gene Expression with Hybrid Weakness in Rice (Oryza sativa L.). Life 2022, 12, 1278. https://doi.org/10.3390/life12081278
Wang Y, Xia J, Huang L, Lin Q, Cai Q, Xie H, He W, Wei Y, Xie H, Tang W, et al. Transcriptome Analyses Indicate Significant Association of Increased Non-Additive and Allele-Specific Gene Expression with Hybrid Weakness in Rice (Oryza sativa L.). Life. 2022; 12(8):1278. https://doi.org/10.3390/life12081278
Chicago/Turabian StyleWang, Yingheng, Jing Xia, Likun Huang, Qiang Lin, Qiuhua Cai, Hongguang Xie, Wei He, Yidong Wei, Huaan Xie, Weiqi Tang, and et al. 2022. "Transcriptome Analyses Indicate Significant Association of Increased Non-Additive and Allele-Specific Gene Expression with Hybrid Weakness in Rice (Oryza sativa L.)" Life 12, no. 8: 1278. https://doi.org/10.3390/life12081278